2. Materials and Methods
2.1. Animals
All experiments were conducted with approval of the Institutional Animal Care and Use Committee at the Indiana University School of Medicine and performed in American Association for Laboratory Animal Care (AALAC)-approved facilities. Male Sprague-Dawley rats were purchased from Envigo (Indianapolis, IN, USA) and received food and water ad libitum. After a period of acclimation, the rats were randomly divided into either control (saline) or treated (METH) groups. Methamphetamine (METH) HCl was from Sigma-Aldrich (St. Louis, MO, USA, Catalog #M-8750). A 10 mg/mL METH solution was prepared the day of injections in sterile-filtered 0.9% w/v sodium chloride (saline). Rats received intraperitoneal (i.p.) injections of either saline or 10 mg/mL METH at a volume equal to 1 mL/kg (10 mg METH/kg body mass). Injections were given at 2 h intervals over 8 h for a total of 4 injections. Rats were housed individually during the injections in plastic shoebox-style containers with a wire lid. Food and water were available ad libitum. A plastic grate was placed on top of the bedding inside the container to prevent aspiration of bedding material. Body temperature was monitored at 15–30 min intervals throughout the injection period and for 1–2 h after the final injection via a subcutaneously-implanted temperature transponder (Bio Medic Data Systems, Inc., Seaford, DE, USA, Catalog # IPTT 300). If a rat’s body temperature exceeded 40 °C, measures were taken (oscillating fan, ice packs on cage lid) to prevent hyperthermia. At either 2 h or 3 days after the last i.p. injection, each rat was deeply anesthetized with an i.p. injection of a ketamine (Henry Schein, Indianapolis, IN USA, KetathesiaTM 100 mg/mL)/xylazine (Akorn Pharmaceuticals, Decatur, IL, USA, AnaSed® Injection 20 mg/mL) 70%/30% v/v mix. Once a rat had no response to a toe-pinch, the chest cavity was opened and 0.35 mL heparin (Sagent Pharmaceuticals, Inc, Schaumburg, IL, USA, 1000 USP units/mL) was injected into the left ventricle, followed by perfusion with ice-cold phosphate-buffered saline (PBS). Blood and PBS exited the body through an incision made in the right atrium. Perfusion continued for 5 min at a flow rate of 20 mL/min. We chose to perfuse with PBS prior to brain extraction to reduce the contribution of circulating macrophages contained within the brain’s capillaries, thereby restricting our collection to Cd11b/c+ cells that reside within the brain parenchyma.
The rat was decapitated via guillotine and the whole brain was removed from the skull. The brain was bisected on ice into its right and left hemispheres via RNase Away®-treated (Thermo Fisher, Waltham, MA, USA, #7005-11) razor blade and the striatum and prefrontal cortex were blunt-dissected from each hemisphere. The two pieces of the same region were combined into a single DNase-free RNase-free 1.5 mL tube containing 200 μL of ice cold dissociation buffer (see below).
The number of rats per group were: 2 h post saline STR (6), 2 h post METH STR (8), 2 h post saline PFC (4), 2 h post METH (5). The respective numbers for the 3 day groups were 6, 7, 6, and 7.
2.2. Buffers and Antibodies for Single-Cell Suspensions
For the mechanical dissociation of STR and PFC to a single-cell suspension, several buffers were prepared. Diethyl pyrocarbonate (DEPC)-phosphate-buffered saline (PBS) pH 7.4 was prepared using DEPC-treated water (Thermo Fisher, AM9922) containing 150 mM sodium chloride (Santa Cruz Biotechnology, Dallas, TX, sc-203274B), 1.5 mM sodium phosphate monobasic (Sigma-Aldrich S-9638), and 17.5 mM sodium phosphate dibasic (Thermo Fisher, S374-1). Dissociation buffer was prepared with DEPC-PBS supplemented with RNase-free DNase (Qiagen, Hilden, Germany) 79254, stock = 2.72 units/μL, final concentration in DEPC-PBS = 40 units/mL) and RNasin® Plus RNase Inhibitor (Promega, Madison, WI, USA, N2615, stock = 40 units/μL, final concentration in DEPC-PBS = 80 units/mL). Blocking buffer consisted of DEPC-PBS with 1% w/v bovine serum albumin (BSA) (Sigma-Aldrich, A3059), 80 units/mL RNasin® Plus RNase Inhibitor and 5% v/v normal mouse serum (Sigma-Aldrich, M5905). Blocked cells were divided, with a fraction of the cells incubated with phycoerythrin (PE)-labeled isotype IgG2a control antibody (Abcam, Cambridge, MA, USA, ab91363) and the remainder of the cells incubated with mouse monoclonal phycoerythrin (PE)-labeled antibody to CD11b/c (Abcam, ab112239) in blocking buffer. For fluorescence activated cell sorting (FACS), stained and washed cells were resuspended in DEPC-PBS with 1% w/v BSA and 80 units/mL RNasin® Plus RNase Inhibitor (Promega).
2.3. Mechanical Dissociation
Keeping the tissue-containing tube on ice, an RNase Away®-treated plastic pestle designed to fit a 1.5 mL tube was used to gently disrupt the tissue to smaller pieces. Another 300 μL ice cold dissociation buffer was added to a final volume of 500 μL. Any remaining tissue chunks were mashed. The suspension was filtered through 100 micron mesh into a clean 1.5 mL DNase-free, RNase-free tube on ice. The filter was washed with another 500 μL dissociation buffer into the same tube. The combined filtrate was centrifuged 5 min 2500× g at 4 °C to pellet the cells. After aspirating the supernatant, the cells were blocked and incubated with PE-labeled CD11b/c+ antibody.
2.4. Blocking and Staining Rat Striatal Cells
One mL of blocking buffer was added to each cell pellet and the cells were resuspended by gentle pipetting. The tubes were closed and rocked for 10 min at room temperature (RT). Fifty μL was removed from each sample and combined in a common tube for gating of cells via flow cytometry (FACS). Another 50 μL was removed from each sample to combine into a negative control for PE nonspecific binding with the PE-labeled isotype control antibody. The remaining 900 μL of cells from each sample were used for PE-labeled CD11b/c antibody labeling. All tubes were spun for 5 min at 2500× g 4 °C to pellet the cells. Supernatant was discarded. Cell pellets from each sample to be incubated with PE-labeled CD11b/c antibody were resuspended in 500 μL of blocking buffer containing 5 μL (0.5 μg) of antibody. A proportionate amount of PE-labeled isotype control antibody was incubated with the resuspended cell pellet for nonspecific PE binding. The resuspended cell pellet that would not receive any antibody was incubated in the same volume of blocking buffer as the isotype control pellet. All tubes were wrapped in foil to protect the fluorescent label from light and rotated end-over-end overnight at 4 °C.
2.5. FACS and RNA Isolation
The next morning, the cells were collected by centrifuging the tubes for 5 min at 2500×
g 4 °C. After aspirating the supernatant, each cell pellet was washed twice by resuspending it in 500 μL DEPC-PBS, centrifuging, and aspirating the DEPC-PBS. The final pellet was resuspended in DEPC-PBS + 1% BSA + RNasin
® Plus RNase Inhibitor for cell sorting on BD Biosciences FACSAria flow cytometers.
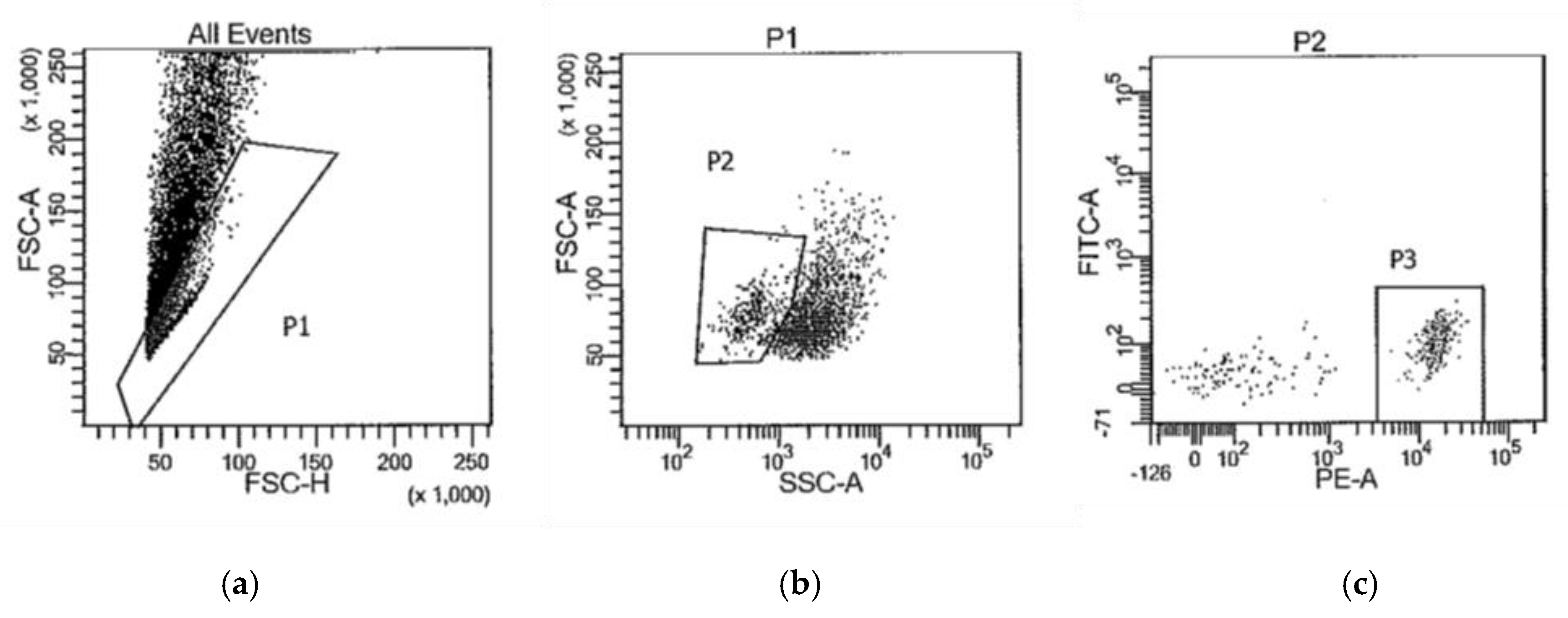
Figure 1 is representative of the gating strategy used to isolate PE-CD11b/c+ cells (P3). The collected CD11b/c+ cells were pelleted by centrifugation at 16,000×
g for 5 min at 4 °C. After aspirating the supernatant, the pellet was resuspended in 50 μL Solution XB (Arcturus
TM PicoPure
TM RNA Isolation Kit, Thermo Fisher, KIT0204) and incubated at 42 °C for 30 min. To remove any insoluble material, the tubes were spun at 3000× g for 2 min and the cleared supernatant was transferred to a clean DNase-free RNase-free 1.5 mL tube. Total RNA was isolated as per the instructions provided with the Arcturus
TM PicoPure
TM RNA Isolation Kit instructions (ThermoFischer), including optional on-column DNase treatment.
2.6. Clontech SMART-Seq V4 Ultra Low Input RNA Methods for Illumina HiSeq 4000 Sequencing
Purified total RNA was first evaluated for its quantity and quality, using Agilent Bioanalyzer 2100 (Agilent, Santa Clara, CA, USA). One nanogram of total RNA per sample was used for library preparation. cDNA was first synthesized using SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing (Takara Clontech Laboratories, Inc., Mountain View, CA, USA). Dual indexed cDNA library was then prepared using Nextera XT DNA Library Prep Kit (Illumina, Inc., San Diego, CA, USA). Each library was quantified and its quality accessed by Qubit and Agilent Bioanalyzer, and multiple libraries were pooled in equal molarity. The average size of the library insert was about 150–200 b. Five microliters of 2 nM pooled libraries per lane were then denatured, neutralized, and applied to the cBot for flow cell deposition and cluster amplification, before loading on to HiSeq 4000 for 75 b paired-end sequencing (Illumina, Inc.). Each lane generated approximately 300–350 million reads. A Phred quality score (Q score) was used to measure the quality of sequencing. More than 90% of the sequencing reads reached Q30 (99.9% base call accuracy).
2.7. Sequence Alignment and Gene Counts
The sequencing data were first assessed using FastQC (Babraham Bioinformatics, Cambridge, UK) for quality control. All sequenced libraries were mapped to the rat genome (mm10) using STAR RNA-seq aligner [
26] with the following parameter: “--outSAMmapqUnique 60”. The reads distribution across the genome was assessed using bamutils (from ngsutils) [
27]. Uniquely mapped sequencing reads were assigned to mm10 refGene genes using featureCounts (from subread) [
28] with the following parameters: “-s 2 –p –Q 10”. Quality control of sequencing and mapping results were summarized using MultiQC [
29]. The data were normalized using the TMM (trimmed mean of M values) method. Differential expression analysis was performed using edgeR [
30,
31]. False discovery rate (FDR) was computed from
p-values using the Benjamini–Hochberg procedure.
2.8. Ingenuity® Pathway Analysis
Ingenuity® Pathway Analysis (IPA®) software (Qiagen, Germantown, MD, USA) was employed to analyze relationships between genes that exhibited significant differential expression (FDR < 0.05) as a result of binge METH treatment.
4. Discussion
Significant changes in gene expression were found in microglial cells of rat STR and PFC following a binge METH dosing paradigm. These changes are detectable at 2 h after the last dose of METH, with some changes persisting for up to 3 days. About one third of genes changed by binge METH at 2 h after METH are shared by the STR and PFC. However, the microglia from the STR appear to exhibit longer-lasting changes than microglia from the PFC given that the PFC had no significantly changed genes at 3 days after METH. Since genes attributed as being unique to macrophages are either not detected or are present only at very low levels, peripheral macrophages appear to have minimal contribution to the number of CD11b/c+ cells collected from rat STR or PFC at 2 h post saline or METH.
Activation of microglia from the steady-state involves differential gene expression patterns that direct the cell into one of at least two polarizations, the most common designations being M1 and M2. At 2 h post-binge METH, microglia appear to be transitioning from a resting state to an M1 inflammatory-reactive state.
Il1b is upregulated 2 h after the last dose of binge METH in both the STR and PFC and emerges as a key marker of such activation. Moreover,
P2ry12 was downregulated in both brain regions at 2 h post-binge METH and is consistent with the finding that microglial activation is associated with its downregulation [
33]. These findings are in contrast to other known markers of M1 activation (
Il6,
Tnf,
Ccl2,
iNos,
Nox) that were not changed after METH and suggest that microglia are only in the early stages of responding to the insult of binge METH. Thus, this study provides insight as to how soon microglia respond to binge METH and that
IL1β is an early inflammatory mediator produced by microglia after METH exposure. These findings add to a growing body of literature that microglial activation can be further differentiated beyond M1 and M2 that depend on the type of insult [
44,
45]. More specifically, microglial activation states also differ in rodent models of aging [
34], retinal degeneration [
38], ischemia [
37,
46], Alzheimer’s disease [
47], Parkinson’s disease [
48,
49], and binge alcohol consumption [
39].
While
Tnf RNA expression was not changed in response to binge METH treatment, there were increases in the expression of a specific TNF superfamily member
Tnfsf9.
Tnfsf9 was upregulated 19.5-fold and 11.5-fold in the STR and PFC, respectively, at 2 h post-binge METH. TNFSF9 is also known as CD137L or 4-1BBL, a transmembrane cytokine expressed on antigen presenting cells (APC), including macrophages [
50]. TNFSF9 interacts with its receptor, TNFRSF9 on activated T-cells, and can generate a signaling cascade. Yeo et al. [
51] demonstrated that interaction of TNFSF9 on BV2 and N9 cells as well as on primary microglia resulted in responses typically associated with activation of microglia to an inflammatory condition, including generation of reactive oxygen species (ROS). Thus, binge METH may produce a specific upregulation in this one TNF superfamily member which contributes to an M1 inflammatory activation state.
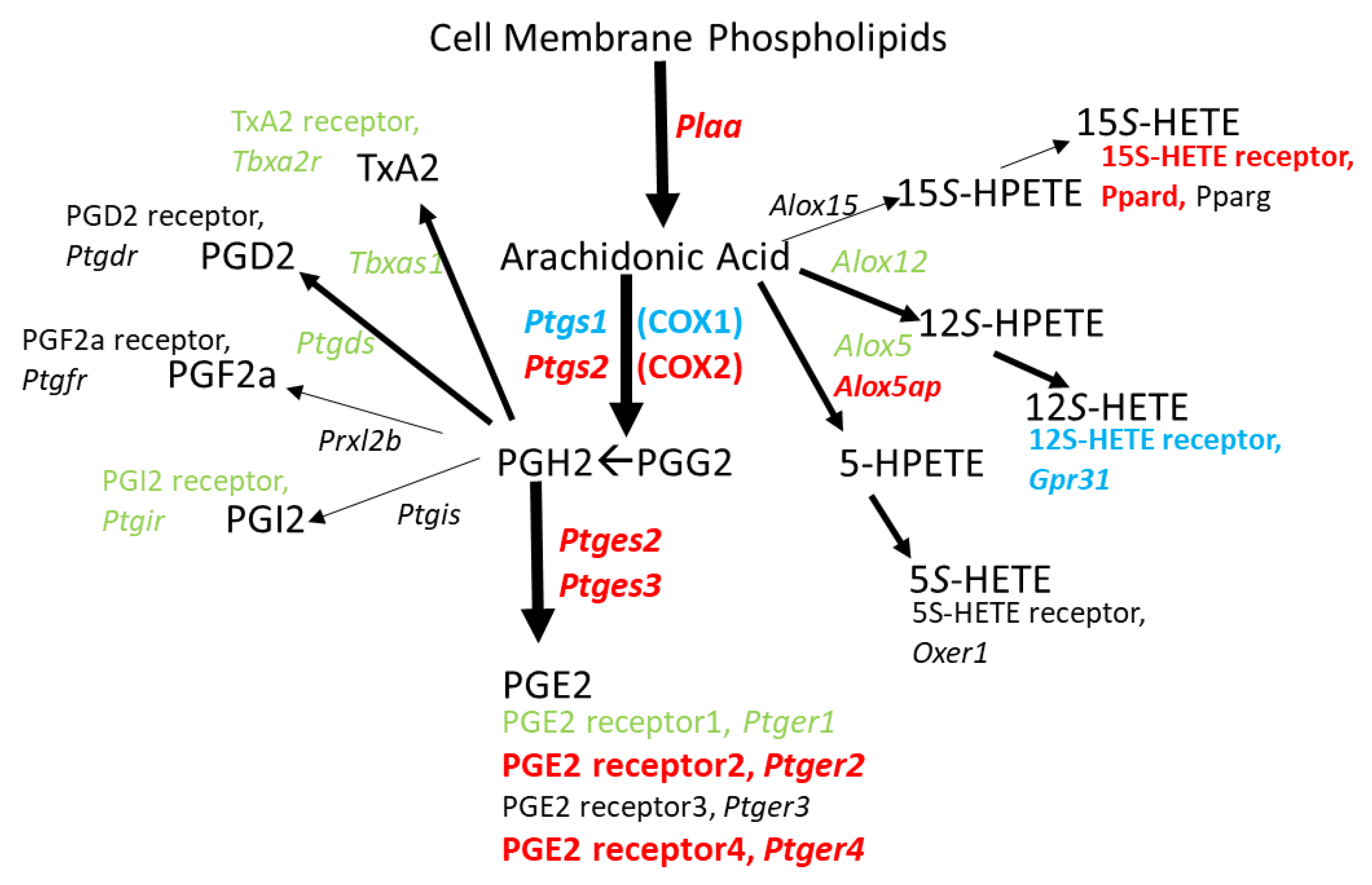
IL1β production after METH can be induced by LPS [
52,
53] that in turn, can increase the release of PGE2 in a manner attenuated by COX inhibitor indomethacin [
54], suggesting of a role for prostaglandins and the arachidonic acid cascade. Furthermore, PGE2 acting through the EP2 receptor was essential for pro-IL1β production induced by LPS in mouse bone-marrow derived macrophages [
55]. Along these lines,
Figure 3 and
Figure 4 illustrate that METH changes expression for a number of genes involved in directing arachidonic acid metabolism toward PGE2 production that could be dependent on LPS derived from a leaky gut. In fact, Persons et al. [
56] showed that METH self-administration in Fischer 344 rats resulted in lower expression of gut tight junction proteins claudin-1 and ZO-1 as well as a morphological disorganization of these proteins in the colon. Additionally, human METH users suffer from bowel ischemia which is associated with loss of gut integrity [
57,
58] that may lead to the entry of LPS into the brain parenchyma as shown in rats following a METH-induced compromise of the blood-brain-barrier [
59]. Thus, high doses of METH may promote PGE2 production in brain microglia that exacerbates LPS-induced IL1β production. The enhanced expression of
Ptger2 suggested by our current RNAseq data could further promote this cycle by allowing PGE2 to act in an autocrine fashion to induce COX2 expression via EP2 receptor activation [
60]. This feed-forward cycle involving LPS, EP2, and COX2 is supported by the observation that EP2-deficient mice have reduced serum levels of inflammatory mediators after the LPS challenge [
61]. Moreover, activation of EP2 in cultures of newborn Sprague-Dawley rat cortical microglia with PGE2 or butaprost upregulates mRNA’s encoding COX2 and IL1β, as well as iNOS, and IL6 [
62].
Il1r2 had a sharp increase in expression 2 h post-binge METH in both the STR (39-fold) and PFC (131-fold). IL1R2 protein is a membrane-bound decoy receptor for IL1 expressed in mouse neutrophils which under LPS-stimulation was increased to its cleaved soluble form by ADAM17 [
63]. Johannson et al. [
64] found that
Il1r2 RNA expression was enhanced in microglia isolated from adult mouse brains exposed to Aβ peptide. The large increase in
Il1r2 expression in microglia 2 h post-binge METH indicates that microglia are similarly responsive to METH. Interestingly,
Il1r2 expression was significantly decreased in microglia from mouse brains exposed to Aβ peptide in EP2 knock-out mice vs. wild type EP2 mice [
64]. In addition, EP2-/- mice had enhanced microglial Aβ phagocytosis characteristic of M2 anti-inflammatory microglia and a lack of Aβ-activated paracrine neurotoxicity [
65]. It is possible that enhanced
Ptger2 expression induced by binge METH treatment may serve to enhance
Il1r2 expression and suppress transition toward an M2 anti-inflammatory state in the early hours after METH.
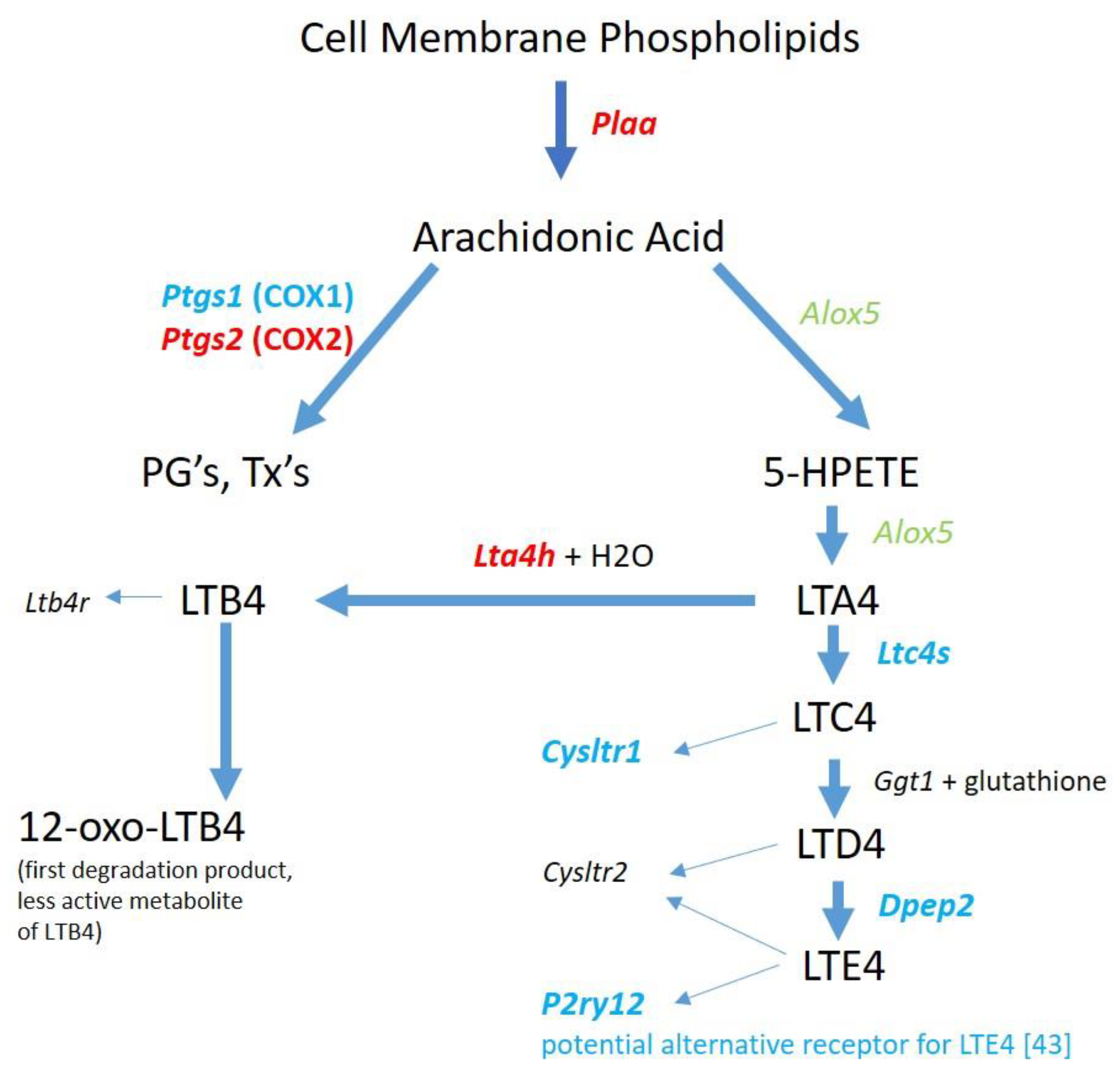
The production of the cysteinyl LT’s from AA may be diminished as illustrated by the gene expression results in
Figure 3. However, it is noted that
Alox5 is present and
Alox5ap is upregulated which could still result in LTA4 synthesis. LTA4 is the precursor to not only the cysteinyl LT’s but also to LTB4. The gene for the enzyme that converts LTA4 to LTB4,
Lta4h, is significantly upregulated. LTB4 longevity could be enhanced via the downregulation of
Ptgr1, the gene for prostaglandin reductase 1, a key enzyme for inactivating LTB4 as an inflammatory mediator [
66]. In models of LPS-induced septic shock, Collin et al. [
67] and Kwon et al. [
68] demonstrated that inhibition of ALOX5 and antagonism LTB4 receptors, respectively, reduced multiple LPS-induced effects. Saiwai et al. [
69] used a model of spinal cord injury and reported that LTB4 receptor inhibition suppressed leukocyte infiltration and attenuated the inflammatory reaction. Since no RNA for LTB4 receptors was detected in the RNAseq data, the potential LTB4 production in microglia from METH-treated rats would not act in an autocrine fashion, but most likely be released to stimulate LTB4 receptors on other cells. Thus, it appears that LTB4 could be a potential contributor to METH-induced inflammation.
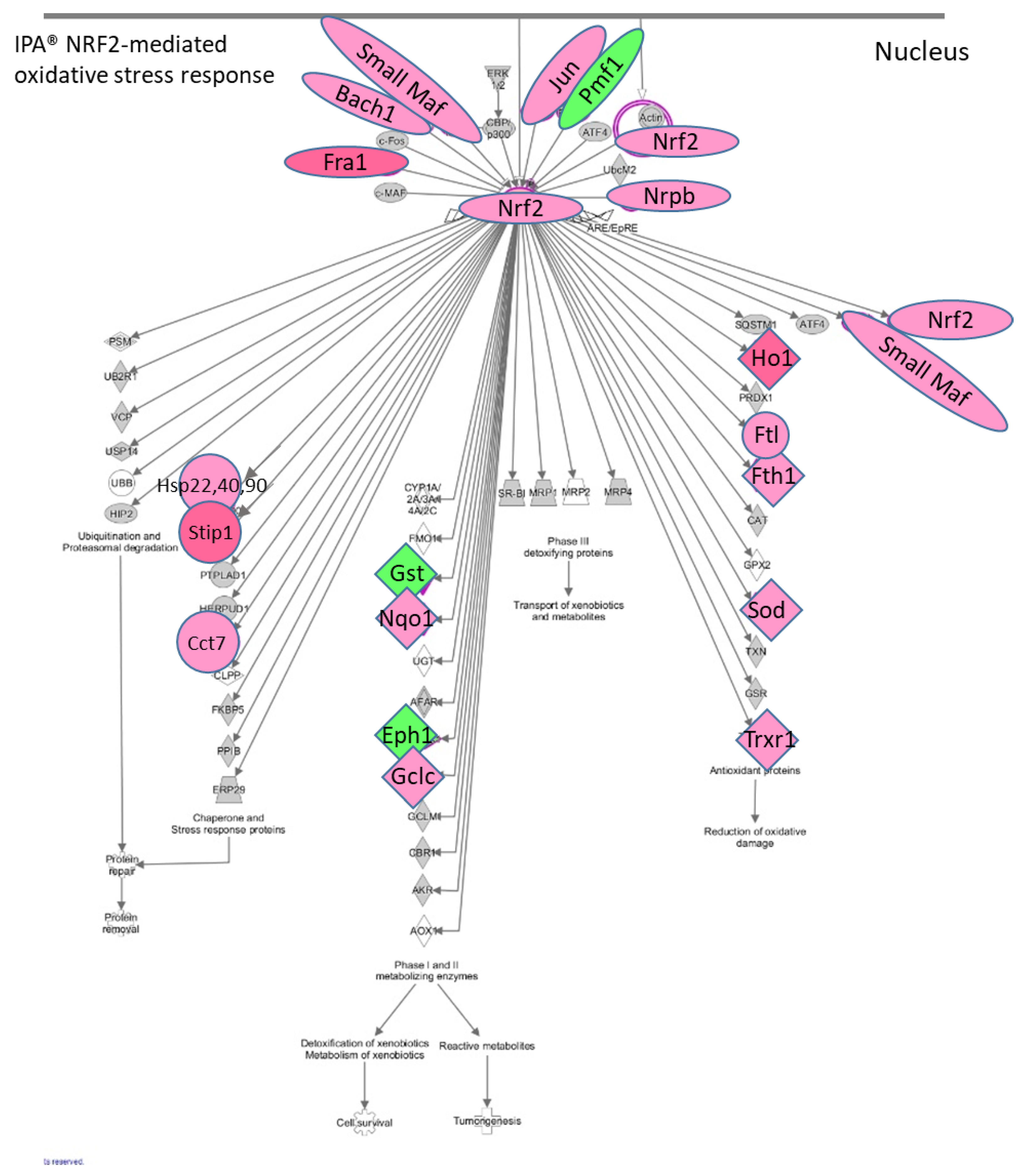
The IPA
® results showed that there is an enhancement of the NRF2 (Nuclear factor erythroid 2-related factor 2) (NFE2L2)-mediated canonical pathway in the STR microglia at 2 h post METH. METH is known to induce production of reactive oxygen species [
13,
70]. The NRF2-mediated pathway is a cellular defense mechanism against oxidative stress (
Figure 5). It is noteworthy that among the elements which can activate this pathway are drugs, cytokines, prostaglandins, and bacterial infections. NRF2 appears to be a key connection in the upregulation of
Hsp’s
22 (Hspb8),
40 and
90,
Stip1,
Cct7,
Nqo,
Gclc,
Hmox-1,
Ftl,
Fth1,
Sod1,
Txnrd1, small Maf’s (
Maff,
Mafg,
Mafk) as well as
Nrf2 itself. Small MAF’s and NRF2 dimerize to form transcriptional activators, suggesting another point of positive feedback in STR after METH and further illustrates the different aspects of microglia activation. More specifically,
Hsp-22,
Hsp-40 family member genes, and
Hsp90 are significantly upregulated 2 h after METH (up to 37-fold) as are
Stip1 (16-fold) and
Fkbp5 (2-fold). HSP’s are critical to protein folding and maturation. The protein product STIP1 is a co-chaperone for HSP90 and serves as a scaffold for the interaction of HSP90 with HSP70. HSP90 ATPase activity is inhibited by PTGES3 by its binding to the closed conformation of dimeric HSP90 and delaying release of a client protein [
71].
Ptges3 is significantly upregulated over 2-fold in both the STR and PFC 2 h post METH (
Table S2). Thus, protein products of
Ptges3 may serve two roles by promoting the synthesis of PGE2 and by influencing the maturation and folding of proteins.
CCT‘s, chaperonin containing TCP1 complexes, are also involved in the folding of proteins, including actin and tubulin.
Cct7, the gene for molecular chaperone subunit 7 of CCT, was significantly upregulated in both the STR and PFC, 3.0- and 2.8-fold, respectively. LPS has been shown to stimulate rearrangement of actin within primary cultures of peritoneal rat macrophages [
72] and rat neonatal microglia [
73], which can be associated with changes in microglial morphology associated with activation. Thus, upregulation of
Cct7 may be associated with the folding of cytoskeletal proteins linked to changes in morphology of microglia after activation.
Several genes upregulated by NFR2 are associated with detoxification of reactive species.
Nqo upregulation would result in expression of a protein that serves to detoxify reactive quinone species. Dopamine quinone formation is induced by METH in dopaminergic neurons [
74,
75] and can activate microglia [
76].
Gclc upregulation can lead to enhanced expression of glutamyl cysteinyl ligase which is the rate limiting enzyme in the synthesis of glutathione, a key molecule involved with the detoxification of reactive species. Upregulation of
Gclc and
Sod1 could be associated with the microglial uptake of glutamine. As shown in
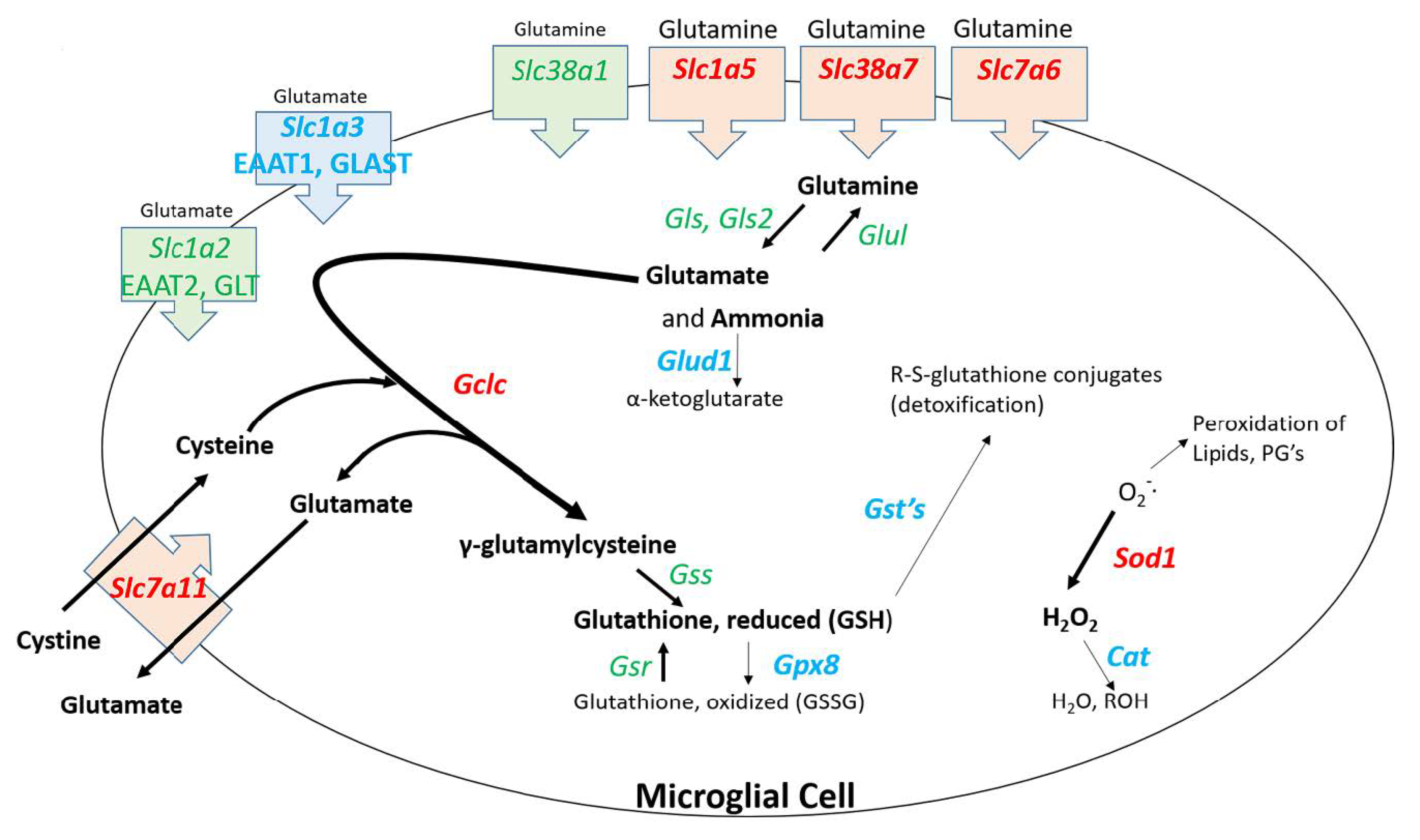
Figure 6, several genes for solute carriers involved with glutamine uptake are significantly upregulated in microglia. It is known that METH causes the release of glutamate from neurons [
77,
78] that is ordinarily taken up by astrocytes [
79] and converted to glutamine [
80], released into the synaptic space and subsequently taken up by neurons where it is converted back glutamate. The RNAseq data indicate that at 2 h post METH, the microglia may be primed to enhance their uptake of glutamine and convert it ultimately to reduced glutathione via the 9-fold increase in
Gclc (
Figure 6). Such a conversion along with the increase in
Sod1 may be associated with a response by microglia to detoxify reactive species but appear to be countered by the downregulation of genes for the enzymes such as glutathione-S-transferases (GST), glutathione peroxidase 8 (GPX8), and catalase (CAT) needed to complete the detoxification cycle. Consequently, the balance of effect could lead to the release and accumulation of pro-oxidant species such as H
2O
2 [
81,
82] to produce the neurotoxicity to METH.
Several reviews point to the evolution from thinking of microglia as either simply M1 or M2 phenotypes to a spectrum of phenotypes or even sub-populations of cells specialized in their response to a given stimulus [
44,
84,
85]. One might envision a subset of microglia poised to respond to the insult of toxic METH by locally generating COX2 expression and subsequently PGE2 production and release. These cells could amplify their initial PGE2 response via autocrine signaling through enhanced EP2 expression, while also permitting activation of a separate subset of microglia poised to respond to PGE2 via upregulated expression of EP4 receptors to enhance phagocytosis [
86], suppress expression of inflammatory mediators [
87,
88] and enhance expression of enzymes to detoxify reactive oxygen species via NRF2 [
89]. METH may serve to disrupt this coordinated effort by the incomplete detoxification of reactive species when
Gclc and
Sod1 are upregulated but
Gpx8 and
Cat are downregulated. Single-cell RNAseq could be a useful tool to determine if such subpopulations of cells could exist.
There are only six genes upregulated >2-fold at both 2 h and 3 days in the STR after METH. The gene for Betaine/GABA transporter 1,
Slc6a12, is one of these genes (
Table 3,
Supplemental Table S5). In a review by Kempson et al. 2014 [
90], the role of this particular GABA transporter in the brain is described as controversial since its expression levels relative to other GABA transporters are very low. While expression levels of genes for GABA transporters in our RNAseq data from microglia are modest,
Scl6a12 is expressed at similar basal levels to the only other detected gene for a GABA transporter,
Slc6a1, which is not changed in the STR at either time point by METH. The role of potential enhanced uptake of an inhibitory neurotransmitter by microglia for at least 3 days after METH has yet to be explored.
Another gene that remains upregulated at 3 days after METH in the STR is
Csf1 or macrophage colony stimulating factor (
Table 3,
Table S6). CSF1 and activation of its receptor CSF1R, regulates the survival, proliferation, and differentiation of macrophages and microglia during development [
91]. The gene for CSF1R is highly expressed in all CD11b/c+ cells analyzed but its expression is not changed by METH (
Table S6). The alternative ligand for CSF1R, IL34, is significantly upregulated 3-fold at 3 days post METH in the STR (
Table S6) and also can stimulate CSF1R. Thus, both ligands elicit a pro-inflammatory phenotype in microglia [
92] that persists for at least 3 days after METH.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}