Novel Type of Complicated Autosomal Dominant Hereditary Spastic Paraplegia Associated with Congenital Distal Arthrogryposis Type I



Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical and Laboratory Studies
2.2. Genetic Analysis
3. Results
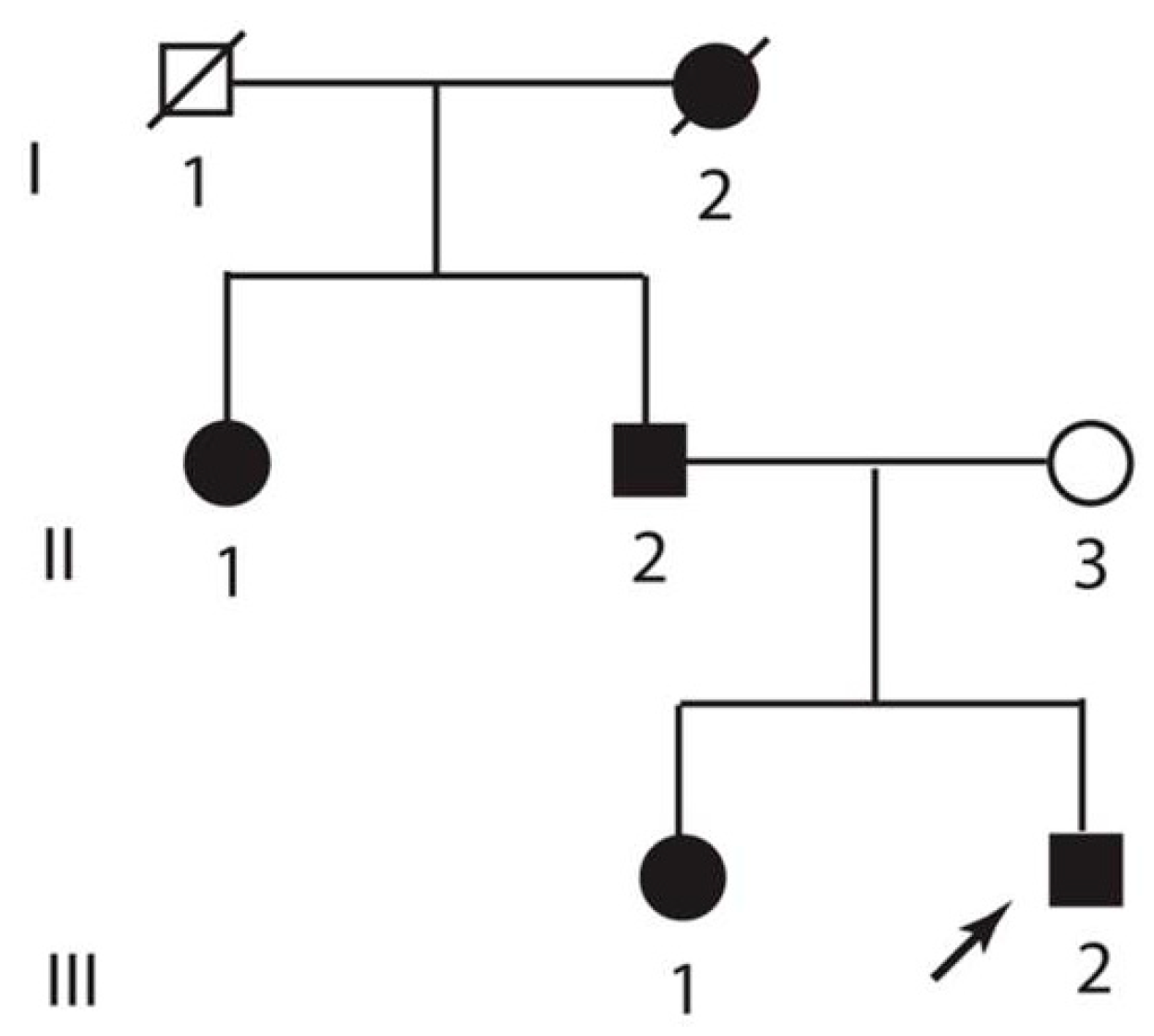
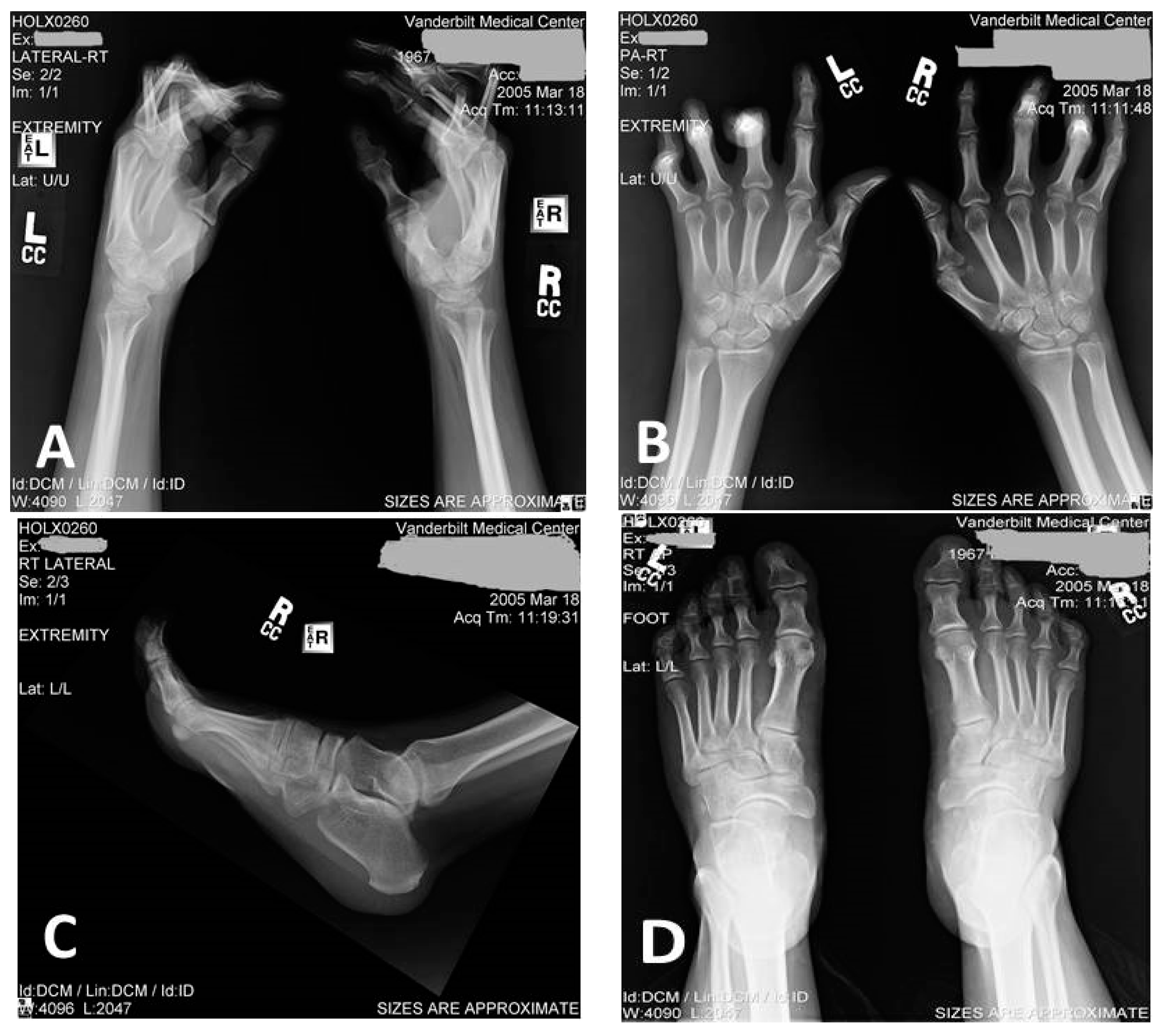
3.1. Clinical Description
3.2. Genetic Analysis
4. Discussion
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- de Souza, P.V.S.; de Rezende Pinto, W.B.V.; de Rezende Batistella, G.N.; Bortholin, T.; Oliveira, A.S.B. Hereditary spastic paraplegia: Clinical and genetic hallmarks. Cerebellum 2017, 16, 525–551. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J.; Löscher, W.; Quasthoff, S.; Wanschitz, J.; Auer-Grumbach, M.; Stevanin, G. Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J. Neurol. Sci. 2012, 318, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Hedera, P. Hereditary myelopathies. Continuum (Minneap Minn). 2018, 24, 523–550. [Google Scholar] [CrossRef] [PubMed]
- Fink, J.K.; Hedera, P. Hereditary spastic paraplegia: Genetic heterogeneity and genotype-phenotype correlation. Semin. Neurol. 1999, 19, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Gorman, G.; Beetz, C.; Byrne, P.; Dytko, M.; McMonagle, P.; Kinsella, K.; Farrell, M.; Hutchinson, M. Dementia in SPG4 hereditary spastic paraplegia: Clinical, genetic, and neuropathologic evidence. Neurology 2009, 73, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.T.; Laurá, M.; Hersheson, J.; Horga, A.; Jaunmuktane, Z.; Brandner, S.; Pittman, A.; Hughes, D.; Polke, J.M.; Sweeney, M.G.; et al. Extended phenotypic spectrum of KIF5A mutations: From spastic paraplegia to axonal neuropathy. Neurology 2014, 83, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Cross, H.; Proukakis, C.; Hershberger, R.; Bork, P.; Ciccarelli, F.D.; Patton, M.A.; McKusick, V.A.; Crosby, A.H. SPG20 is mutated in Troyer syndrome, an hereditary spastic paraplegia. Nat. Genet. 2002, 31, 347–348. [Google Scholar] [CrossRef] [PubMed]
- Proukakis, C.; Cross, H.; Patel, H.; Patton, M.A.; Valentine, A.; Crosby, A.H. Troyer syndrome revisited. A clinical and radiological study of a complicated hereditary spastic paraplegia. J. Neurol. 2004, 251, 1105–1110. [Google Scholar] [CrossRef] [PubMed]
- Rainier, S.; Bui, M.; Mark, E.; Thomas, D.; Tokarz, D.; Ming, L.; Delaney, C.; Richardson, R.J.; Albers, J.W.; Matsunami, N.; et al. Neuropathy target esterase gene mutations cause motor neuron disease. Am. J. Hum. Genet. 2008, 82, 780–785. [Google Scholar] [CrossRef] [PubMed]
- Hufnagel, R.B.; Arno, G.; Hein, N.D.; Hersheson, J.; Prasad, M.; Anderson, Y.; Krueger, L.A.; Gregory, L.C.; Stoetzel, C.; Jaworek, T.J.; et al. Neuropathy target esterase impairments cause Oliver-McFarlane and Laurence-Moon syndromes. J. Med. Genet. 2015, 52, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Rink, B.D. Arthrogryposis: A review and approach to prenatal diagnosis. Obstet. Gynecol. Surv. 2011, 66, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Haliloglu, G.; Topaloglu, H. Arthrogryposis and fetal hypomobility syndrome. Handb. Clin. Neurol. 2013, 113, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Tajsharghi, H.; Oldfors, A. Myosinopathies: Pathology and mechanisms. Acta Neuropathol. 2013, 125, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Kee, A.J.; Hardeman, E.C. Tropomyosins in skeletal muscle diseases. Adv. Exp. Med. Biol. 2008, 644, 143–157. [Google Scholar] [PubMed]
- Hedera, P.; Fenichel, G.M.; Blair, M.; Haines, J.L. Novel mutation in the SPG3A gene in an African American family with an early onset of hereditary spastic paraplegia. Arch. Neurol. 2004, 61, 1600–1603. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Vemula, S.R.; Xiao, J.; Valente, E.M.; Defazio, G.; Petrucci, S.; Gigante, A.F.; Rudzińska-Bar, M.; Wszolek, Z.K.; Kennelly, K.D.; et al. Whole-exome sequencing for variant discovery in blepharospasm. Mol. Genet. Genom. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from next-generation sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jian, X.; Boerwinkle, E. dbNSFP: Lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat. 2011, 32, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [PubMed]
- Novarino, G.; Fenstermaker, A.G.; Zaki, M.S.; Hofree, M.; Silhavy, J.L.; Heiberg, A.D.; Abdellateef, M.; Rosti, B.; Scott, E.; Mansour, L.; et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science 2014, 343, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Tesson, C.; Koht, J.; Stevanin, G. Delving into the complexity of hereditary spastic paraplegias: How unexpected phenotypes and inheritance modes are revolutionizing their nosology. Hum. Genet. 2015, 134, 511–538. [Google Scholar] [CrossRef] [PubMed]
- Panza, E.; Pippucci, T.; Cusano, R.; Lo Nigro, C.; Pradella, L.; Contardi, S.; Rouleau, G.A.; Stevanin, G.; Ravazzolo, R.; Liguori, R.; et al. Refinement of the SPG9 locus on chromosome 10q23.3-24.2 and exclusion of candidate genes. Eur. J. Neurol. 2008, 5, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Coutelier, M.; Goizet, C.; Durr, A.; Habarou, F.; Morais, S.; Dionne-Laporte, A.; Tao, F.; Konop, J.; Stoll, M.; Charles, P.; et al. Alteration of ornithine metabolism leads to dominant and recessive hereditary spastic paraplegia. Brain 2015, 138, 2191–2205. [Google Scholar] [CrossRef] [PubMed]
- Hamann, G.; Zankl, M.; Schimrigk, K.; Kloss, R. Spastic disorder in patients with hereditary multiple exostoses, but without spinal cord compression: A new syndrome? J. Med. Genet. 1992, 29, 494–496. [Google Scholar] [PubMed]
- Mannan, A.U.; Krawen, P.; Sauter, S.M.; Boehm, J.; Chronowska, A.; Paulus, W.; Neesen, J.; Engel, W. ZFYVE27 (SPG33), a novel spastin-binding protein, is mutated in hereditary spastic paraplegia. Am. J. Hum. Genet. 2006, 79, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Martignoni, M.; Riano, E.; Rugarli, E.I. The role of ZFYVE27/protrudin in hereditary spastic paraplegia. Am. J. Hum. Genet. 2008, 83, 127–128. [Google Scholar] [CrossRef] [PubMed]
- Blackstone, C.; O’Kane, C.J.; Reid, E. Hereditary spastic paraplegias: Membrane traffic and the motor pathway. Nat. Rev. Neurosci. 2011, 12, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Salinas, S.; Proukakis, C.; Crosby, A.; Warner, T.T. Hereditary spastic paraplegia: Clinical features and pathogenetic mechanisms. Lancet. Neurol. 2008, 7, 1127–1138. [Google Scholar] [CrossRef]
- Blackstone, C. Cellular pathways of hereditary spastic paraplegia. Annu. Rev. Neurosci. 2012, 35, 25–47. [Google Scholar] [CrossRef] [PubMed]
- Horokawa, N.; Niwa, S.; Tanaka, Y. Molecular motors in neurons: Transport mechanisms and roles in brain function and disease. Neuron 2010, 68, 610–638. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Subject | Skeletal Abnormalities | Spastic Gait | Age of Onset of Paraparesis (Years) | Rhabdomyolysis |
|---|---|---|---|---|
| I/2 | Both hands and feet with bilateral pes equinovarus | Yes, with wheelchair-dependency in her 50s | 30s | Unknown |
| II/1 | Mild hand deformities | Yes, required an assistive device in her 50s and wheelchair in her 60s | 30 | Yes (history of passing very dark urine) |
| II/2 | Severe hand deformities, mild feet deformities | Yes, required assistive device in his 60s, wheelchair in his 70s | 39 | Yes (history of passing very dark urine) |
| III/1 | Mild hand deformities | Yes, abnormal but independent gait at age 47 years | 27 | Yes (documented elevation of CPK and myoglobinuria) |
| III/2 | Severe hand deformities, mild feet deformities | Yes, abnormal but independent gait at age of 51 years | 32 | Yes (documented elevation of CPK and myoglobinuria) |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hedera, P.; Moretti, P.; Howard, J.; Zhao, J. Novel Type of Complicated Autosomal Dominant Hereditary Spastic Paraplegia Associated with Congenital Distal Arthrogryposis Type I. Brain Sci. 2018, 8, 136. https://doi.org/10.3390/brainsci8070136
Hedera P, Moretti P, Howard J, Zhao J. Novel Type of Complicated Autosomal Dominant Hereditary Spastic Paraplegia Associated with Congenital Distal Arthrogryposis Type I. Brain Sciences. 2018; 8(7):136. https://doi.org/10.3390/brainsci8070136
Chicago/Turabian StyleHedera, Peter, Paolo Moretti, Jane Howard, and Jiali Zhao. 2018. "Novel Type of Complicated Autosomal Dominant Hereditary Spastic Paraplegia Associated with Congenital Distal Arthrogryposis Type I" Brain Sciences 8, no. 7: 136. https://doi.org/10.3390/brainsci8070136
APA StyleHedera, P., Moretti, P., Howard, J., & Zhao, J. (2018). Novel Type of Complicated Autosomal Dominant Hereditary Spastic Paraplegia Associated with Congenital Distal Arthrogryposis Type I. Brain Sciences, 8(7), 136. https://doi.org/10.3390/brainsci8070136