
The Effects of Hypoxia and Inflammation on Synaptic Signaling in the CNS


Abstract
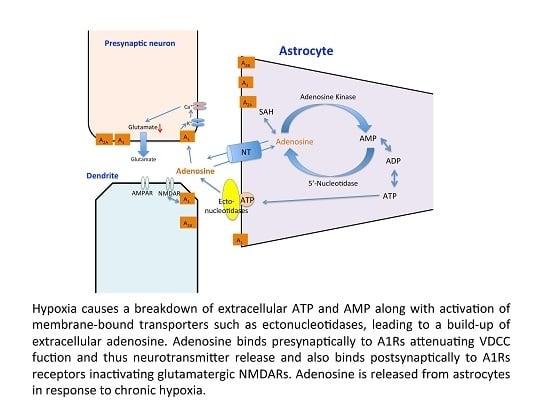
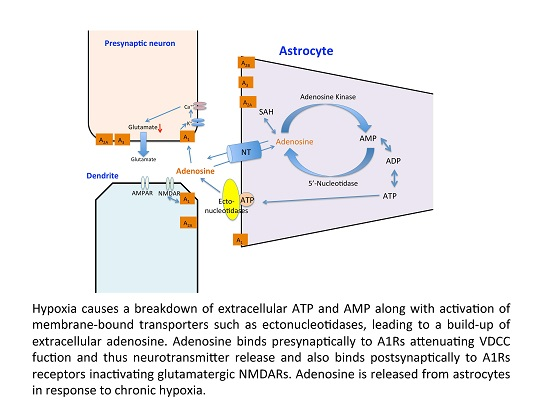
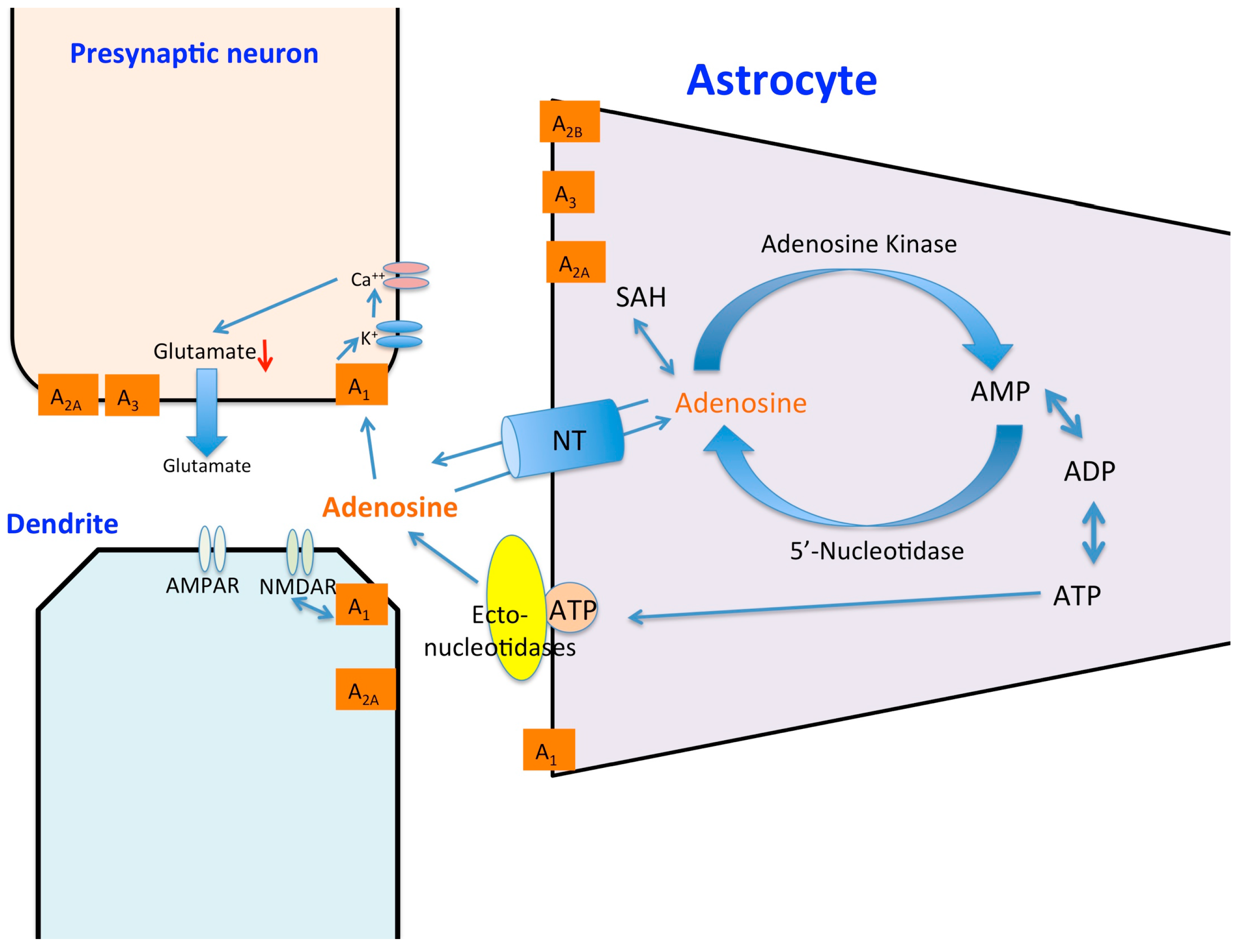
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Hypoxia and Synaptic Signaling
3. Hypoxia and Synaptic Plasticity
4. Hypoxia and Neuroinflammation in the CNS
5. TNF-α and Hypoxia
6. TNF-α and Synaptic Plasticity
7. TNF-α, Hypoxia and Synaptic Plasticity
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| A1Rs | Adenosine A1 receptors |
| AMP | adenosine monophosphate |
| AMPARs | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors |
| ATP | adenosine triphosphate |
| BDNF | brain-derived neurotrophic factor |
| CaMKII | calmodulin-dependent protein kinase II |
| CIH | chronic intermittent hypoxia |
| CNS | central nervous system |
| CREB | cAMP response element-binding protein |
| DMOG | dimethyloxaloglycine |
| DPCPX | 8-cyclopentyl-1,3-dipropylxanthine |
| EPSP | excitatory post-synaptic potential |
| ERK | extracellular signal-regulated kinases |
| HIF | hypoxia inducible factors |
| HRE | hypoxic responsive element |
| IL-1β | interleukin-1beta |
| iNOS | inducible nitric oxide synthase |
| LPS | lipopolysaccharide |
| LTP | long-term potentiation |
| NFkB | nuclear factor kB |
| NMDAR | N-methyl-d-aspartate receptors |
| PHDs | prolyl hydroxyl domains |
| PI3K | phosphatidyl-inositol-3 kinase |
| ROCK | Rho-associated protein kinase |
| ROS | reactive oxygen species |
| TNF-α | tumor necrosis factor alpha |
References
- Sharp, F.R.; Bernaudin, M. HIF1 and oxygen sensing in the brain. Nat. Rev. Neurosci. 2004, 5, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Bickler, P.E.; Donohoe, P.H. Adaptive responses of vertebrate neurons to hypoxia. J. Exp. Biol. 2002, 205, 3579–3586. [Google Scholar]
- Eltzschig, H.K.; Bratton, D.L.; Colgan, S.P. Targeting hypoxia signaling for the treatment of ischaemic and inflammatory diseases. Nat. Rev. Drug Discov. 2014, 13, 852–869. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, A.; O’Connor, J.J. Hypoxia-inducible factor signaling mechanisms in the central nervous system. Acta Physiol. 2013, 208, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Moroz, E.; Carlin, S.; Dyomina, K.; Burke, S.; Thaler, H.T.; Blasberg, R.; Serganova, I. Real-time imaging of HIF-1α stabilization and degradation. PLoS ONE 2009, 4, e5077. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.D.; Coleman, M.L.; Pugh, C.W. Hypoxia, hypoxia-inducible factors (HIF), HIF hydroxylases and oxygen sensing. Cell. Mol. Life Sci. 2009, 66, 3539–3554. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 2014, 9, 47–71. [Google Scholar] [CrossRef] [PubMed]
- Kamel, H.; Iadecola, C. Brain-immune interactions and ischemic stroke: Clinical implications. Arch Neurol. 2012, 69, 576–581. [Google Scholar] [PubMed]
- Amantea, D.; Micieli, G.; Tassorelli, C.; Cuartero, M.I.; Ballesteros, I.; Certo, M.; Moro, M.A.; Lizasoain, I.; Bagetta, G. Rational modulation of the innate immune system for neuroprotection in ischemic stroke. Front. Neurosci. 2015, 9, 147. [Google Scholar] [CrossRef] [PubMed]
- Lipton, P.; Whittingham, T.S. The effect of hypoxia on evoked potentials in the in vitro hippocampus. J. Physiol. 1979, 287, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Björklund, O.; Shang, M.; Tonazzini, I.; Daré, E.; Fredholm, B.B. Adenosine A1 and A3 receptors protect astrocytes from hypoxic damage. Eur. J. Pharmacol. 2008, 596, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Palmer, T.M.; Stiles, G.L. Adenosine receptors. Neuropharmacology 1995, 34, 683–694. [Google Scholar] [CrossRef]
- Reppert, S.M.; Weaver, D.R.; Stehle, J.H.; Rivkees, S.A. Molecular cloning and characterization of a rat A1-adenosine receptor that is widely expressed in brain and spinal cord. Mol. Endocrinol. 1991, 5, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- McCool, B.A.; Farroni, J.S. A1 adenosine receptors inhibit multiple voltage-gated Ca2+ channel subtypes in acutely isolated rat basolateral amygdala neurons. Br. J. Pharmacol. 2001, 132, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Boison, D. Adenosine kinase, epilepsy and stroke: Mechanisms and therapies. Trends Pharmacol. Sci. 2006, 27, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Görlach, A. Control of adenosine transport by hypoxia. Circ. Res. 2005, 97, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Otsuguro, K.; Ohta, T.; Ito, S. Adenosine and inosine release during hypoxia in the isolated spinal cord of neonatal rats. Br. J. Pharmacol. 2010, 161, 1806–1816. [Google Scholar] [CrossRef] [PubMed]
- Morote-Garcia, J.C.; Rosenberger, P.; Nivillac, N.M.; Coe, I.R.; Eltzschig, H.K. Hypoxia-inducible factor-dependent repression of equilibrative nucleoside transporter 2 attenuates mucosal inflammation during intestinal hypoxia. Gastroenterology 2009, 136, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.S.; Cho, J.H.; Lee, M.G.; Jang, I.S. Enzymatic conversion of ATP to adenosine contributes to ATP-induced inhibition of glutamate release in rat medullary dorsal horn neurons. Neuropharmacology 2015, 93, 94–102. [Google Scholar] [CrossRef] [PubMed]
- De Mendonça, A.; Sebastião, A.M.; Ribeiro, J.A. Inhibition of NMDA receptor-mediated currents in isolated rat hippocampal neurones by adenosine A1 receptor activation. Neuroreport 1995, 6, 1097–1100. [Google Scholar] [CrossRef] [PubMed]
- Arrigoni, E.; Crocker, A.J.; Saper, C.B.; Greene, R.W.; Scammell, T.E. Deletion of presynaptic adenosine A1 receptors impairs the recovery of synaptic transmission after hypoxia. Neuroscience 2005, 132, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Bai, X.; Li, T.; Chen, F.; Dong, Q.; Zhao, Y.; Liu, X. Decreased extracellular adenosine levels lead to loss of hypoxia-induced neuroprotection after repeated episodes of exposure to hypoxia. PLoS ONE 2013, 8, e57065. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Malik, A.; Choi, H.B.; Ko, R.W.; Dissing-Olesen, L.; MacVicar, B.A. Microglial CR3 activation triggers long-term synaptic depression in the hippocampus via NADPH oxidase. Neuron 2014, 82, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.K.; Vozdek, R.; Bhatla, N.; Horvitz, H.R. CYSL-1 interacts with the O2-sensing hydroxylase EGL-9 to promote H2S-modulated hypoxia-induced behavioral plasticity in C. elegans. Neuron 2012, 73, 925–940. [Google Scholar] [CrossRef] [PubMed]
- Park, E.C.; Ghose, P.; Shao, Z.; Ye, Q.; Kang, L.; Xu, X.Z.; Powell-Coffman, J.A.; Rongo, C. Hypoxia regulates glutamate receptor trafficking through an HIF-independent mechanism. EMBO J. 2012, 31, 1379–1393. [Google Scholar] [CrossRef] [PubMed]
- Epstein, A.C.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef]
- Wang, J.; Ming, H.; Chen, R.; Ju, J.M.; Peng, W.D.; Zhang, G.X.; Liu, C.F. CIH-induced neurocognitive impairments are associated with hippocampal Ca2+ overload, apoptosis, and dephosphorylation of ERK1/2 and CREB that are mediated by overactivation of NMDARs. Brain Res. 2015, 1625, 64–72. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.W.; Johnston, M.V. Physiological and pathophysiological roles of excitatory amino acids during central nervous system development. Brain Res. Brain Res. Rev. 1990, 15, 41–70. [Google Scholar] [CrossRef]
- Wall, A.M.; Corcoran, A.E.; O’Halloran, K.D.; O’Connor, J.J. Effects of prolyl-hydroxylase inhibition and chronic intermittent hypoxia on synaptic transmission and plasticity in the rat CA1 and dentate gyrus. Neurobiol. Dis. 2014, 62, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Leung, K.L.; Chen, L.; Chan, Y.S.; Ng, P.C.; Fok, T.F.; Wing, Y.K.; Ke, Y.; Li, A.M.; Yung, W.H. Brain-derived neurotrophic factor rescues and prevents chronic intermittent hypoxia-induced impairment of hippocampal long-term synaptic plasticity. Neurobiol. Dis. 2010, 40, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Jensen, F.E.; Wang, C.; Stafstrom, C.E.; Liu, Z.; Geary, C.; Stevens, M.C. Acute and chronic increases in excitability in rat hippocampal slices after perinatal hypoxia in vivo. J. Neurophysiol. 1998, 79, 73–81. [Google Scholar] [PubMed]
- Zhou, C.; Lippman, J.J.; Sun, H.; Jensen, F.E. Hypoxia-induced neonatal seizures diminish silent synapses and long-term potentiation in hippocampal CA1 neurons. J. Neurosci. 2011, 31, 18211–18222. [Google Scholar] [CrossRef] [PubMed]
- Kerchner, G.A.; Nicoll, R.A. Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nat. Rev. Neurosci. 2008, 9, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Chiu, G.S.; Chatterjee, D.; Darmody, P.T.; Walsh, J.P.; Meling, D.D.; Johnson, R.W.; Freund, G.G. Hypoxia/reoxygenation impairs memory formation via adenosine-dependent activation of caspase 1. J. Neurosci. 2012, 32, 13945–13955. [Google Scholar] [CrossRef] [PubMed]
- Row, B.W.; Liu, R.; Xu, W.; Kheirandish, L.; Gozal, D. Intermittent hypoxia is associated with oxidative stress and spatial learning deficits in the rat. Am. J. Respir. Crit. Care Med. 2003, 167, 1548–1553. [Google Scholar] [CrossRef] [PubMed]
- Arai, A.; Kessler, M.; Lynch, G. The effects of adenosine on the development of long-term potentiation. Neurosci. Lett. 1990, 119, 41–44. [Google Scholar] [CrossRef]
- Arai, A.; Vanderklish, P.; Kessler, M.; Lee, K.; Lynch, G. A brief period of hypoxia causes proteolysis of cytoskeletal proteins in hippocampal slices. Brain Res. 1990, 555, 276–280. [Google Scholar] [CrossRef]
- Lyubkin, M.; Durand, D.M.; Haxhiu, M.A. Interaction between tetanus long-term potentiation and hypoxia-induced potentiation in the rat hippocampus. J. Neurophysiol. 1997, 78, 2475–2482. [Google Scholar] [PubMed]
- Hammond, C.; Crépel, V.; Gozlan, H.; Ben-Ari, Y. Anoxic LTP sheds light on the multiple facets of NMDA receptors. Trends Neurosci. 1994, 17, 497–503. [Google Scholar] [CrossRef]
- Hsu, K.S.; Huang, C.C. Characterization of the anoxia-induced long-term synaptic potentiation in area CA1 of the rat hippocampus. Br. J. Pharmacol. 1997, 122, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Hsu, K.S. Nitric oxide signaling is required for the generation of anoxia-induced long-term potentiation in the hippocampus. Eur. J. Neurosci. 1997, 9, 2202–2206. [Google Scholar] [CrossRef] [PubMed]
- Weilinger, N.L.; Tang, P.L.; Thompson, R.J. Anoxia-induced NMDA receptor activation opens pannexin channels via Src family kinases. J. Neurosci. 2012, 32, 12579–12588. [Google Scholar] [CrossRef] [PubMed]
- Kass, I.S.; Lipton, T.P. Calcium and Long-term transmission damage following anoxia in dentate gyrus and CA1 regions of the rat hippocampal slice. J. Physiol. 1986, 378, 313–334. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, P.; Humeres, A.; Elgueta, C.; Kirkwood, A.; Hidalgo, C.; Núñez, M.T. Iron mediates N-methyl-d-aspartate receptor-dependent stimulation of calcium-induced pathways and hippocampal synaptic plasticity. J. Biol. Chem. 2011, 286, 13382–13392. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, A.; Kunze, R.; Harney, S.C.; Breier, G.; Marti, H.H.; O’Connor, J.J. A role for prolyl hydroxylase domain proteins in hippocampal synaptic plasticity. Hippocampus 2013, 872, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Petrovic, J.M.; Callaghan, D.; Jones, A.; Cui, H.; Howlett, C.; Stanimirovic, D. Evidence that hypoxia-inducible factor-1 (HIF-1) mediates transcriptional activation of interleukin-1beta (IL-1β) in astrocyte cultures. J. Neuroimmunol. 2006, 174, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Batti, L.; Taylor, C.T.; O’Connor, J.J. Hydroxylase inhibition reduces synaptic transmission and protects against a glutamate-induced ischemia in the CA1 region of the rat hippocampus. Neuroscience 2010, 167, 1014–1024. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.R.; Ma, Q.; Ding, A.S.; Zeng, B.X.; Wang, F.Z. Effects of intermittent hypoxia on long-term potentiation in hippocampal dentate gyrus of rat. Zhongguo Ying Yong Sheng Li Xue Za Zhi 2001, 17, 18–20. [Google Scholar] [PubMed]
- Payne, R.S.; Goldbart, A.; Gozal, D.; Schurr, A. Effect of intermittent hypoxia on long-term potentiation in rat hippocampal slices. Brain Res. 2004, 1029, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Yung, W. Chronic intermittent hypoxia-induced deficits in synaptic plasticity and neurocognitive functions: A role for brain-derived neurotrophic factor. Acta Pharmacol. Sin. 2012, 33, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Domac, F.M.; Somay, G.; Misirli, H.; Erenoglu, N.Y. Tumor necrosis factor alpha serum levels and inflammatory response in acute ischemic stroke. Neurosciences 2007, 12, 25–30. [Google Scholar] [PubMed]
- Viviani, B.; Bartesaghi, S.; Gardoni, F.; Vezzani, A.; Behrens, M.M.; Bartfai, T.; Binaglia, M.; Corsini, E.; Di Luca, M.; Galli, C.L.; Marinovich, M. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J. Neurosci. 2003, 23, 8692–8700. [Google Scholar] [PubMed]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [PubMed]
- Stanimirovic, D.; Zhang, W.; Howlett, C.; Lemieux, P.; Smith, C. Inflammatory gene transcription in human astrocytes exposed to hypoxia: Roles of the nuclear factor-kappaB and autocrine stimulation. J. Neuroimmunol. 2001, 119, 365–376. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.-G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Nishio, K.; Ogawa, Y.; Kimata, J.; Kinumi, T.; Yoshida, Y.; Noguchi, N.; Niki, E. Turning point in apoptosis/necrosis induced by hydrogen peroxide. Free Radic. Res. 2006, 40, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M.; Shirato, I.; Kobayashi, M.; Endou, H. Hydrogen peroxide induces necrosis, apoptosis, oncosis and apoptotic oncosis of mouse terminal proximal straight tubule cells. Nephron 1999, 81, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; Friedle, S.A.; Watters, J.J. Chronic intermittent hypoxia exerts CNS region-specific effects on rat microglial inflammatory and TLR4 gene expression. PLoS ONE 2013, 8, e81584. [Google Scholar] [CrossRef] [PubMed]
- Udayabanu, M.; Kumaran, D.; Nair, R.U.; Srinivas, P.; Bhagat, N.; Aneja, R.; Katyal, A. Nitric oxide associated with iNOS expression inhibits acetylcholinesterase activity and induces memory impairment during acute hypobaric hypoxia. Brain Res. 2008, 1230, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Saha, R.N.; Pahan, K. Regulation of Inducible Nitric Oxide Synthase Gene in Glial Cells. Antioxid. Redox Signal. 2006, 8, 929–947. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Wang, Y.; Feng, J.; Cao, J.; Chen, B. Intermittent hypoxia from obstructive sleep apnea may cause neuronal impairment and dysfunction in central nervous system: The potential roles played by microglia. Neuropsychiatr. Dis. Treat. 2013, 9, 1077–1086. [Google Scholar] [PubMed]
- Ding, J.; Li, Q.Y.; Wang, X.; Sun, C.H.; Lu, C.Z.; Xiao, B.G. Fasudil protects hippocampal neurons against hypoxia-reoxygenation injury by suppressing microglial inflammatory responses in mice. J. Neurochem. 2010, 114, 1619–1629. [Google Scholar] [CrossRef] [PubMed]
- Yeh, N.C.; Tien, K.J.; Yang, C.M.; Wang, J.J.; Weng, S.F. Increased risk of Parkinson’s disease in patients with obstructive sleep apnea: A population-based, propensity score-matched, longitudinal follow-up study. Medicine 2016, 95, e2293. [Google Scholar] [CrossRef] [PubMed]
- Baune, B.T.; Camara, M.-L.; Eyre, H.; Jawahar, C.; Anscomb, H.; Körner, H. Tumour necrosis factor—Alpha mediated mechanisms of cognitive dysfunction. Transl. Neurosci. 2012, 3, 263–277. [Google Scholar] [CrossRef]
- Watters, O.; O’Connor, J.J. A role for tumor necrosis factor-α in ischemia and ischemic preconditioning. J. Neuroinflamm. 2011, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Goeddel, D.V. TNF-R1 signaling: A beautiful pathway. Science 2002, 296, 1634–1635. [Google Scholar] [CrossRef] [PubMed]
- Glazner, G.W.; Mattson, M.P. Differential effects of BDNF, ADNF9, and TNFα on levels of NMDA receptor subunits, calcium homeostasis, and neuronal vulnerability to excitotoxicity. Exp. Neurol. 2000, 161, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, L.; Klein, M.; Schlett, K.; Pfizenmaier, K.; Eisel, U.L. Tumor necrosis factor (TNF)-mediated neuroprotection against glutamate-induced excitotoxicity is enhanced by N-methyl-d-aspartate receptor activation. Essential role of a TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-kappa B pathway. J. Biol. Chem. 2004, 279, 32869–32881. [Google Scholar] [CrossRef] [PubMed]
- Veroni, C.; Gabriele, L.; Canini, I.; Castiello, L.; Coccia, E.; Remoli, M.E.; Columba-Cabezas, S.; Aricò, E.; Aloisi, F. Agresti, C. Activation of TNF receptor 2 in microglia promotes induction of anti-inflammatory pathways. Mol. Cell. Neurosci. 2010, 45, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Belarbi, K.; Jopson, T.; Tweedie, D.; Arellano, C.; Luo, W.; Greig, N.H.; Rosi, S. TNF-α protein synthesis inhibitor restores neuronal function and reverses cognitive deficits induced by chronic neuroinflammation. J. Neuroinflamm. 2012, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Marousi, S.; Antonacopoulou, A.; Kalofonos, H.; Papathanasopoulos, P.; Karakantza, M.; Ellul, J. Functional inflammatory genotypes in ischemic stroke: Could we use them to predict age of onset and long-term outcome? Stroke Res. Treat. 2011, 2011, 792923. [Google Scholar] [CrossRef] [PubMed]
- Klein, B.D.; White, H.S.; Callahan, K.S. Cytokine and intracellular signaling regulation of tissue factor expression in astrocytes. Neurochem. Int. 2000, 36, 441–449. [Google Scholar] [CrossRef]
- Tsoi, L.-M.; Wong, K.-Y.; Liu, Y.-M.; Ho, Y.-Y. Apoprotein E isoform-dependent expression and secretion of pro-inflammatory cytokines TNF-alpha and IL-6 in macrophages. Arch. Biochem. Biophys. 2007, 460, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Lull, M.E.; Block, M.L. Microglial activation and chronic neurodegeneration. Neurotherapeutics 2010, 7, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Barichello, T.; dos Santos, I.; Savi, G.D.; Simões, L.R.; Silvestre, T.; Comim, C.M.; Sachs, D.; Teixeira, M.M.; Teixeira, A.L.; Quevedo, J. TNF-α, IL-1β, IL-6, and cinc-1 levels in rat brain after meningitis induced by Streptococcus pneumoniae. J. Neuroimmunol. 2010, 221, 42–45. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Clark, R.K.; McDonnell, P.C.; Young, P.R.; White, R.F.; Barone, F.C.; Feuerstein, G.Z. Tumor necrosis factor-alpha expression in ischemic neurons. Stroke 1994, 25, 1481–1488. [Google Scholar] [CrossRef] [PubMed]
- Batti, L.; O’Connor, J.J. Tumor necrosis factor-alpha impairs the recovery of synaptic transmission from hypoxia in rat hippocampal slices. J. Neuroimmunol. 2010, 218, 21–27. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, J.J. Targeting tumour necrosis factor-α in hypoxia and synaptic signaling. Ir. J. Med. Sci. 2013, 182, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Hindryckx, P.; De Vos, M.; Jacques, P.; Ferdinande, L.; Peeters, H.; Olievier, K.; Bogaert, S.; Brinkman, B.; Vandenabeele, P.; Elewaut, D. Hydroxylase inhibition abrogates TNF-α-induced intestinal epithelial damage by hypoxia-inducible factor-1-dependent repression of FADD. J. Immunol. 2010, 185, 6306–6316. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, H.L.; Minami, M.; Lessov, N.S.; Coste, S.C.; Stevens, S.L.; Henshall, D.C.; Meller, R.; Simon, R.P.; Stenzel-Poore, M.P. Endotoxin preconditioning protects against the cytotoxic effects of TNFα after stroke: A novel role for TNFalpha in LPS-ischemic tolerance. J. Cereb Blood Flow Metab. 2007, 27, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Cook, J.A. Molecular mechanisms of endotoxin tolerance. J. Endotoxin Res. 2004, 10, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Stetler, R.A.; Leak, R.K.; Gan, Y.; Li, P.; Zhang, F.; Hu, X.; Jing, Z.; Chen, J.; Zigmond, M.J.; Gao, Y. Preconditioning provides neuroprotection in models of CNS disease: Paradigms and clinical significance. Prog. Neurobiol. 2014, 114, 58–83. [Google Scholar] [CrossRef] [PubMed]
- Bastide, M.; Gelé, P.; Pétrault, O.; Pu, Q.; Caliez, A.; Robin, E.; Deplanque, D.; Duriez, P.; Bordet, R. Delayed cerebrovascular protective effect of lipopolysaccharide in parallel to brain ischemic tolerance. J. Cereb. Blood Flow Metab. 2003, 23, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Karikó, K.; Weissman, D.; Welsh, F.A. Inhibition of toll-like receptor and cytokine signaling—A unifying theme in ischemic tolerance. J. Cereb. Blood Flow Metab. 2004, 24, 1288–1304. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; Cartmell, T.; Molina-Holgado, F. Endotoxin preconditioning protects neurones from in vitro ischemia: Role of endogenous IL-1beta and TNF-alpha. J. Neuroimmunol. 2006, 173, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Pickering, M.; O’Connor, J.J. Pro-inflammatory cytokines and their effects in the dentate gyrus. Prog. Brain Res. 2007, 163, 339–354. [Google Scholar] [PubMed]
- Cumiskey, D.; Butler, M.P.; Moynagh, P.N.; O’Connor, J.J. Evidence for a role for the group I metabotropic glutamate receptor in the inhibitory effect of tumor necrosis factor-alpha on long-term potentiation. Brain Res. 2007, 1136, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.P.; O’Connor, J.J.; Moynagh, P.N. Dissection of tumor-necrosis factor-alpha inhibition of long-term potentiation (LTP) reveals a p38 mitogen-activated protein kinase-dependent mechanism which maps to early-but not late-phase LTP. Neuroscience 2004, 124, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, A.J.; Murray, C.A.; O’Neill, L.A.J.; Lynch, M.A.; O’Connor, J.J. Interleukin-1β (IL-1β) and tumour necrosis factor (TNF) inhibit long-term potentiation in the rat dentate gyrus in vitro. Neurosci. Lett. 1996, 203, 17–20. [Google Scholar] [CrossRef]
- Tancredi, V.; D’Arcangelo, G.; Grassi, F.; Tarroni, P.; Palmieri, G.; Santoni, A.; Eusebi, F. Tumor necrosis factor alters synaptic transmission in rat hippocampal slices. Neurosci. Lett. 1992, 146, 176–178. [Google Scholar] [CrossRef]
- Curran, B.; Murray, H.; O’Connor, J.J. A role for c-jun n-terminal kinase in the inhibition of long-term potentiation by interleukin-1β and long-term depression in the rat dentate gyrus in vitro. Neuroscience 2003, 118, 347–357. [Google Scholar] [CrossRef]
- Stellwagen, D.; Malenka, R.C. Synaptic scaling mediated by glial TNF-alpha. Nature 2006, 440, 1054–1059. [Google Scholar] [CrossRef] [PubMed]
- Beattie, E.C.; Stellwagen, D.; Morishita, W.; Bresnahan, J.C.; Ha, B.K.; Von Zastrow, M.; Beattie, M.S.; Malenka, R.C. Control of synaptic strength by glial TNFalpha. Science 2002, 295, 2282–2285. [Google Scholar] [CrossRef] [PubMed]
- Stellwagen, D.; Beattie, E.C.; Seo, J.Y.; Malenka, R.C. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J. Neurosci. 2005, 25, 3219–3228. [Google Scholar] [CrossRef] [PubMed]
- Vesce, S.; Rossi, D.; Brambilla, L.; Volterra, A. Glutamate release from astrocytes in physiological conditions and in neurodegenerative disorders characterized by neuroinflammation. Int. Rev. Neurobiol. 2007, 82, 57–71. [Google Scholar] [PubMed]
- Korn, T.; Magnus, T.; Jung, S. Autoantigen specific T cells inhibit glutamate uptake in astrocytes by decreasing expression of astrocytic glutamate transporter GLAST: A mechanism mediated by tumor necrosis factor-alpha. FASEB J. 2005, 19, 1878–1880. [Google Scholar] [CrossRef] [PubMed]
- Boycott, H.E.; Wilkinson, J.A.; Boyle, J.P.; Pearson, H.A.; Peers, C. Differential involvement of TNF alpha in hypoxic suppression of astrocyte glutamate transporters. Glia 2008, 56, 998–1004. [Google Scholar] [CrossRef] [PubMed]
- Carmen, J.; Rothstein, J.D.; Kerr, D.A. Tumor necrosis factor-alpha modulates glutamate transport in the CNS and is a critical determinant of outcome from viral encephalomyelitis. Brain Res. 2009, 1263, 143–154. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Liu, Q.; Wu, J.; Shen, Y. Genetic deletion of TNF receptor suppresses excitatory synaptic transmission via reducing AMPA receptor synaptic localization in cortical neurons. FASEB J. 2012, 26, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Santello, M.; Bezzi, P.; Volterra, A. TNF-α controls glutamatergic gliotransmission in the hippocampal dentate gyrus. Neuron 2011, 69, 988–1001. [Google Scholar] [CrossRef] [PubMed]
- Arai, A.; Lynch, G. Factors regulating the magnitude of long-term potentiation induced by theta pattern stimulation. Brain Res. 1992, 598, 173–184. [Google Scholar] [CrossRef]
- Wall, A.M.; Mukandala, G.; Nigel, H.; O’Connor, J.J. Tumor necrosis factor-α potentiates long-term potentiation in the rat dentate gyrus after acute hypoxia. J. Neurosci. Res. 2015, 93, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Culver, C.; Sundqvist, A.; Mudie, S.; Melvin, A.; Xirodimas, D.; Rocha, S. Mechanism of hypoxia-induced NF-kappaB. Mol. Cell. Biol. 2010, 30, 4901–4921. [Google Scholar] [CrossRef] [PubMed]
- Pickering, M.; Cumiskey, D.; O’Connor, J.J. Actions of TNF-α on glutamatergic synaptic transmission in the central nervous system. Exp. Physiol. 2005, 90, 663–670. [Google Scholar] [CrossRef] [PubMed]
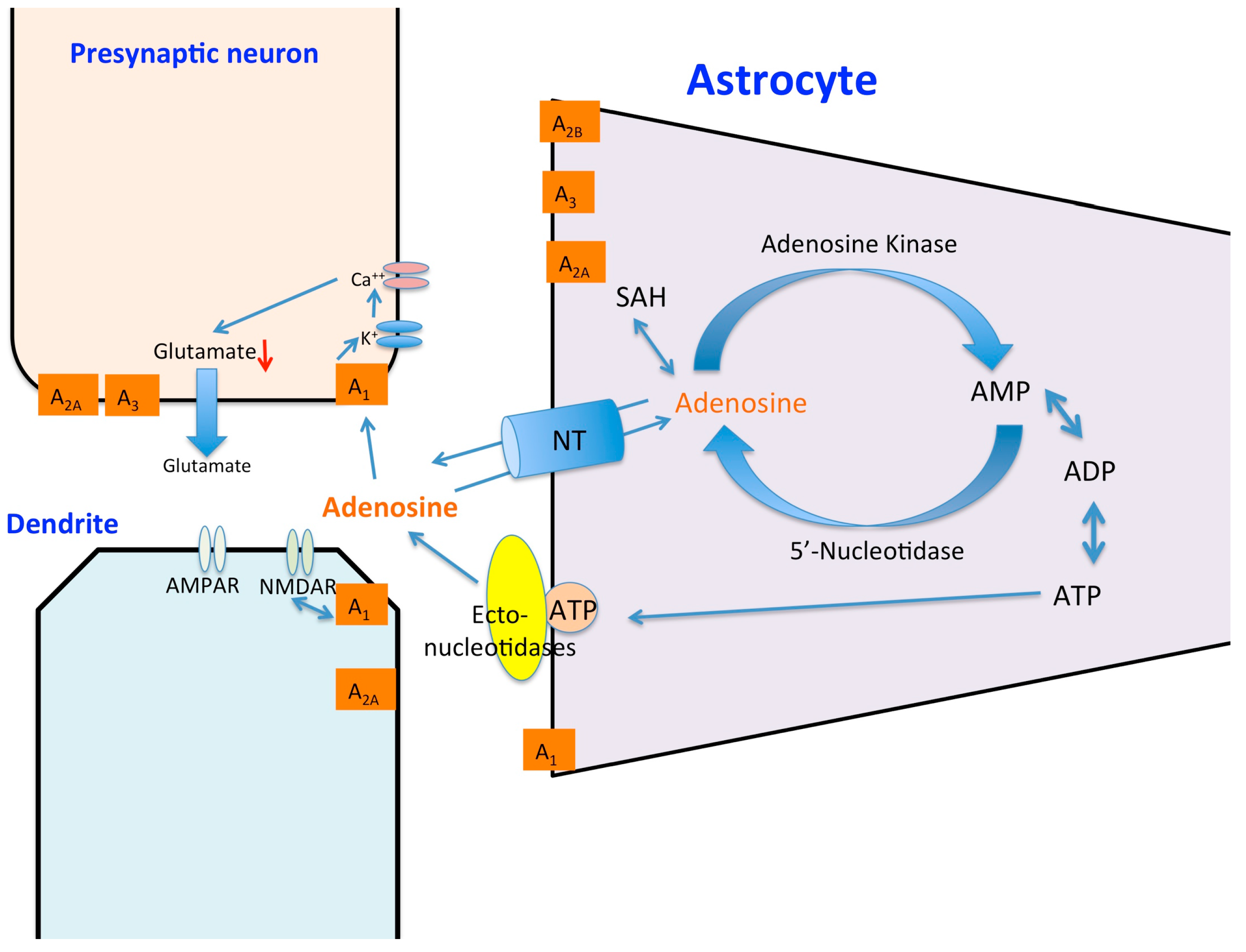
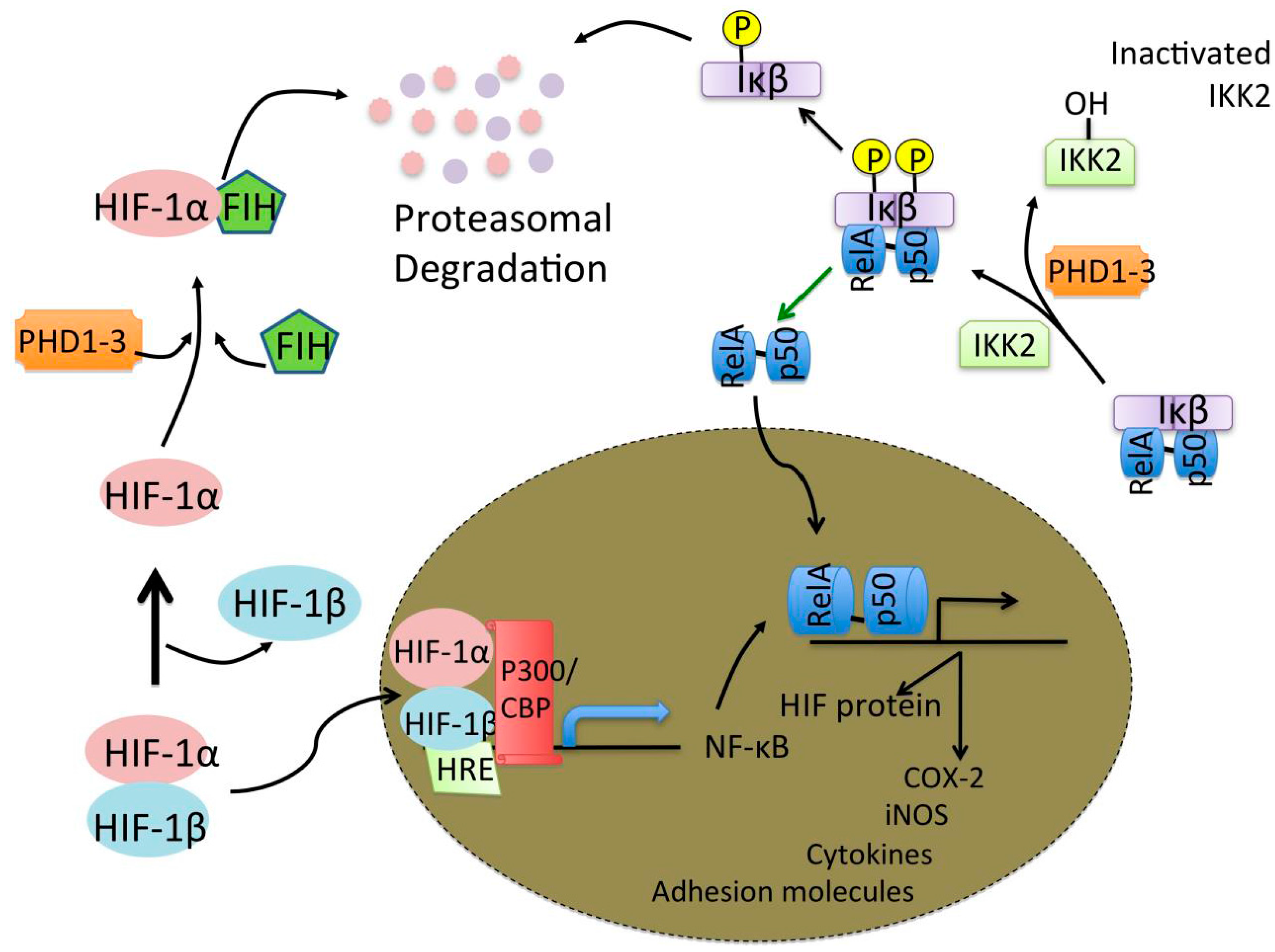
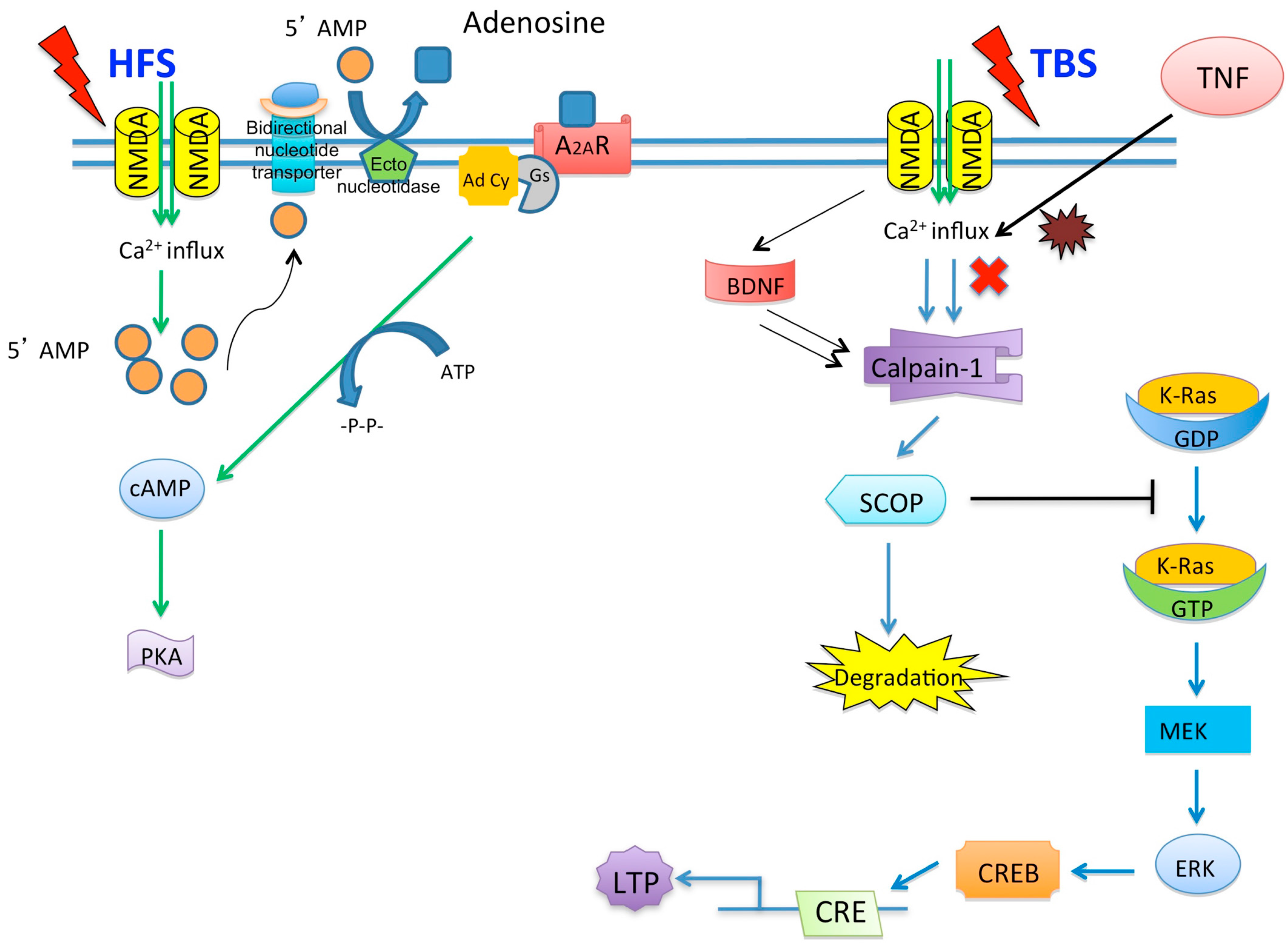
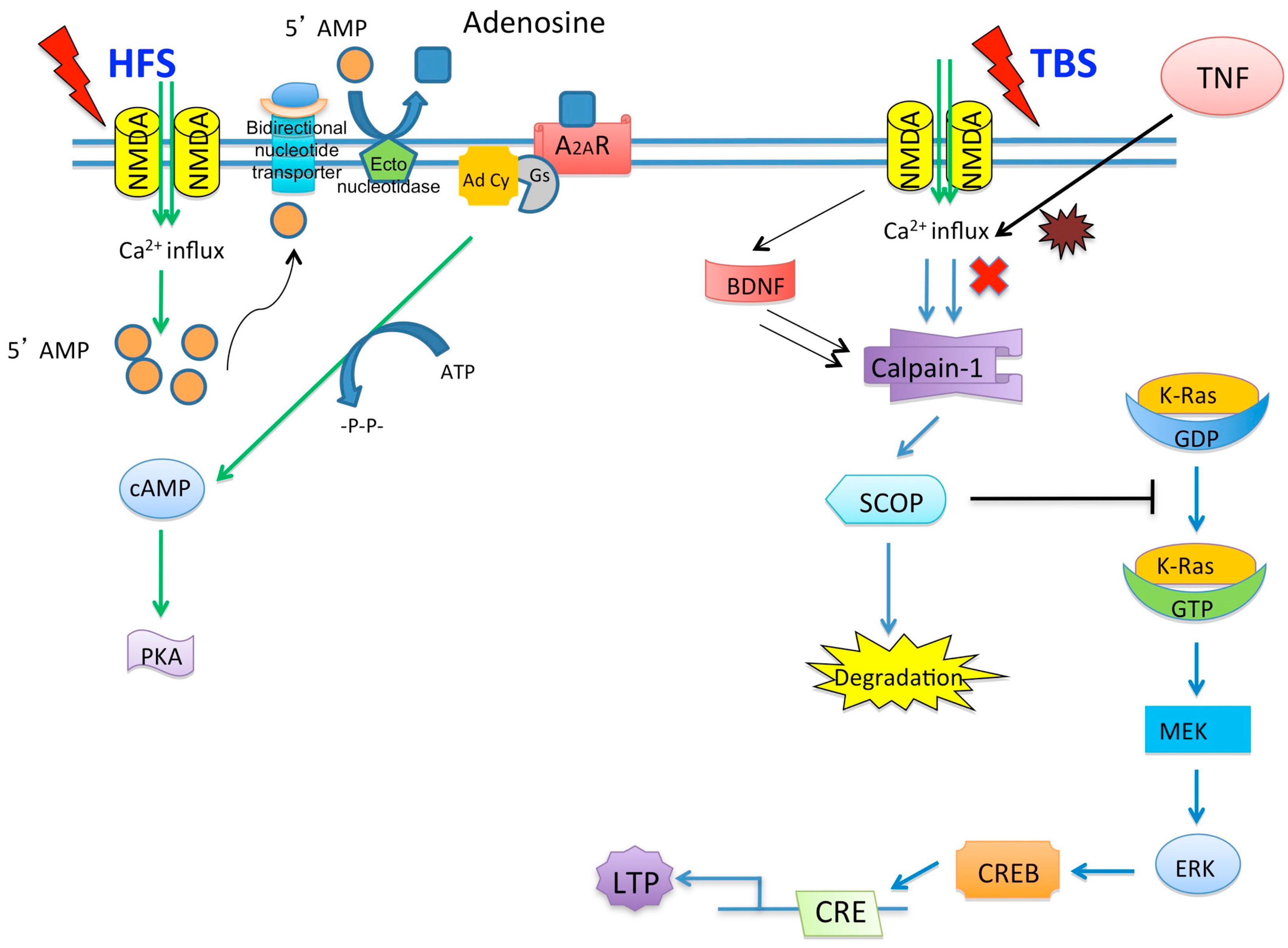
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mukandala, G.; Tynan, R.; Lanigan, S.; O’Connor, J.J. The Effects of Hypoxia and Inflammation on Synaptic Signaling in the CNS. Brain Sci. 2016, 6, 6. https://doi.org/10.3390/brainsci6010006
Mukandala G, Tynan R, Lanigan S, O’Connor JJ. The Effects of Hypoxia and Inflammation on Synaptic Signaling in the CNS. Brain Sciences. 2016; 6(1):6. https://doi.org/10.3390/brainsci6010006
Chicago/Turabian StyleMukandala, Gatambwa, Ronan Tynan, Sinead Lanigan, and John J. O’Connor. 2016. "The Effects of Hypoxia and Inflammation on Synaptic Signaling in the CNS" Brain Sciences 6, no. 1: 6. https://doi.org/10.3390/brainsci6010006
APA StyleMukandala, G., Tynan, R., Lanigan, S., & O’Connor, J. J. (2016). The Effects of Hypoxia and Inflammation on Synaptic Signaling in the CNS. Brain Sciences, 6(1), 6. https://doi.org/10.3390/brainsci6010006