DYRK1A Protein, A Promising Therapeutic Target to Improve Cognitive Deficits in Down Syndrome



Abstract
1. Introduction
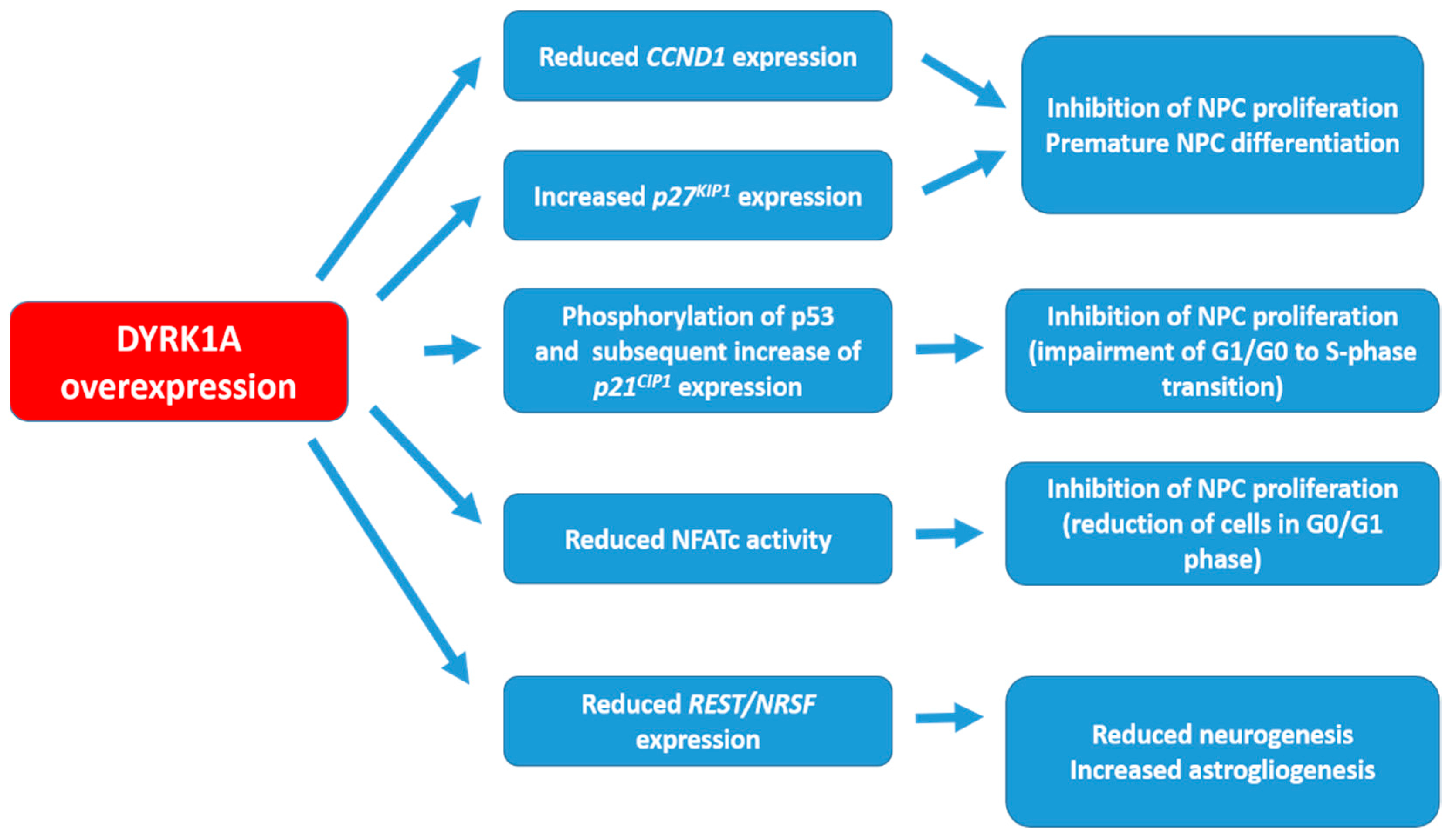
2. DYRK1A Targets and the Possible Mechanisms of Action
3. Preclinical Studies
3.1. Results from Mouse Models
3.2. Results from Human Cells
4. Clinical Studies
5. Conclusions and Challenges
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Antonarakis, S.E. Down syndrome and the complexity of genome dosage imbalance. Nat. Rev. Genet. 2017, 18, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Dierssen, M. Down syndrome: The brain in trisomic mode. Nat. Rev. Neurosci. 2012, 13, 844–858. [Google Scholar] [CrossRef] [PubMed]
- Ballard, C.; Mobley, W.; Hardy, J.; Williams, G.; Corbett, A. Dementia in down’s syndrome. Lancet Neurol. 2016, 15, 622–636. [Google Scholar] [CrossRef]
- Becker, W.; Sippl, W. Activation, regulation, and inhibition of dyrk1a. FEBS J. 2011, 278, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Tejedor, F.J.; Hämmerle, B. Mnb/dyrk1a as a multiple regulator of neuronal development. FEBS J. 2011, 278, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Dowjat, W.K.; Adayev, T.; Kuchna, I.; Nowicki, K.; Palminiello, S.; Hwang, Y.W.; Wegiel, J. Trisomy-driven overexpression of dyrk1a kinase in the brain of subjects with down syndrome. Neurosci. Lett. 2007, 413, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Guimera, J.; Casas, C.; Estivill, X.; Pritchard, M. Humanminibrainhomologue (mnbh/dyrk1): Characterization, alternative splicing, differential tissue expression, and overexpression in down syndrome. Genomics 1999, 57, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Møller, R.S.; Kübart, S.; Hoeltzenbein, M.; Heye, B.; Vogel, I.; Hansen, C.P.; Menzel, C.; Ullmann, R.; Tommerup, N.; Ropers, H.-H.; et al. Truncation of the down syndrome candidate gene dyrk1a in two unrelated patients with microcephaly. Am. J. Human Genet. 2008, 82, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Shimojima, K.; Nishizawa, T.; Matsuo, M.; Ito, M.; Imai, K. Clinical manifestations of the deletion of down syndrome critical region including dyrk1a and kcnj6. Am. J. Med. Genet. Part A 2011, 155, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Oegema, R.; De Klein, A.; Verkerk, A.J.; Schot, R.; Dumee, B.; Douben, H.; Eussen, B.; Dubbel, L.; Poddighe, P.J.; Van der Laar, I.; et al. Distinctive phenotypic abnormalities associated with submicroscopic 21q22 deletion including dyrk1a. Mol. Syndromol. 2010, 1, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Van Bon, B.W.M.; Coe, B.P.; Bernier, R.; Green, C.; Gerdts, J.; Witherspoon, K.; Kleefstra, T.; Willemsen, M.H.; Kumar, R.; Bosco, P.; et al. Disruptive de novo mutations of dyrk1a lead to a syndromic form of autism and id. Mol. Psychiatry 2015, 21, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Valetto, A.; Orsini, A.; Bertini, V.; Toschi, B.; Bonuccelli, A.; Simi, F.; Sammartino, I.; Taddeucci, G.; Simi, P.; Saggese, G. Molecular cytogenetic characterization of an interstitial deletion of chromosome 21 (21q22.13q22.3) in a patient with dysmorphic features, intellectual disability and severe generalized epilepsy. Eur. J. Med. Genet. 2012, 55, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Courcet, J.-B.; Faivre, L.; Malzac, P.; Masurel-Paulet, A.; Lopez, E.; Callier, P.; Lambert, L.; Lemesle, M.; Thevenon, J.; Gigot, N.; et al. The dyrk1a gene is a cause of syndromic intellectual disability with severe microcephaly and epilepsy. J. Med. Genet. 2012, 49, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Bronicki, L.M.; Redin, C.; Drunat, S.; Piton, A.; Lyons, M.; Passemard, S.; Baumann, C.; Faivre, L.; Thevenon, J.; Rivière, J.-B.; et al. Ten new cases further delineate the syndromic intellectual disability phenotype caused by mutations in dyrk1a. Eur. J. Human Genet. 2015, 23, 1482–1487. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Lee, H.; Argiropoulos, B.; Dorrani, N.; Mann, J.; Martinez-Agosto, J.A.; Gomez-Ospina, N.; Gallant, N.; Bernstein, J.A.; Hudgins, L.; et al. Dyrk1a haploinsufficiency causes a new recognizable syndrome with microcephaly, intellectual disability, speech impairment, and distinct facies. Eur. J. Human Genet. 2015, 23, 1473–1481. [Google Scholar] [CrossRef] [PubMed]
- Redin, C.; Gérard, B.; Lauer, J.; Herenger, Y.; Muller, J.; Quartier, A.; Masurel-Paulet, A.; Willems, M.; Lesca, G.; El-Chehadeh, S.; et al. Efficient strategy for the molecular diagnosis of intellectual disability using targeted high-throughput sequencing. J. Med. Genet. 2014, 51, 724–736. [Google Scholar] [CrossRef] [PubMed]
- Luco, S.M.; Pohl, D.; Sell, E.; Wagner, J.D.; Dyment, D.A.; Daoud, H. Case report of novel dyrk1a mutations in 2 individuals with syndromic intellectual disability and a review of the literature. BMC Med. Genet. 2016, 17, 15. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, N.; Miya, F.; Tsunoda, T.; Kato, M.; Saitoh, S.; Yamasaki, M.; Shimizu, A.; Torii, C.; Kanemura, Y.; Kosaki, K. Targeted next-generation sequencing in the diagnosis of neurodevelopmental disorders. Clin. Genet. 2015, 88, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Ruaud, L.; Mignot, C.; Guët, A.; Ohl, C.; Nava, C.; Héron, D.; Keren, B.; Depienne, C.; Benoit, V.; Maystadt, I.; et al. Dyrk1a mutations in two unrelated patients. Eur. J. Med. Genet. 2015, 58, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Benavides-Piccione, R.; Dierssen, M.; Ballesteros-Yáñez, I.; Martínez de Lagrán, M.; Arbonés, M.L.; Fotaki, V.; DeFelipe, J.; Elston, G.N. Alterations in the phenotype of neocortical pyramidal cells in the dyrk1a+/− mouse. Neurobiol. Dis. 2005, 20, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Fotaki, V.; Dierssen, M.; Alcántara, S.; Martínez, S.; Martí, E.; Casas, C.; Visa, J.; Soriano, E.; Estivill, X.; Arbonés, M.L. Dyrk1a haploinsufficiency affects viability and causes developmental delay and abnormal brain morphology in mice. Mol. Cell. Biol. 2002, 22, 6636–6647. [Google Scholar] [CrossRef] [PubMed]
- Guedj, F.; Pereira, P.L.; Najas, S.; Barallobre, M.-J.; Chabert, C.; Souchet, B.; Sebrie, C.; Verney, C.; Herault, Y.; Arbones, M.; et al. Dyrk1a: A master regulatory protein controlling brain growth. Neurobiol. Dis. 2012, 46, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Altafaj, X.; Dierssen, M.; Baamonde, C.; Martí, E.; Visa, J.; Guimerà, J.; Oset, M.; González, J.R.; Flórez, J.; Fillat, C.; et al. Neurodevelopmental delay, motor abnormalities and cognitive deficits in transgenic mice overexpressing dyrk1a (minibrain), a murine model of down’s syndrome. Human Mol. Genet. 2001, 10, 1915–1923. [Google Scholar] [CrossRef]
- Ahn, K.-J.; Jeong, H.K.; Choi, H.-S.; Ryoo, S.-R.; Kim, Y.J.; Goo, J.-S.; Choi, S.-Y.; Han, J.-S.; Ha, I.; Song, W.-J. Dyrk1a bac transgenic mice show altered synaptic plasticity with learning and memory defects. Neurobiol. Dis. 2006, 22, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Das, I.; Reeves, R.H. The use of mouse models to understand and improve cognitive deficits in down syndrome. Dis. Mod. Mech. 2011, 4, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Roubertoux, P.L.; Baril, N.; Cau, P.; Scajola, C.; Ghata, A.; Bartoli, C.; Bourgeois, P.; Di Christofaro, J.; Tordjman, S.; Carlier, M. Differential brain, cognitive and motor profiles associated with partial trisomy. Modeling down syndrome in mice. Behav. Genet. 2017, 47, 305–322. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.J.; Stevens, M.E.; Sudanagunta, S.P.; Bronson, R.T.; Makhinson, M.; Watabe, A.M.; O’Dell, T.J.; Fung, J.; Weier, H.U.; Cheng, J.F.; et al. Functional screening of 2 mb of human chromosome 21q22.2 in transgenic mice implicates minibrain in learning defects associated with down syndrome. Nat. Genet. 1997, 16, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Duchon, A.; Herault, Y. Dyrk1a, a dosage-sensitive gene involved in neurodevelopmental disorders, is a target for drug development in down syndrome. Front. Behav. Neurosci. 2016, 10, 104. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.L.; Fruit, C.; Hérault, Y.; Meijer, L.; Besson, T. Dual-specificity tyrosine phosphorylation-regulated kinase 1a (dyrk1a) inhibitors: A survey of recent patent literature. Expert Opin. Ther. Patents 2017, 27, 1183–1199. [Google Scholar] [CrossRef] [PubMed]
- Nakano-Kobayashi, A.; Awaya, T.; Kii, I.; Sumida, Y.; Okuno, Y.; Yoshida, S.; Sumida, T.; Inoue, H.; Hosoya, T.; Hagiwara, M. Prenatal neurogenesis induction therapy normalizes brain structure and function in down syndrome mice. Proc. Natl. Acad. Sci. USA 2017, 114, 10268–10273. [Google Scholar] [CrossRef] [PubMed]
- Hämmerle, B.; Ulin, E.; Guimera, J.; Becker, W.; Guillemot, F.; Tejedor, F.J. Transient expression of mnb/dyrk1a couples cell cycle exit and differentiation of neuronal precursors by inducing p27kip1 expression and suppressing notch signaling. Development 2011, 138, 2543–2554. [Google Scholar] [CrossRef] [PubMed]
- Litovchick, L.; Florens, L.A.; Swanson, S.K.; Washburn, M.P.; DeCaprio, J.A. Dyrk1a protein kinase promotes quiescence and senescence through dream complex assembly. Genes Develop. 2011, 25, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Oh, Y.; Yoo, L.; Jung, M.-S.; Song, W.-J.; Lee, S.-H.; Seo, H.; Chung, K.C. Dyrk1a phosphorylates p53 and inhibits proliferation of embryonic neuronal cells. J. Biol. Chem. 2010, 285, 31895–31906. [Google Scholar] [CrossRef] [PubMed]
- Yabut, O.; Domogauer, J.; D’Arcangelo, G. Dyrk1a overexpression inhibits proliferation and induces premature neuronal differentiation of neural progenitor cells. J. Neurosci. 2010, 30, 4004–4014. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-Y.; Lin, J.-R.; Tsai, F.-C.; Meyer, T. Dosage of dyrk1a shifts cells within a p21-cyclin d1 signaling map to control the decision to enter the cell cycle. Mol. Cell 2013, 52, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, T.; Hsieh, J.; Nakashima, K.; Taira, K.; Gage, F.H. A small modulatory dsrna specifies the fate of adult neural stem cells. Cell 2004, 116, 779–793. [Google Scholar] [CrossRef]
- Ballas, N.; Grunseich, C.; Lu, D.D.; Speh, J.C.; Mandel, G. Rest and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell 2005, 121, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Abrajano, J.J.; Qureshi, I.A.; Gokhan, S.; Zheng, D.; Bergman, A.; Mehler, M.F. Rest and corest modulate neuronal subtype specification, maturation and maintenance. PLoS ONE 2009, 4, e7936. [Google Scholar] [CrossRef] [PubMed]
- Bahn, S.; Mimmack, M.; Ryan, M.; Caldwell, M.; Jauniaux, E.; Starkey, M.; Svendsen, C.; Emson, P. Neuronal target genes of the neuron-restrictive silencer factor in neurospheres derived from fetuses with down’s syndrome: A gene expression study. Lancet 2002, 359, 310–315. [Google Scholar] [CrossRef]
- Hibaoui, Y.; Grad, I.; Letourneau, A.; Sailani, M.R.; Dahoun, S.; Santoni, F.A.; Gimelli, S.; Guipponi, M.; Pelte, M.-F.; Béna, F.; et al. Modelling and rescuing neurodevelopmental defect of down syndrome using induced pluripotent stem cells from monozygotic twins discordant for trisomy 21. EMBO Mol. Med. 2014, 6, 259–277. [Google Scholar] [CrossRef] [PubMed]
- Kurabayashi, N.; Nguyen, M.D.; Sanada, K. Dyrk1a overexpression enhances stat activity and astrogliogenesis in a down syndrome mouse model. EMBO Rep. 2015, 16, 1548–1562. [Google Scholar] [CrossRef] [PubMed]
- Arron, J.R.; Winslow, M.M.; Polleri, A.; Chang, C.-P.; Wu, H.; Gao, X.; Neilson, J.R.; Chen, L.; Heit, J.J.; Kim, S.K.; et al. Nfat dysregulation by increased dosage of dscr1 and dyrk1a on chromosome 21. Nature 2006, 441, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pérez, M.C.; Fernández, M.; Neria, F.; Berjón-Otero, M.; Doncel-Pérez, E.; Cano, E.; Tranque, P. Nfat transcription factors regulate survival, proliferation, migration, and differentiation of neural precursor cells. Glia 2015, 63, 987–1004. [Google Scholar] [CrossRef] [PubMed]
- Lott, I.T.; Dierssen, M. Cognitive deficits and associated neurological complications in individuals with down’s syndrome. Lancet Neurol. 2010, 9, 623–633. [Google Scholar] [CrossRef]
- Wegiel, J.; Gong, C.-X.; Hwang, Y.-W. The role of dyrk1a in neurodegenerative diseases. FEBS J. 2011, 278, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Stringer, M.; Abeysekera, I.; Dria, K.J.; Roper, R.J.; Goodlett, C.R. Low dose egcg treatment beginning in adolescence does not improve cognitive impairment in a down syndrome mouse model. Pharmacol. Biochem. Behav. 2015, 138, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Stringer, M.; Abeysekera, I.; Thomas, J.; LaCombe, J.; Stancombe, K.; Stewart, R.J.; Dria, K.J.; Wallace, J.M.; Goodlett, C.R.; Roper, R.J. Epigallocatechin-3-gallate (egcg) consumption in the ts65dn model of down syndrome fails to improve behavioral deficits and is detrimental to skeletal phenotypes. Physiol. Behav. 2017, 177, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Altafaj, X.; Martín, E.D.; Ortiz-Abalia, J.; Valderrama, A.; Lao-Peregrín, C.; Dierssen, M.; Fillat, C. Normalization of dyrk1a expression by aav2/1-shdyrk1a attenuates hippocampal-dependent defects in the ts65dn mouse model of down syndrome. Neurobiol. Dis. 2013, 52, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.M.; Block, A.; Tong, S.; Davisson, M.T.; Gardiner, K.J. Age exacerbates abnormal protein expression in a mouse model of down syndrome. Neurobiol. Aging 2017, 57, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.M.; Sturgeon, X.; Ellison, M.; Davisson, M.T.; Gardiner, K.J. Loss of correlations among proteins in brains of the ts65dn mouse model of down syndrome. J. Proteome Res. 2012, 11, 1251–1263. [Google Scholar] [CrossRef] [PubMed]
- García-Cerro, S.; Martínez, P.; Vidal, V.; Corrales, A.; Flórez, J.; Vidal, R.; Rueda, N.; Arbonés, M.L.; Martínez-Cué, C. Overexpression of dyrk1a is implicated in several cognitive, electrophysiological and neuromorphological alterations found in a mouse model of down syndrome. PLoS ONE 2014, 9, e106572. [Google Scholar] [CrossRef] [PubMed]
- García-Cerro, S.; Vidal, V.; Lantigua, S.; Berciano, M.T.; Lafarga, M.; Ramos-Cabrer, P.; Padro, D.; Rueda, N.; Martínez-Cué, C. Cerebellar alterations in a model of down syndrome: The role of the dyrk1a gene. Neurobiol. Dis. 2018, 110, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Souchet, B.; Guedj, F.; Sahún, I.; Duchon, A.; Daubigney, F.; Badel, A.; Yanagawa, Y.; Barallobre, M.J.; Dierssen, M.; Yu, E.; et al. Excitation/inhibition balance and learning are modified by dyrk1a gene dosage. Neurobiol. Dis. 2014, 69, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, A.; Lacroix, T.; Stasko, M.R.; Scott-McKean, J.J.; Costa, A.C.S.; Gardiner, K.J. Molecular responses of the ts65dn and ts1cje mouse models of down syndrome to mk-801. Genes Brain Behav. 2008, 7, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, K.J. Pharmacological approaches to improving cognitive function in down syndrome: Current status and considerations. Drug Des. Devel. Ther. 2014, 9, 103–125. [Google Scholar] [CrossRef] [PubMed]
- Stagni, F.; Giacomini, A.; Guidi, S.; Ciani, E.; Bartesaghi, R. Timing of therapies for down syndrome: The sooner, the better. Front. Behav. Neurosci. 2015, 9, 265. [Google Scholar] [CrossRef] [PubMed]
- Guedj, F.; Sébrié, C.; Rivals, I.; Ledru, A.; Paly, E.; Bizot, J.C.; Smith, D.; Rubin, E.; Gillet, B.; Arbones, M.; et al. Green tea polyphenols rescue of brain defects induced by overexpression of Dyrk1a. PLoS ONE 2009, 4, e4606. [Google Scholar] [CrossRef] [PubMed]
- Pons-Espinal, M.; Martinez de Lagran, M.; Dierssen, M. Environmental enrichment rescues dyrk1a activity and hippocampal adult neurogenesis in tgdyrk1a. Neurobiol. Dis. 2013, 60, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Thomazeau, A.; Lassalle, O.; Iafrati, J.; Souchet, B.; Guedj, F.; Janel, N.; Chavis, P.; Delabar, J.; Manzoni, O.J. Prefrontal deficits in a murine model overexpressing the down syndrome candidate gene Dyrk1a. J. Neurosci. 2014, 34, 1138–1147. [Google Scholar] [CrossRef] [PubMed]
- Souchet, B.; Guedj, F.; Penke-Verdier, Z.; Daubigney, F.; Duchon, A.; Herault, Y.; Bizot, J.-C.; Janel, N.; Créau, N.; Delatour, B.; et al. Pharmacological correction of excitation/inhibition imbalance in down syndrome mouse models. Front. Behav. Neurosci. 2015, 9, 267. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, R.; De Sola, S.; Pons, M.; Duchon, A.; de Lagran, M.M.; Farré, M.; Fitó, M.; Benejam, B.; Langohr, K.; Rodriguez, J.; et al. Epigallocatechin-3-gallate, a dyrk1a inhibitor, rescues cognitive deficits in down syndrome mouse models and in humans. Mol. Nut. Food Res. 2014, 58, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Abalia, J.; Sahún, I.; Altafaj, X.; Andreu, N.; Estivill, X.; Dierssen, M.; Fillat, C. Targeting dyrk1a with aavshrna attenuates motor alterations in tgdyrk1a, a mouse model of down syndrome. Am. J. Human Genet. 2008, 83, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Catuara-Solarz, S.; Espinosa-Carrasco, J.; Erb, I.; Langohr, K.; Notredame, C.; Gonzalez, J.R.; Dierssen, M. Principal component analysis of the effects of environmental enrichment and (-)-epigallocatechin-3-gallate on age-associated learning deficits in a mouse model of down syndrome. Front. Behav. Neurosci. 2015, 9, 330. [Google Scholar] [CrossRef] [PubMed]
- Stagni, F.; Giacomini, A.; Emili, M.; Trazzi, S.; Guidi, S.; Sassi, M.; Ciani, E.; Rimondini, R.; Bartesaghi, R. Short- and long-term effects of neonatal pharmacotherapy with epigallocatechin-3-gallate on hippocampal development in the Ts65dn mouse model of down syndrome. Neurosci. 2016, 333, 277–301. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Ramakrishna, N.; Wieraszko, A.; Hwang, Y.W. Promotion of neuronal plasticity by (-)-epigallocatechin-3-gallate. Neurochem. Res. 2008, 33, 776–783. [Google Scholar] [CrossRef] [PubMed]
- Hibaoui, Y.; Feki, A. Human pluripotent stem cells: Applications and challenges in neurological diseases. Front. Physiol. 2012, 3, 267. [Google Scholar] [CrossRef] [PubMed]
- Hibaoui, Y.; Feki, A. Concise review: Methods and cell types used to generate down syndrome induced pluripotent stem cells. J. Clin. Med. 2015, 4, 696–714. [Google Scholar] [CrossRef] [PubMed]
- Hibaoui, Y.; Grad, I.; Letourneau, A.; Santoni, F.A.; Antonarakis, S.E.; Feki, A. Data in brief: Transcriptome analysis of induced pluripotent stem cells from monozygotic twins discordant for trisomy 21. Genom. Data 2014, 2, 226–229. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, R.; de Sola, S.; Hernandez, G.; Farré, M.; Pujol, J.; Rodriguez, J.; Espadaler, J.M.; Langohr, K.; Cuenca-Royo, A.; Principe, A.; et al. Safety and efficacy of cognitive training plus epigallocatechin-3-gallate in young adults with down’s syndrome (tesdad): A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016, 15, 801–810. [Google Scholar] [CrossRef]
- Noll, C.; Planque, C.; Ripoll, C.; Guedj, F.; Diez, A.; Ducros, V.; Belin, N.; Duchon, A.; Paul, J.-L.; Badel, A.; et al. Dyrk1a, a novel determinant of the methionine-homocysteine cycle in different mouse models overexpressing this down-syndrome-associated kinase. PLoS ONE 2009, 4, e7540. [Google Scholar] [CrossRef] [PubMed]
- Grskovic, M.; Javaherian, A.; Strulovici, B.; Daley, G.Q. Induced pluripotent stem cells—Opportunities for disease modelling and drug discovery. Nat. Rev. Drug Discov. 2011, 10, 915–929. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.; Brown, D.; Alexander, R.; March, R.; Morgan, P.; Satterthwaite, G.; Pangalos, M.N. Lessons learned from the fate of astrazeneca’s drug pipeline: A five-dimensional framework. Nat. Rev. Drug Discov. 2014, 13, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, J.; Miller, P. Phase II and phase III attrition rates 2011–2012. Nat. Rev. Drug Discov. 2013, 12, 569. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Model | Species | Dyrk1a Aneuploidies or Mutations | Phenotypes | References |
|---|---|---|---|---|
| mBACtgDyrk1a | Mouse | Triplication of the mouse Dyrk1a gene | Alterations in brain size and neuronal density. Neurodevelopmental delays, motor abnormalities, altered synaptic plasticity, learning and memory deficits. | [22,23,24] |
| YACtg152F7 (versus YACtg141G6) | Mouse | Yeast artificial chromosome containing PIGP, TTC3, DSCR9, DSCR3 and DYRK1A (for YACtg152F7) versus YAC containing PIGP, TTC3, DSCR9, DSCR3 but not DYRK1A (for YACtg141G6) | Reduced performance in Morris water-maze and fear-conditioning tests consistent with learning and memory defects. Normal hippocampal long term potentiation. | [26] |
| Dyrk1a−/− | Mouse | Loss of function of Dyrk1a | Mid-gestational death (between E10.5 and E13.5 periods). Before death, embryos showed reduction of brain size (30%), growth retardation, morphological developmental delay in the primitive organs. | [21] |
| Dyrk1a−/+ | Mouse | Dyrk1a haploinsufficiency | Reduced brain size and alterations in the density of neurons in various brain regions. The pyramidal cells from the cortex are smaller, with less branching and dentritic spines. Decreased viability, pre- and post-natal growth retardation, developmental delays, motor and learning difficulties. Atypical behaviors including increased anxiety, impaired reactivity to stress. | [20,21] |
| 152F7, 230E8, 141G6, 285E6 and Ts65Dn | Mouse | Segmental trisomies 21 produced by inserting human contiguous fragments in the D21S17-ETS2 region of HSA21 | Only the 152F7 mouse strain which contains a triplication of Dyrk1a, is closer to Ts65Dn mice for reference memory: learning slope and probe test in the Morris water maze. The other cognitive processes such as working, discriminating and episodic memory are not affected in the 152F7 mice. | [27] |
| Individuals with DYRK1A haploinsufficiency | Human | DYRK1A haploinsufficiency resulting from deletions, translocations, frameshift, splice site, nonsense, misense in DYRK1A gene | Intellectual disability, microcephaly, autism spectrum disorder, speech and motor delays, gait disturbances, facial dysmorphology and short stature is common to all individuals. Seizures, feeding difficulties, vision abnormalities and intrauterine growth restriction are present in ~2/3 of all individuals. | [8,9,10,11,12,13,14,15,16,17,18,19] |
| Age of Ts65Dn Mice | Brain Regions | DYRK1A Expression or Activity | References |
|---|---|---|---|
| 2.25 month | Cerebellum, Hippocampus | 1.2-fold increase of Dyrk1a activity but not significant 1.4-fold increase of Dyrk1a activity but not significant | [46] |
| 2.25 month | Cerebellum, Cortex, Hippocampus | Decreased Dyrk1a protein expression (−60%) No difference in Dyrk1a expression No difference in Dyrk1a expression | [47] |
| 3.5 month | Hippocampus | Increased Dyrk1a protein expression (+26%) | [48] |
| 4.4–7.8 month | Cerebellum, Cortex, Hippocampus | Increased Dyrk1a protein expression (+24%) Increased Dyrk1a protein expression (+58%) Increased Dyrk1a protein expression (+31%) | [50] |
| 5–6 month | Cerebellum, Hippocampus | Increased Dyrk1a protein expression (+60%) Increased Dyrk1a protein expression (+58%) | [51,52] |
| 5–6 month | Cerebellum, Cortex, Hippocampus | Increased Dyrk1a protein expression (+60.3%) Increased Dyrk1a protein expression (+64.3%) Increased Dyrk1a protein expression (+68%) | [53] |
| ~6 month | Cerebellum, Cortex, Hippocampus | Increased Dyrk1a protein expression (+22%) Increased Dyrk1a protein expression (+58%) Increased Dyrk1a protein expression (+30%) | [49] |
| 7–8 month | Cortex, Hippocampus | Increased Dyrk1a protein expression (+32%) Increased Dyrk1a protein expression (+41%) | [54] |
| ~12 month | Cerebellum, Cortex, Hippocampus | Increased Dyrk1a protein expression (+98%) Increased Dyrk1a protein expression (+98%) Increased Dyrk1a protein expression (+100%) | [49] |
| Model | Species | Treatment, Intervention | Effect on Brain Structures and Behavior | References |
|---|---|---|---|---|
| TgDyrk1a | Mouse | Normalisation of Dyrk1a through Dyrk1a shRNA in the striatum of 2–3 month-old mice. | Attenuation of the hyperactive behavior, improvement of motor coordination (treadmill test) and PPI (prepulse inhibition) of startle reflex. | [62] |
| TgDyrk1a | Mouse | Decaffeinated MGTE in drinking water (EGCG concentration of 90 mg/mL for a dose of 2–3 mg/day) for 1 month in 3 week-old mice. | Improvement of hippocampal cell proliferation. | [58] |
| TgDyrk1a | Mouse | MGTE lightly caffeinated (45% EGCG) in drinking water (EGCG concentration of 90 mg/mL for a dose of 2–3 mg/day) for 1 month in 3 month-old mice. | Improvement of the MWM spatial learning tasks and NOR test. | [61] |
| mBACtgDyrk1a | Mouse | Green tea extract (45% EGCG) in drinking water with an equivalent dose of 120–200 mg/kg/day EGCG, for 4–6 weeks in 3–4 month-old mice. | Improvement of spine density in prefrontal cortex pyramidal neurons and normalization of LTP. | [59] |
| mBACtgDyrk1a | Mouse | MGTE lightly caffeinated (45% EGCG) in food supplementation (EGCG dose of 10 mg/kg/day or 60 mg/kg/day or 360 mg/kg/day) for 4 weeks in 3–4 month-old mice. | 60 mg/kg/day appeared to be the best compromise in enhancing glutaminergic markers without enhancing GABAergic markers expression in cortex. Rescue of glutaminergic markers expression (but not of GABAergic markers) with all doses in hippocampus. Improvement of the rate of spontaneous alternation. | [60] |
| YACtg152F7 | Mouse | Green tea in drinking water with an equivalent dose of 0.6–1 mg/day EGCG or polyphenon 60 with an equivalent dose of 1.2 mg/day from gestation to adulthood. | Rescue of brain weight and volume (and volume of hypothalamus/thalamus). Improvement of NOR test. | [57] |
| Ts65Dn | Mouse | Normalisation of Dyrk1a through Dyrk1a shRNA in the hippocampus. | Improvement of LTP and initial thigmotaxis but not later thigmotaxic behavior. No improvement of MWM Latency. | [48] |
| Ts65Dn | Mouse | Ts65Dn crossed with Dyrk1a+/− mice. | Improvement of the MWM, fear conditioning test and LTP. Do not improve the density of mature hippocampal granule cells, dentate gyrus volume and subgranular zone area. Do not rescue behavioral alterations (hyperactivity/attention). | [51] |
| Ts65Dn | Mouse | MGTE lightly caffeinated (45% EGCG) in drinking water (EGCG concentration of 90 mg/mL for a dose of 2–3 mg/day) for 1 month in 3 month-old mice. | Improvement of the MWM spatial learning tasks and NOR test. | [61] |
| Ts65Dn | Mouse | Decaffeinated MGTE in drinking water (EGCG dose of 30 mg/kg/day) for 1 month in 5–6 month-old mice. | No improvement in spatial and memory performance. Improvement of the Gallagher index and the thigmotaxis along learning sessions (but no improvement in the latency to reach the escape platform). | [63] |
| Ts65Dn | Mouse | Pure EGCG in drinking water at ~20 mg/kg/day starting from 24 days of age for 3 or 7 weeks. | No improvement in the MCSF, the MWM spatial learning tasks, NOR or balance beam tasks. | [46] |
| Ts65Dn | Mouse | Pure EGCG in drinking water at ~50 mg/kg/day starting from 24 days of age for 7 weeks. | No improvement in the MCSF, the MWM spatial learning tasks, NOR or balance beam tasks. | [47] |
| Ts65Dn | Mouse | Polyphenon 60 * in drinking water at 225 mg/kg/day, containing 27% EGCG (~60mg/kg/day) for 6 weeks in 3–4 month-old mice. | Rescue of GABAergic and glutaminergic markers expression in the cortex and the hippocampus (but not in the cerebellum). Improvement in the Y-maze test. | [60] |
| Ts65Dn | Mouse | Pure EGCG in drinking water at ~25 mg/kg/day starting from postnatal day 3 to postnatal day 15. | Improvement of the proliferation and connectivity in neocortex and hippocampus at P15. However, these improvements measured at P15 disappeared at P45. No improvements in Y-maze and MWM at P45. | [64] |
| NPCs and neurons derived from DS-iPSCs | Human | Normalisation of DYRK1A through DYRK1A shRNA or treatment with 10 µM EGCG of NPCs and neurons derived from DS-iPSCs. | Improvement of proliferation and decrease of apoptosis of NPCs derived from DS-iPSCs. Rescue of neurogenesis impairment of NPCs and neurons derived from DS-iPSCs. Improvement of REST/NRSF, NOTCH and WNT signaling in NPCs derived from DS-iPSCs. | [40] |
| NPCs derived from DS-iPSCs | Human | Treatment of NPCs derived from DS-iPSCs with 5µM ALGERNON (#688). | Increased of proliferation of NPCs derived from DS-iPSCs and increased proportion of these NPCs in G1-phase. | [30] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feki, A.; Hibaoui, Y. DYRK1A Protein, A Promising Therapeutic Target to Improve Cognitive Deficits in Down Syndrome. Brain Sci. 2018, 8, 187. https://doi.org/10.3390/brainsci8100187
Feki A, Hibaoui Y. DYRK1A Protein, A Promising Therapeutic Target to Improve Cognitive Deficits in Down Syndrome. Brain Sciences. 2018; 8(10):187. https://doi.org/10.3390/brainsci8100187
Chicago/Turabian StyleFeki, Anis, and Youssef Hibaoui. 2018. "DYRK1A Protein, A Promising Therapeutic Target to Improve Cognitive Deficits in Down Syndrome" Brain Sciences 8, no. 10: 187. https://doi.org/10.3390/brainsci8100187
APA StyleFeki, A., & Hibaoui, Y. (2018). DYRK1A Protein, A Promising Therapeutic Target to Improve Cognitive Deficits in Down Syndrome. Brain Sciences, 8(10), 187. https://doi.org/10.3390/brainsci8100187