Protein Misfolding and Aggregation as a Therapeutic Target for Polyglutamine Diseases


Abstract
1. The Polyglutamine Diseases
2. Pathogenic Mechanism of Polyglutamine Diseases
2.1. A Gain of Toxicity in PolyQ Diseases
2.2. Expansion Mutation of the PolyQ Tract in Pathogenesis
2.3. Inclusion Bodies and Aggregates of Proteins with Expanded PolyQ Tracts
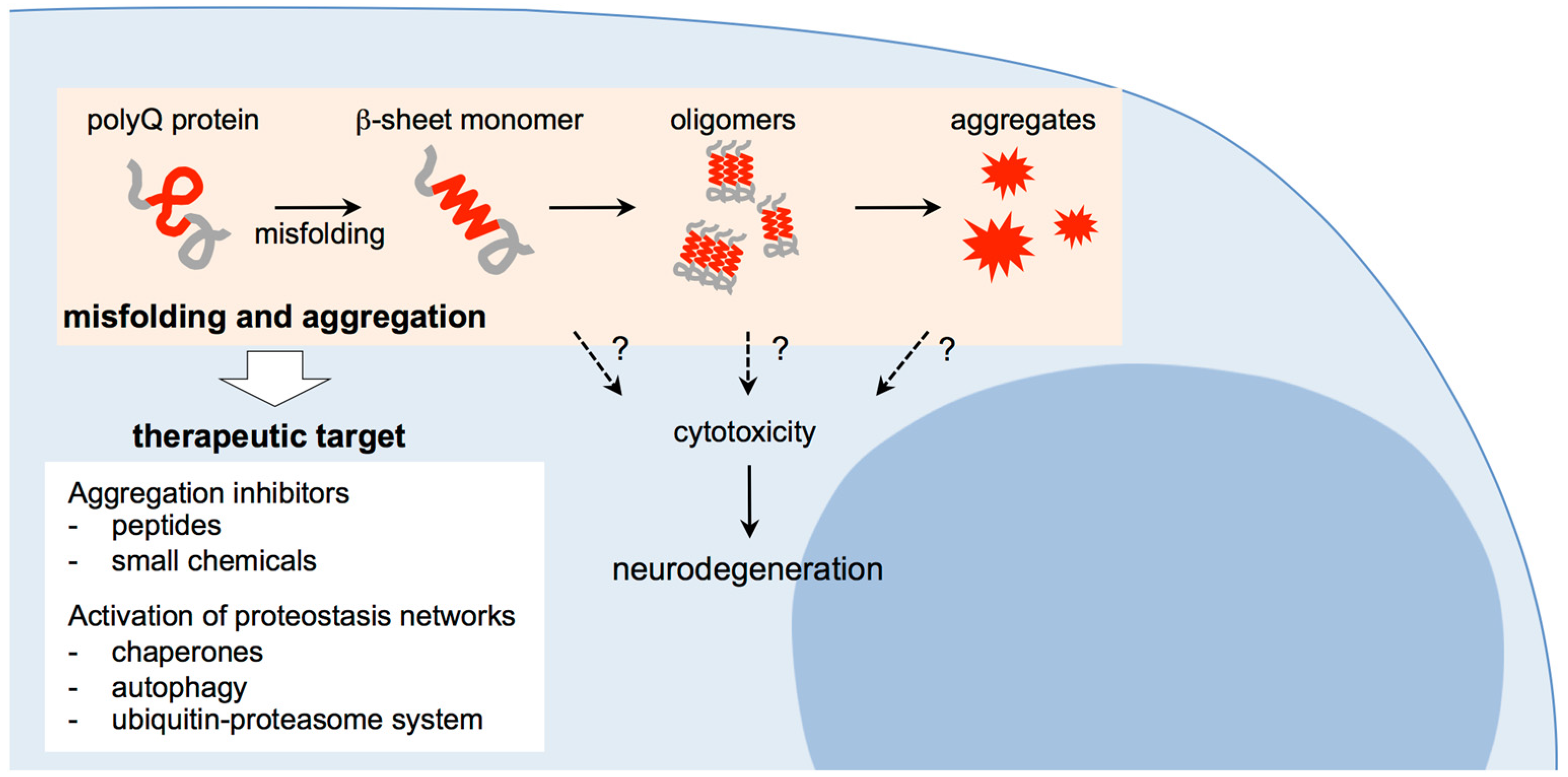
2.4. Abnormal Conformational Changes of Expanded PolyQ Proteins
3. Therapeutic Approaches for Polyglutamine Diseases: Targeting Misfolding and Aggregation of Expanded Polyglutamine Proteins
3.1. Suppression of Polyglutamine Aggregation by Inhibitor Peptides and Chemicals
3.2. Suppression of Polyglutamine Aggregation by Activation of Cellular Proteostasis Networks
4. Gene Silencing: An Emerging Approach Targeting Upstream of Pathological Protein Accumulation
5. Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Gusella, J.F.; MacDonald, M.E. Molecular genetics: Unmasking polyglutamine triggers in neurodegenerative disease. Nat. Rev. Neurosci. 2000, 1, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Orr, H.T.; Zoghbi, H.Y. Trinucleotide repeat disorders. Annu. Rev. Neurosci. 2007, 30, 575–621. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Popiel, H.A. Conformational changes and aggregation of expanded polyglutamine proteins as therapeutic targets of the polyglutamine diseases: Exposed beta-sheet hypothesis. Curr. Pharm. Des. 2008, 14, 3267–3279. [Google Scholar] [CrossRef] [PubMed]
- La Spada, A.R.; Wilson, E.M.; Lubahn, D.B.; Harding, A.E.; Fischbeck, K.H. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 1991, 352, 77–79. [Google Scholar] [CrossRef] [PubMed]
- The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar]
- Banfi, S.; Servadio, A.; Chung, M.Y.; Kwiatkowski, T.J., Jr.; McCall, A.E.; Duvick, L.A.; Shen, Y.; Roth, E.J.; Orr, H.T.; Zoghbi, H.Y. Identification and characterization of the gene causing type 1 spinocerebellar ataxia. Nat. Genet. 1994, 7, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Nagafuchi, S.; Yanagisawa, H.; Ohsaki, E.; Shirayama, T.; Tadokoro, K.; Inoue, T.; Yamada, M. Structure and expression of the gene responsible for the triplet repeat disorder, dentatorubral and pallidoluysian atrophy (DRPLA). Nat. Genet. 1994, 8, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Wexler, N.S.; Young, A.B.; Tanzi, R.E.; Travers, H.; Starosta-Rubinstein, S.; Penney, J.B.; Snodgrass, S.R.; Shoulson, I.; Gomez, F.; Ramos Arroyo, M.A.; et al. Homozygotes for Huntington’s disease. Nature 1987, 326, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Myers, R.H.; Leavitt, J.; Farrer, L.A.; Jagadeesh, J.; McFarlane, H.; Mastromauro, C.A.; Mark, R.J.; Gusella, J.F. Homozygote for Huntington disease. Am. J. Hum. Genet. 1989, 45, 615–618. [Google Scholar] [PubMed]
- Squitieri, F.; Gellera, C.; Cannella, M.; Mariotti, C.; Cislaghi, G.; Rubinsztein, D.C.; Almqvist, E.W.; Turner, D.; Bachoud-Levi, A.C.; Simpson, S.A.; et al. Homozygosity for CAG mutation in Huntington disease is associated with a more severe clinical course. Brain 2003, 126, 946–955. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.E.; Rogaeva, E.A.; Tsuda, T.; Hutterer, J.; St George-Hyslop, P. Homozygous inheritance of the Machado-Joseph disease gene. Ann. Neurol. 1994, 36, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, R.; Futamura, N.; Fujimoto, Y.; Yanagimoto, S.; Horikawa, H.; Suzumura, A.; Takayanagi, T. Spinocerebellar ataxia type 6. Molecular and clinical features of 35 Japanese patients including one homozygous for the CAG repeat expansion. Neurology 1997, 49, 1238–1243. [Google Scholar] [CrossRef] [PubMed]
- Gandelman, K.Y.; Gibson, L.; Meyn, M.S.; Yang-Feng, T.L. Molecular definition of the smallest region of deletion overlap in the Wolf-Hirschhorn syndrome. Am. J. Hum. Genet. 1992, 51, 571–578. [Google Scholar] [PubMed]
- Quigley, C.A.; Friedman, K.J.; Johnson, A.; Lafreniere, R.G.; Silverman, L.M.; Lubahn, D.B.; Brown, T.R.; Wilson, E.M.; Willard, H.F.; French, F.S. Complete deletion of the androgen receptor gene: Definition of the null phenotype of the androgen insensitivity syndrome and determination of carrier status. J. Clin. Endocrinol. Metab. 1992, 74, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Duyao, M.P.; Auerbach, A.B.; Ryan, A.; Persichetti, F.; Barnes, G.T.; McNeil, S.M.; Ge, P.; Vonsattel, J.P.; Gusella, J.F.; Joyner, A.L.; et al. Inactivation of the mouse Huntington’s disease gene homolog Hdh. Science 1995, 269, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, S.; Tanno, Y.; Onodera, O.; Yamazaki, M.; Sato, S.; Ishikawa, A.; Miyatani, N.; Nagashima, M.; Ishikawa, Y.; Sahashi, K.; et al. Strong correlation between the number of CAG repeats in androgen receptor genes and the clinical onset of features of spinal and bulbar muscular atrophy. Neurology 1992, 42, 2300–2302. [Google Scholar] [CrossRef] [PubMed]
- Doyu, M.; Sobue, G.; Mukai, E.; Kachi, T.; Yasuda, T.; Mitsuma, T.; Takahashi, A. Severity of X-linked recessive bulbospinal neuronopathy correlates with size of the tandem CAG repeat in androgen receptor gene. Ann. Neurol. 1992, 32, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Duyao, M.; Ambrose, C.; Myers, R.; Novelletto, A.; Persichetti, F.; Frontali, M.; Folstein, S.; Ross, C.; Franz, M.; Abbott, M.; et al. Trinucleotide repeat length instability and age of onset in Huntington’s disease. Nat. Genet. 1993, 4, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Andrew, S.E.; Goldberg, Y.P.; Kremer, B.; Telenius, H.; Theilmann, J.; Adam, S.; Starr, E.; Squitieri, F.; Lin, B.; Kalchman, M.A.; et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat. Genet. 1993, 4, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Koide, R.; Ikeuchi, T.; Onodera, O.; Tanaka, H.; Igarashi, S.; Endo, K.; Takahashi, H.; Kondo, R.; Ishikawa, A.; Hayashi, T.; et al. Unstable expansion of CAG repeat in hereditary dentatorubral-pallidoluysian atrophy (DRPLA). Nat. Genet. 1994, 6, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Jodice, C.; Malaspina, P.; Persichetti, F.; Novelletto, A.; Spadaro, M.; Giunti, P.; Morocutti, C.; Terrenato, L.; Harding, A.E.; Frontali, M. Effect of trinucleotide repeat length and parental sex on phenotypic variation in spinocerebellar ataxia I. Am. J. Hum. Genet. 1994, 54, 959–965. [Google Scholar] [PubMed]
- Ranum, L.P.; Chung, M.Y.; Banfi, S.; Bryer, A.; Schut, L.J.; Ramesar, R.; Duvick, L.A.; McCall, A.; Subramony, S.H.; Goldfarb, L.; et al. Molecular and clinical correlations in spinocerebellar ataxia type I: Evidence for familial effects on the age at onset. Am. J. Hum. Genet. 1994, 55, 244–252. [Google Scholar] [PubMed]
- Kawaguchi, Y.; Okamoto, T.; Taniwaki, M.; Aizawa, M.; Inoue, M.; Katayama, S.; Kawakami, H.; Nakamura, S.; Nishimura, M.; Akiguchi, I.; et al. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat. Genet. 1994, 8, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Burright, E.N.; Clark, H.B.; Servadio, A.; Matilla, T.; Feddersen, R.M.; Yunis, W.S.; Duvick, L.A.; Zoghbi, H.Y.; Orr, H.T. SCA1 transgenic mice: A model for neurodegeneration caused by an expanded CAG trinucleotide repeat. Cell 1995, 82, 937–948. [Google Scholar] [CrossRef]
- Ikeda, H.; Yamaguchi, M.; Sugai, S.; Aze, Y.; Narumiya, S.; Kakizuka, A. Expanded polyglutamine in the Machado-Joseph disease protein induces cell death in vitro and in vivo. Nat. Genet. 1996, 13, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Warrick, J.M.; Paulson, H.L.; Gray-Board, G.L.; Bui, Q.T.; Fischbeck, K.H.; Pittman, R.N.; Bonini, N.M. Expanded polyglutamine protein forms nuclear inclusions and causes neural degeneration in Drosophila. Cell 1998, 93, 939–949. [Google Scholar] [CrossRef]
- Faber, P.W.; Alter, J.R.; MacDonald, M.E.; Hart, A.C. Polyglutamine-mediated dysfunction and apoptotic death of a Caenorhabditis elegans sensory neuron. Proc. Natl. Acad. Sci. USA 1999, 96, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Tomioka, I.; Ishibashi, H.; Minakawa, E.N.; Motohashi, H.H.; Takayama, O.; Saito, Y.; Popiel, H.A.; Puentes, S.; Owari, K.; Nakatani, T.; et al. Transgenic Monkey Model of the Polyglutamine Diseases Recapitulating Progressive Neurological Symptoms. eNeuro 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Li, L.B.; Yu, Z.; Teng, X.; Bonini, N.M. RNA toxicity is a component of ataxin-3 degeneration in Drosophila. Nature 2008, 453, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Banez-Coronel, M.; Porta, S.; Kagerbauer, B.; Mateu-Huertas, E.; Pantano, L.; Ferrer, I.; Guzman, M.; Estivill, X.; Marti, E. A pathogenic mechanism in Huntington’s disease involves small CAG-repeated RNAs with neurotoxic activity. PLoS Genet. 2012, 8, e1002481. [Google Scholar] [CrossRef] [PubMed]
- Banez-Coronel, M.; Ayhan, F.; Tarabochia, A.D.; Zu, T.; Perez, B.A.; Tusi, S.K.; Pletnikova, O.; Borchelt, D.R.; Ross, C.A.; Margolis, R.L.; et al. RAN Translation in Huntington Disease. Neuron 2015, 88, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Marti, E. RNA toxicity induced by expanded CAG repeats in Huntington’s disease. Brain Pathol. 2016, 26, 779–786. [Google Scholar] [CrossRef] [PubMed]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef] [PubMed]
- Becher, M.W.; Kotzuk, J.A.; Sharp, A.H.; Davies, S.W.; Bates, G.P.; Price, D.L.; Ross, C.A. Intranuclear neuronal inclusions in Huntington’s disease and dentatorubral and pallidoluysian atrophy: Correlation between the density of inclusions and IT15 CAG triplet repeat length. Neurobiol. Dis. 1998, 4, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Paulson, H.L.; Perez, M.K.; Trottier, Y.; Trojanowski, J.Q.; Subramony, S.H.; Das, S.S.; Vig, P.; Mandel, J.L.; Fischbeck, K.H.; Pittman, R.N. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron 1997, 19, 333–344. [Google Scholar] [CrossRef]
- Onodera, O.; Burke, J.R.; Miller, S.E.; Hester, S.; Tsuji, S.; Roses, A.D.; Strittmatter, W.J. Oligomerization of expanded-polyglutamine domain fluorescent fusion proteins in cultured mammalian cells. Biochem. Biophys. Res. Commun. 1997, 238, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Scherzinger, E.; Lurz, R.; Turmaine, M.; Mangiarini, L.; Hollenbach, B.; Hasenbank, R.; Bates, G.P.; Davies, S.W.; Lehrach, H.; Wanker, E.E. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 1997, 90, 549–558. [Google Scholar] [CrossRef]
- Davies, S.W.; Turmaine, M.; Cozens, B.A.; DiFiglia, M.; Sharp, A.H.; Ross, C.A.; Scherzinger, E.; Wanker, E.E.; Mangiarini, L.; Bates, G.P. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 1997, 90, 537–548. [Google Scholar] [CrossRef]
- Cummings, C.J.; Mancini, M.A.; Antalffy, B.; DeFranco, D.B.; Orr, H.T.; Zoghbi, H.Y. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat. Genet. 1998, 19, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Bence, N.F.; Sampat, R.M.; Kopito, R.R. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 2001, 292, 1552–1555. [Google Scholar] [CrossRef] [PubMed]
- Steffan, J.S.; Kazantsev, A.; Spasic-Boskovic, O.; Greenwald, M.; Zhu, Y.Z.; Gohler, H.; Wanker, E.E.; Bates, G.P.; Housman, D.E.; Thompson, L.M. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. USA 2000, 97, 6763–6768. [Google Scholar] [CrossRef] [PubMed]
- Gutekunst, C.A.; Li, S.H.; Yi, H.; Mulroy, J.S.; Kuemmerle, S.; Jones, R.; Rye, D.; Ferrante, R.J.; Hersch, S.M.; Li, X.J. Nuclear and neuropil aggregates in Huntington’s disease: Relationship to neuropathology. J. Neurosci. 1999, 19, 2522–2534. [Google Scholar] [PubMed]
- Kuemmerle, S.; Gutekunst, C.A.; Klein, A.M.; Li, X.J.; Li, S.H.; Beal, M.F.; Hersch, S.M.; Ferrante, R.J. Huntington aggregates may not predict neuronal death in Huntington’s disease. Ann. Neurol. 1999, 46, 842–849. [Google Scholar] [CrossRef]
- Huynh, D.P.; Figueroa, K.; Hoang, N.; Pulst, S.M. Nuclear localization or inclusion body formation of ataxin-2 are not necessary for SCA2 pathogenesis in mouse or human. Nat. Genet. 2000, 26, 44–50. [Google Scholar] [PubMed]
- Saudou, F.; Finkbeiner, S.; Devys, D.; Greenberg, M.E. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 1998, 95, 55–66. [Google Scholar] [CrossRef]
- Muchowski, P.J.; Ning, K.; D’Souza-Schorey, C.; Fields, S. Requirement of an intact microtubule cytoskeleton for aggregation and inclusion body formation by a mutant huntingtin fragment. Proc. Natl. Acad. Sci. USA 2002, 99, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Ordway, J.M.; Tallaksen-Greene, S.; Gutekunst, C.A.; Bernstein, E.M.; Cearley, J.A.; Wiener, H.W.; Dure, L.S., IV; Lindsey, R.; Hersch, S.M.; Jope, R.S.; et al. Ectopically expressed CAG repeats cause intranuclear inclusions and a progressive late onset neurological phenotype in the mouse. Cell 1997, 91, 753–763. [Google Scholar] [CrossRef]
- Kopito, R.R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000, 10, 524–530. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. Opinion: What is the role of protein aggregation in neurodegeneration? Nat. Rev. Mol. Cell Biol. 2005, 6, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Arrasate, M.; Mitra, S.; Schweitzer, E.S.; Segal, M.R.; Finkbeiner, S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 2004, 431, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Perutz, M.F.; Johnson, T.; Suzuki, M.; Finch, J.T. Glutamine repeats as polar zippers: Their possible role in inherited neurodegenerative diseases. Proc. Natl. Acad. Sci. USA 1994, 91, 5355–5358. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Berthelier, V.; Hamilton, J.B.; O’Nuallain, B.; Wetzel, R. Amyloid-like features of polyglutamine aggregates and their assembly kinetics. Biochemistry 2002, 41, 7391–7399. [Google Scholar] [CrossRef] [PubMed]
- Poirier, M.A.; Li, H.; Macosko, J.; Cai, S.; Amzel, M.; Ross, C.A. Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrilization. J. Biol. Chem. 2002, 277, 41032–41037. [Google Scholar] [CrossRef] [PubMed]
- Nucifora, L.G.; Burke, K.A.; Feng, X.; Arbez, N.; Zhu, S.; Miller, J.; Yang, G.; Ratovitski, T.; Delannoy, M.; Muchowski, P.J.; et al. Identification of novel potentially toxic oligomers formed in vitro from mammalian-derived expanded huntingtin exon-1 protein. J. Biol. Chem. 2012, 287, 16017–16028. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Okamoto, Y.; Popiel, H.A.; Fujikake, N.; Toda, T.; Kinjo, M.; Nagai, Y. Detection of polyglutamine protein oligomers in cells by fluorescence correlation spectroscopy. J. Biol. Chem. 2007, 282, 24039–24048. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Kikuchi, S.; Katada, S.; Nagai, Y.; Nishizawa, M.; Onodera, O. Soluble polyglutamine oligomers formed prior to inclusion body formation are cytotoxic. Hum. Mol. Genet. 2008, 17, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Olshina, M.A.; Angley, L.M.; Ramdzan, Y.M.; Tang, J.; Bailey, M.F.; Hill, A.F.; Hatters, D.M. Tracking mutant huntingtin aggregation kinetics in cells reveals three major populations that include an invariant oligomer pool. J. Biol. Chem. 2010, 285, 21807–21816. [Google Scholar] [CrossRef] [PubMed]
- Legleiter, J.; Mitchell, E.; Lotz, G.P.; Sapp, E.; Ng, C.; DiFiglia, M.; Thompson, L.M.; Muchowski, P.J. Mutant huntingtin fragments form oligomers in a polyglutamine length-dependent manner in vitro and in vivo. J. Biol. Chem. 2010, 285, 14777–14790. [Google Scholar] [CrossRef] [PubMed]
- Sathasivam, K.; Lane, A.; Legleiter, J.; Warley, A.; Woodman, B.; Finkbeiner, S.; Paganetti, P.; Muchowski, P.J.; Wilson, S.; Bates, G.P. Identical oligomeric and fibrillar structures captured from the brains of R6/2 and knock-in mouse models of Huntington’s disease. Hum. Mol. Genet. 2010, 19, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Tucker, T.; Ren, H.; Kenan, D.J.; Henderson, B.S.; Keene, J.D.; Strittmatter, W.J.; Burke, J.R. Inhibition of polyglutamine protein aggregation and cell death by novel peptides identified by phage display screening. J. Biol. Chem. 2000, 275, 10437–10442. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Inui, T.; Popiel, H.A.; Fujikake, N.; Hasegawa, K.; Urade, Y.; Goto, Y.; Naiki, H.; Toda, T. A toxic monomeric conformer of the polyglutamine protein. Nat. Struct. Mol. Biol. 2007, 14, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.; Arrasate, M.; Brooks, E.; Libeu, C.P.; Legleiter, J.; Hatters, D.; Curtis, J.; Cheung, K.; Krishnan, P.; Mitra, S.; et al. Identifying polyglutamine protein species in situ that best predict neurodegeneration. Nat. Chem. Biol. 2011, 7, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Trottier, Y.; Lutz, Y.; Stevanin, G.; Imbert, G.; Devys, D.; Cancel, G.; Saudou, F.; Weber, C.; David, G.; Tora, L.; et al. Polyglutamine expansion as a pathological epitope in Huntington’s disease and four dominant cerebellar ataxias. Nature 1995, 378, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Legleiter, J.; Lotz, G.P.; Miller, J.; Ko, J.; Ng, C.; Williams, G.L.; Finkbeiner, S.; Patterson, P.H.; Muchowski, P.J. Monoclonal antibodies recognize distinct conformational epitopes formed by polyglutamine in a mutant huntingtin fragment. J. Biol. Chem. 2009, 284, 21647–21658. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Fujikake, N.; Ohno, K.; Higashiyama, H.; Popiel, H.A.; Rahadian, J.; Yamaguchi, M.; Strittmatter, W.J.; Burke, J.R.; Toda, T. Prevention of polyglutamine oligomerization and neurodegeneration by the peptide inhibitor QBP1 in Drosophila. Hum. Mol. Genet. 2003, 12, 1253–1259. [Google Scholar] [CrossRef] [PubMed]
- Popiel, H.A.; Nagai, Y.; Fujikake, N.; Toda, T. Delivery of the aggregate inhibitor peptide QBP1 into the mouse brain using PTDs and its therapeutic effect on polyglutamine disease mice. Neurosci. Lett. 2009, 449, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Heiser, V.; Scherzinger, E.; Boeddrich, A.; Nordhoff, E.; Lurz, R.; Schugardt, N.; Lehrach, H.; Wanker, E.E. Inhibition of huntingtin fibrillogenesis by specific antibodies and small molecules: Implications for Huntington’s disease therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 6739–6744. [Google Scholar] [CrossRef] [PubMed]
- Lecerf, J.M.; Shirley, T.L.; Zhu, Q.; Kazantsev, A.; Amersdorfer, P.; Housman, D.E.; Messer, A.; Huston, J.S. Human single-chain Fv intrabodies counteract in situ huntingtin aggregation in cellular models of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 4764–4769. [Google Scholar] [CrossRef] [PubMed]
- Khoshnan, A.; Ko, J.; Patterson, P.H. Effects of intracellular expression of anti-huntingtin antibodies of various specificities on mutant huntingtin aggregation and toxicity. Proc. Natl. Acad. Sci. USA 2002, 99, 1002–1007. [Google Scholar] [CrossRef] [PubMed]
- Colby, D.W.; Chu, Y.; Cassady, J.P.; Duennwald, M.; Zazulak, H.; Webster, J.M.; Messer, A.; Lindquist, S.; Ingram, V.M.; Wittrup, K.D. Potent inhibition of huntingtin aggregation and cytotoxicity by a disulfide bond-free single-domain intracellular antibody. Proc. Natl. Acad. Sci. USA 2004, 101, 17616–17621. [Google Scholar] [CrossRef] [PubMed]
- Wolfgang, W.J.; Miller, T.W.; Webster, J.M.; Huston, J.S.; Thompson, L.M.; Marsh, J.L.; Messer, A. Suppression of Huntington’s disease pathology in Drosophila by human single-chain Fv antibodies. Proc. Natl. Acad. Sci. USA 2005, 102, 11563–11568. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.E.; Zhou, H.; McGuire, J.R.; Cerullo, V.; Lee, B.; Li, S.H.; Li, X.J. Suppression of neuropil aggregates and neurological symptoms by an intracellular antibody implicates the cytoplasmic toxicity of mutant huntingtin. J. Cell Biol. 2008, 181, 803–816. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wu, J.; Luo, Y.; Liang, X.; Supnet, C.; Kim, M.W.; Lotz, G.P.; Yang, G.; Muchowski, P.J.; Kodadek, T.; et al. Expanded polyglutamine-binding peptoid as a novel therapeutic agent for treatment of Huntington’s disease. Chem. Biol. 2011, 18, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, I.; Mahlke, C.; Yuan, J. Pivotal role of oligomerization in expanded polyglutamine neurodegenerative disorders. Nature 2003, 421, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Wood, N.I.; Pallier, P.N.; Wanderer, J.; Morton, A.J. Systemic administration of Congo red does not improve motor or cognitive function in R6/2 mice. Neurobiol. Dis. 2007, 25, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Heiser, V.; Engemann, S.; Brocker, W.; Dunkel, I.; Boeddrich, A.; Waelter, S.; Nordhoff, E.; Lurz, R.; Schugardt, N.; Rautenberg, S.; et al. Identification of benzothiazoles as potential polyglutamine aggregation inhibitors of Huntington’s disease by using an automated filter retardation assay. Proc. Natl. Acad. Sci. USA 2002, 99 (Suppl. 4), 16400–16406. [Google Scholar] [CrossRef] [PubMed]
- Hockly, E.; Tse, J.; Barker, A.L.; Moolman, D.L.; Beunard, J.L.; Revington, A.P.; Holt, K.; Sunshine, S.; Moffitt, H.; Sathasivam, K.; et al. Evaluation of the benzothiazole aggregation inhibitors riluzole and PGL-135 as therapeutics for Huntington’s disease. Neurobiol. Dis. 2006, 21, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Ehrnhoefer, D.E.; Duennwald, M.; Markovic, P.; Wacker, J.L.; Engemann, S.; Roark, M.; Legleiter, J.; Marsh, J.L.; Thompson, L.M.; Lindquist, S.; et al. Green tea (-)-epigallocatechin-gallate modulates early events in huntingtin misfolding and reduces toxicity in Huntington’s disease models. Hum. Mol. Genet. 2006, 15, 2743–2751. [Google Scholar] [CrossRef] [PubMed]
- Bonanomi, M.; Natalello, A.; Visentin, C.; Pastori, V.; Penco, A.; Cornelli, G.; Colombo, G.; Malabarba, M.G.; Doglia, S.M.; Relini, A.; et al. Epigallocatechin-3-gallate and tetracycline differently affect ataxin-3 fibrillogenesis and reduce toxicity in spinocerebellar ataxia type 3 model. Hum. Mol. Genet. 2014, 23, 6542–6552. [Google Scholar] [CrossRef] [PubMed]
- Visentin, C.; Pellistri, F.; Natalello, A.; Vertemara, J.; Bonanomi, M.; Gatta, E.; Penco, A.; Relini, A.; De Gioia, L.; Airoldi, C.; et al. Epigallocatechin-3-gallate and related phenol compounds redirect the amyloidogenic aggregation pathway of ataxin-3 towards non-toxic aggregates and prevent toxicity in neural cells and Caenorhabditis elegans animal model. Hum. Mol. Genet. 2017, 26, 3271–3284. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.; Koppenhafer, S.L.; Bonini, N.M.; Paulson, H.L. Analysis of the role of heat shock protein (Hsp) molecular chaperones in polyglutamine disease. J. Neurosci. 1999, 19, 10338–10347. [Google Scholar] [PubMed]
- Warrick, J.M.; Chan, H.Y.; Gray-Board, G.L.; Chai, Y.; Paulson, H.L.; Bonini, N.M. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat. Genet. 1999, 23, 425–428. [Google Scholar] [PubMed]
- Jana, N.R.; Dikshit, P.; Goswami, A.; Kotliarova, S.; Murata, S.; Tanaka, K.; Nukina, N. Co-chaperone CHIP associates with expanded polyglutamine protein and promotes their degradation by proteasomes. J. Biol. Chem. 2005, 280, 11635–11640. [Google Scholar] [CrossRef] [PubMed]
- Miller, V.M.; Nelson, R.F.; Gouvion, C.M.; Williams, A.; Rodriguez-Lebron, E.; Harper, S.Q.; Davidson, B.L.; Rebagliati, M.R.; Paulson, H.L. CHIP suppresses polyglutamine aggregation and toxicity in vitro and in vivo. J. Neurosci. 2005, 25, 9152–9161. [Google Scholar] [CrossRef] [PubMed]
- Al-Ramahi, I.; Lam, Y.C.; Chen, H.K.; de Gouyon, B.; Zhang, M.; Perez, A.M.; Branco, J.; de Haro, M.; Patterson, C.; Zoghbi, H.Y.; et al. CHIP protects from the neurotoxicity of expanded and wild-type ataxin-1 and promotes their ubiquitination and degradation. J. Biol. Chem. 2006, 281, 26714–26724. [Google Scholar] [CrossRef] [PubMed]
- Adachi, H.; Waza, M.; Tokui, K.; Katsuno, M.; Minamiyama, M.; Tanaka, F.; Doyu, M.; Sobue, G. CHIP overexpression reduces mutant androgen receptor protein and ameliorates phenotypes of the spinal and bulbar muscular atrophy transgenic mouse model. J. Neurosci. 2007, 27, 5115–5126. [Google Scholar] [CrossRef] [PubMed]
- Wyttenbach, A.; Sauvageot, O.; Carmichael, J.; Diaz-Latoud, C.; Arrigo, A.P.; Rubinsztein, D.C. Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum. Mol. Genet. 2002, 11, 1137–1151. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, K.; Nakayama, H.; Akagi, T.; Nekooki, M.; Ohtawa, K.; Takio, K.; Hashikawa, T.; Nukina, N. Purification of polyglutamine aggregates and identification of elongation factor-1alpha and heat shock protein 84 as aggregate-interacting proteins. J. Neurosci. 2002, 22, 9267–9277. [Google Scholar] [PubMed]
- Ishihara, K.; Yamagishi, N.; Saito, Y.; Adachi, H.; Kobayashi, Y.; Sobue, G.; Ohtsuka, K.; Hatayama, T. Hsp105alpha suppresses the aggregation of truncated androgen receptor with expanded CAG repeats and cell toxicity. J. Biol. Chem. 2003, 278, 25143–25150. [Google Scholar] [CrossRef] [PubMed]
- Carra, S.; Sivilotti, M.; Chavez Zobel, A.T.; Lambert, H.; Landry, J. HspB8, a small heat shock protein mutated in human neuromuscular disorders, has in vivo chaperone activity in cultured cells. Hum. Mol. Genet. 2005, 14, 1659–1669. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.J.; Zijlstra, M.P.; Kanon, B.; van Waarde-Verhagen, M.A.; Brunt, E.R.; Oosterveld-Hut, H.M.; Carra, S.; Sibon, O.C.; Kampinga, H.H. HSPB7 is the most potent polyQ aggregation suppressor within the HSPB family of molecular chaperones. Hum. Mol. Genet. 2010, 19, 4677–4693. [Google Scholar] [CrossRef] [PubMed]
- Akerfelt, M.; Morimoto, R.I.; Sistonen, L. Heat shock factors: Integrators of cell stress, development and lifespan. Nat. Rev. Mol. Cell. Biol. 2010, 11, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Sittler, A.; Lurz, R.; Lueder, G.; Priller, J.; Lehrach, H.; Hayer-Hartl, M.K.; Hartl, F.U.; Wanker, E.E. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington’s disease. Hum. Mol. Genet. 2001, 10, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Hay, D.G.; Sathasivam, K.; Tobaben, S.; Stahl, B.; Marber, M.; Mestril, R.; Mahal, A.; Smith, D.L.; Woodman, B.; Bates, G.P. Progressive decrease in chaperone protein levels in a mouse model of Huntington’s disease and induction of stress proteins as a therapeutic approach. Hum. Mol. Genet. 2004, 13, 1389–1405. [Google Scholar] [CrossRef] [PubMed]
- Tokui, K.; Adachi, H.; Waza, M.; Katsuno, M.; Minamiyama, M.; Doi, H.; Tanaka, K.; Hamazaki, J.; Murata, S.; Tanaka, F.; et al. 17-DMAG ameliorates polyglutamine-mediated motor neuron degeneration through well-preserved proteasome function in an SBMA model mouse. Hum. Mol. Genet. 2009, 18, 898–910. [Google Scholar] [CrossRef] [PubMed]
- Katsuno, M.; Sang, C.; Adachi, H.; Minamiyama, M.; Waza, M.; Tanaka, F.; Doyu, M.; Sobue, G. Pharmacological induction of heat-shock proteins alleviates polyglutamine-mediated motor neuron disease. Proc. Natl. Acad. Sci. USA 2005, 102, 16801–16806. [Google Scholar] [CrossRef] [PubMed]
- Fujikake, N.; Nagai, Y.; Popiel, H.A.; Okamoto, Y.; Yamaguchi, M.; Toda, T. Heat shock transcription factor 1-activating compounds suppress polyglutamine-induced neurodegeneration through induction of multiple molecular chaperones. J. Biol. Chem. 2008, 283, 26188–26197. [Google Scholar] [CrossRef] [PubMed]
- Popiel, H.A.; Takeuchi, T.; Fujita, H.; Yamamoto, K.; Ito, C.; Yamane, H.; Muramatsu, S.; Toda, T.; Wada, K.; Nagai, Y. Hsp40 Gene Therapy Exerts Therapeutic Effects on Polyglutamine Disease Mice via a Non-Cell Autonomous Mechanism. PLoS ONE 2012, 7, e51069. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Suzuki, M.; Fujikake, N.; Popiel, H.A.; Kikuchi, H.; Futaki, S.; Wada, K.; Nagai, Y. Intercellular chaperone transmission via exosomes contributes to maintenance of protein homeostasis at the organismal level. Proc. Natl. Acad. Sci. USA 2015, 112, E2497–E2506. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.K.; Bauer, P.O.; Kurosawa, M.; Goswami, A.; Washizu, C.; Machida, Y.; Tosaki, A.; Yamada, M.; Knopfel, T.; Nakamura, T.; et al. Blocking acid-sensing ion channel 1 alleviates Huntington’s disease pathology via an ubiquitin-proteasome system-dependent mechanism. Hum. Mol. Genet. 2008, 17, 3223–3235. [Google Scholar] [CrossRef] [PubMed]
- Bauer, P.O.; Wong, H.K.; Oyama, F.; Goswami, A.; Okuno, M.; Kino, Y.; Miyazaki, H.; Nukina, N. Inhibition of Rho kinases enhances the degradation of mutant huntingtin. J. Biol. Chem. 2009, 284, 13153–13164. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.H.; Wang, Y.; Kegel, K.B.; Kazantsev, A.; Apostol, B.L.; Thompson, L.M.; Yoder, J.; Aronin, N.; DiFiglia, M. Autophagy regulates the processing of amino terminal huntingtin fragments. Hum. Mol. Genet. 2003, 12, 3231–3244. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, Y.; Fujikake, N.; Okamoto, Y.; Popiel, H.A.; Hatanaka, Y.; Ueyama, M.; Suzuki, M.; Gaumer, S.; Murata, M.; Wada, K.; et al. p62 plays a protective role in the autophagic degradation of polyglutamine protein oligomers in polyglutamine disease model flies. J. Biol. Chem. 2015, 290, 1442–1453. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; Rubinsztein, D.C. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Huebener, J.; Renna, M.; Bonin, M.; Riess, O.; Rubinsztein, D.C. Autophagy induction reduces mutant ataxin-3 levels and toxicity in a mouse model of spinocerebellar ataxia type 3. Brain 2010, 133, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Machida, Y.; Niu, S.; Ikeda, T.; Jana, N.R.; Doi, H.; Kurosawa, M.; Nekooki, M.; Nukina, N. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat. Med. 2004, 10, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnacliffe, A.; Rubinsztein, D.C. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J. Biol. Chem. 2007, 282, 5641–5652. [Google Scholar] [CrossRef] [PubMed]
- Kruger, U.; Wang, Y.; Kumar, S.; Mandelkow, E.M. Autophagic degradation of tau in primary neurons and its enhancement by trehalose. Neurobiol. Aging 2012, 33, 2291–2305. [Google Scholar] [CrossRef] [PubMed]
- Gomes, C.; Escrevente, C.; Costa, J. Mutant superoxide dismutase 1 overexpression in NSC-34 cells: Effect of trehalose on aggregation, TDP-43 localization and levels of co-expressed glycoproteins. Neurosci. Lett. 2010, 475, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Aguib, Y.; Heiseke, A.; Gilch, S.; Riemer, C.; Baier, M.; Schatzl, H.M.; Ertmer, A. Autophagy induction by trehalose counteracts cellular prion infection. Autophagy 2009, 5, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Shoji-Kawata, S.; Sumpter, R.; Leveno, M.; Campbell, G.R.; Zou, Z.; Kinch, L.; Wilkins, A.D.; Sun, Q.; Pallauf, K.; MacDuff, D.; et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 2013, 494, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Tohnai, G.; Adachi, H.; Katsuno, M.; Doi, H.; Matsumoto, S.; Kondo, N.; Miyazaki, Y.; Iida, M.; Nakatsuji, H.; Qiang, Q.; et al. Paeoniflorin eliminates a mutant AR via NF-YA-dependent proteolysis in spinal and bulbar muscular atrophy. Hum. Mol. Genet. 2014, 23, 3552–3565. [Google Scholar] [CrossRef] [PubMed]
- Bauer, P.O.; Goswami, A.; Wong, H.K.; Okuno, M.; Kurosawa, M.; Yamada, M.; Miyazaki, H.; Matsumoto, G.; Kino, Y.; Nagai, Y.; et al. Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat. Biotechnol. 2010, 28, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Keiser, M.S.; Kordasiewicz, H.B.; McBride, J.L. Gene suppression strategies for dominantly inherited neurodegenerative diseases: Lessons from Huntington’s disease and spinocerebellar ataxia. Hum. Mol. Genet. 2016, 25, R53–R64. [Google Scholar] [CrossRef] [PubMed]
- Fiszer, A.; Krzyzosiak, W.J. Oligonucleotide-based strategies to combat polyglutamine diseases. Nucleic. Acids Res. 2014, 42, 6787–6810. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.; Lucas, J.J.; Hen, R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell 2000, 101, 57–66. [Google Scholar] [CrossRef]
- Wang, Y.L.; Liu, W.; Wada, E.; Murata, M.; Wada, K.; Kanazawa, I. Clinico-pathological rescue of a model mouse of Huntington’s disease by siRNA. Neurosci. Res. 2005, 53, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Harper, S.Q.; Staber, P.D.; He, X.; Eliason, S.L.; Martins, I.H.; Mao, Q.; Yang, L.; Kotin, R.M.; Paulson, H.L.; Davidson, B.L. RNA interference improves motor and neuropathological abnormalities in a Huntington’s disease mouse model. Proc. Natl. Acad. Sci. USA 2005, 102, 5820–5825. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Lebron, E.; Denovan-Wright, E.M.; Nash, K.; Lewin, A.S.; Mandel, R.J. Intrastriatal rAAV-mediated delivery of anti-huntingtin shRNAs induces partial reversal of disease progression in R6/1 Huntington’s disease transgenic mice. Mol. Ther. 2005, 12, 618–633. [Google Scholar] [CrossRef] [PubMed]
- Scoles, D.R.; Meera, P.; Schneider, M.D.; Paul, S.; Dansithong, W.; Figueroa, K.P.; Hung, G.; Rigo, F.; Bennett, C.F.; Otis, T.S.; et al. Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature 2017, 544, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.R.; Rajpal, G.; Dillingham, I.T.; Qutob, M.; Blumenstein, K.G.; Gattis, D.; Hung, G.; Kordasiewicz, H.B.; Paulson, H.L.; McLoughlin, H.S. Evaluation of Antisense Oligonucleotides Targeting ATXN3 in SCA3 Mouse Models. Mol. Ther. Nucleic. Acids 2017, 7, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Sahashi, K.; Katsuno, M.; Hung, G.; Adachi, H.; Kondo, N.; Nakatsuji, H.; Tohnai, G.; Iida, M.; Bennett, C.F.; Sobue, G. Silencing neuronal mutant androgen receptor in a mouse model of spinal and bulbar muscular atrophy. Hum. Mol. Genet. 2015, 24, 5985–5994. [Google Scholar] [CrossRef] [PubMed]
- Kordasiewicz, H.B.; Stanek, L.M.; Wancewicz, E.V.; Mazur, C.; McAlonis, M.M.; Pytel, K.A.; Artates, J.W.; Weiss, A.; Cheng, S.H.; Shihabuddin, L.S.; et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron 2012, 74, 1031–1044. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, A.P.; Yu, Z.; Murray, S.; Peralta, R.; Low, A.; Guo, S.; Yu, X.X.; Cortes, C.J.; Bennett, C.F.; Monia, B.P.; et al. Peripheral androgen receptor gene suppression rescues disease in mouse models of spinal and bulbar muscular atrophy. Cell Rep. 2014, 7, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Lebron, E.; Costa Mdo, C.; Luna-Cancalon, K.; Peron, T.M.; Fischer, S.; Boudreau, R.L.; Davidson, B.L.; Paulson, H.L. Silencing mutant ATXN3 expression resolves molecular phenotypes in SCA3 transgenic mice. Mol. Ther. 2013, 21, 1909–1918. [Google Scholar] [CrossRef] [PubMed]
- Pourshafie, N.; Lee, P.R.; Chen, K.L.; Harmison, G.G.; Bott, L.C.; Katsuno, M.; Sobue, G.; Burnett, B.G.; Fischbeck, K.H.; Rinaldi, C. MiR-298 Counteracts Mutant Androgen Receptor Toxicity in Spinal and Bulbar Muscular Atrophy. Mol. Ther. 2016, 24, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Zhang, L.; Long, Z.; Chen, Z.; Hou, X.; Wang, C.; Peng, H.; Wang, J.; Li, J.; Duan, R.; et al. miR-25 alleviates polyQ-mediated cytotoxicity by silencing ATXN3. FEBS Lett. 2014, 588, 4791–4798. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Adachi, H.; Katsuno, M.; Minamiyama, M.; Jiang, Y.M.; Huang, Z.; Doi, H.; Matsumoto, S.; Kondo, N.; Iida, M.; et al. Viral delivery of miR-196a ameliorates the SBMA phenotype via the silencing of CELF2. Nat. Med. 2012, 18, 1136–1141. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Du, X.; Muramatsu, S.; Gomez, C.M. An miRNA-mediated therapy for SCA6 blocks IRES-driven translation of the CACNA1A second cistron. Sci. Transl. Med. 2016, 8, 347ra94. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Watanabe, S.; Murata, M.; Furuya, H.; Kanazawa, I.; Wada, K.; Hohjoh, H. Tailor-made RNAi knockdown against triplet repeat disease-causing alleles. Proc. Natl. Acad. Sci. USA 2010, 107, 21731–21736. [Google Scholar] [CrossRef] [PubMed]
- Drouet, V.; Ruiz, M.; Zala, D.; Feyeux, M.; Auregan, G.; Cambon, K.; Troquier, L.; Carpentier, J.; Aubert, S.; Merienne, N.; et al. Allele-specific silencing of mutant huntingtin in rodent brain and human stem cells. PLoS ONE 2014, 9, e99341. [Google Scholar] [CrossRef] [PubMed]
- Nobrega, C.; Nascimento-Ferreira, I.; Onofre, I.; Albuquerque, D.; Deglon, N.; de Almeida, L.P. RNA interference mitigates motor and neuropathological deficits in a cerebellar mouse model of Machado-Joseph disease. PLoS ONE 2014, 9, e100086. [Google Scholar] [CrossRef] [PubMed]
- Rue, L.; Banez-Coronel, M.; Creus-Muncunill, J.; Giralt, A.; Alcala-Vida, R.; Mentxaka, G.; Kagerbauer, B.; Zomeno-Abellan, M.T.; Aranda, Z.; Venturi, V.; et al. Targeting CAG repeat RNAs reduces Huntington’s disease phenotype independently of huntingtin levels. J. Clin. Investig. 2016, 126, 4319–4330. [Google Scholar] [CrossRef] [PubMed]
- Watson, L.M.; Wood, M.J. RNA therapy for polyglutamine neurodegenerative diseases. Expert Rev. Mol. Med. 2012, 14, e3. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Nagai, Y.; Tucker, T.; Strittmatter, W.J.; Burke, J.R. Amino acid sequence requirements of peptides that inhibit polyglutamine-protein aggregation and cell death. Biochem. Biophys. Res. Commun. 2001, 288, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Popiel, H.A.; Nagai, Y.; Toda, T.; Yoshimitsu, Y.; Ohno, H.; Oishi, S.; Fujii, N. Structure-activity relationship study on polyglutamine binding peptide QBP1. Bioorg. Med. Chem. 2009, 17, 1259–1263. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Martin, F.; Hervas, R.; Carrion-Vazquez, M.; Laurents, D.V. NMR spectroscopy reveals a preferred conformation with a defined hydrophobic cluster for polyglutamine binding peptide 1. Arch. Biochem. Biophys. 2014, 558, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Katsuno, M.; Adachi, H.; Doyu, M.; Minamiyama, M.; Sang, C.; Kobayashi, Y.; Inukai, A.; Sobue, G. Leuprorelin rescues polyglutamine-dependent phenotypes in a transgenic mouse model of spinal and bulbar muscular atrophy. Nat. Med. 2003, 9, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Banno, H.; Katsuno, M.; Suzuki, K.; Takeuchi, Y.; Kawashima, M.; Suga, N.; Takamori, M.; Ito, M.; Nakamura, T.; Matsuo, K.; et al. Phase 2 trial of leuprorelin in patients with spinal and bulbar muscular atrophy. Ann. Neurol. 2009, 65, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Katsuno, M.; Banno, H.; Suzuki, K.; Takeuchi, Y.; Kawashima, M.; Yabe, I.; Sasaki, H.; Aoki, M.; Morita, M.; Nakano, I.; et al. Efficacy and safety of leuprorelin in patients with spinal and bulbar muscular atrophy (JASMITT study): A multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2010, 9, 875–884. [Google Scholar] [CrossRef]
- Fernandez-Rhodes, L.E.; Kokkinis, A.D.; White, M.J.; Watts, C.A.; Auh, S.; Jeffries, N.O.; Shrader, J.A.; Lehky, T.J.; Li, L.; Ryder, J.E.; et al. Efficacy and safety of dutasteride in patients with spinal and bulbar muscular atrophy: A randomised placebo-controlled trial. Lancet Neurol. 2011, 10, 140–147. [Google Scholar] [CrossRef]
- Hashizume, A.; Katsuno, M.; Suzuki, K.; Hirakawa, A.; Hijikata, Y.; Yamada, S.; Inagaki, T.; Banno, H.; Sobue, G. Long-term treatment with leuprorelin for spinal and bulbar muscular atrophy: Natural history-controlled study. J. Neurol. Neurosurg. Psychiatry 2017. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Disease | Gene | CAG Repeat | |
|---|---|---|---|
| Normal | Disease | ||
| Spinal and bulbar muscular atrophy (SBMA) | androgen receptor | 9–36 | 38–65 |
| Huntington’s disease (HD) | huntingtin | 6–35 | 36–180 |
| Spinocerebeller ataxia type 1 (SCA1) | ataxin-1 | 6–39 | 39–83 |
| Spinocerebeller ataxia type 2 (SCA2) | ataxin-2 | 14–32 | 32–200 |
| Spinocerebeller ataxia type 3 (SCA3) | ataxin-3 | 12–41 | 55–84 |
| Spinocerebeller ataxia type 6 (SCA6) | α1A calcium channel | 4–19 | 20–33 |
| Spinocerebeller ataxia type 7 (SCA7) | ataxin-7 | 4–35 | 37–306 |
| Spinocerebeller ataxia type 17 (SCA17) | TATA-binding protein | 25–44 | 46–63 |
| Dentatorubral pallidoluysian atrophy (DRPLA) | atrophin-1 | 6–36 | 49–88 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takeuchi, T.; Nagai, Y. Protein Misfolding and Aggregation as a Therapeutic Target for Polyglutamine Diseases. Brain Sci. 2017, 7, 128. https://doi.org/10.3390/brainsci7100128
Takeuchi T, Nagai Y. Protein Misfolding and Aggregation as a Therapeutic Target for Polyglutamine Diseases. Brain Sciences. 2017; 7(10):128. https://doi.org/10.3390/brainsci7100128
Chicago/Turabian StyleTakeuchi, Toshihide, and Yoshitaka Nagai. 2017. "Protein Misfolding and Aggregation as a Therapeutic Target for Polyglutamine Diseases" Brain Sciences 7, no. 10: 128. https://doi.org/10.3390/brainsci7100128
APA StyleTakeuchi, T., & Nagai, Y. (2017). Protein Misfolding and Aggregation as a Therapeutic Target for Polyglutamine Diseases. Brain Sciences, 7(10), 128. https://doi.org/10.3390/brainsci7100128