An Update and Perspectives on Mitochondrial Membrane Protein-Associated Neurodegeneration and C19orf12 Research



Abstract
1. Neurodegeneration with Brain Iron Accumulation Disorders
2. MPAN Disorder
2.1. Clinical Phenotype
2.2. MRI and Spectroscopy Findings
2.3. Neuropathology
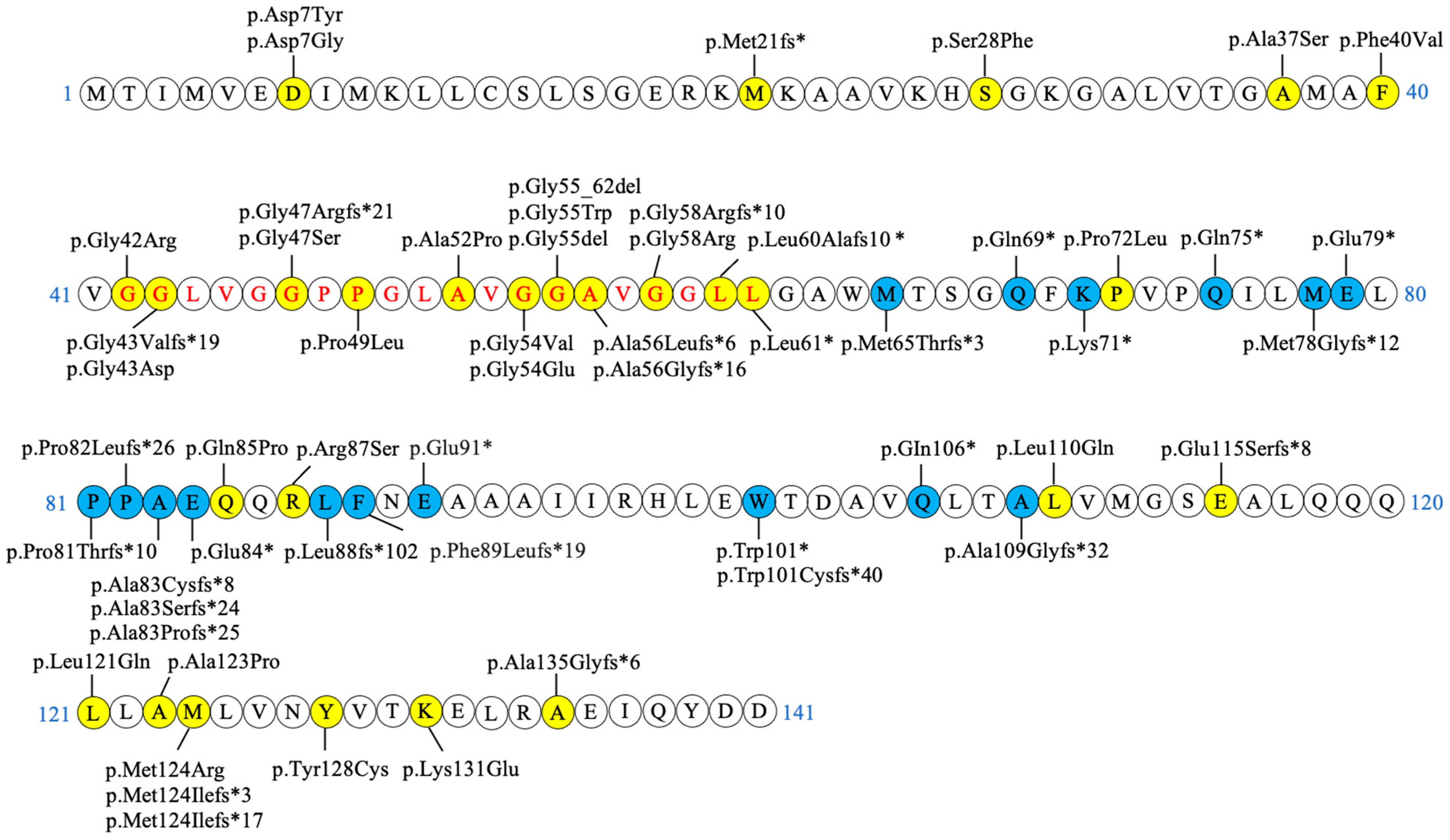
2.4. Genetics
2.5. Therapy
3. C19orf12 Gene and Protein
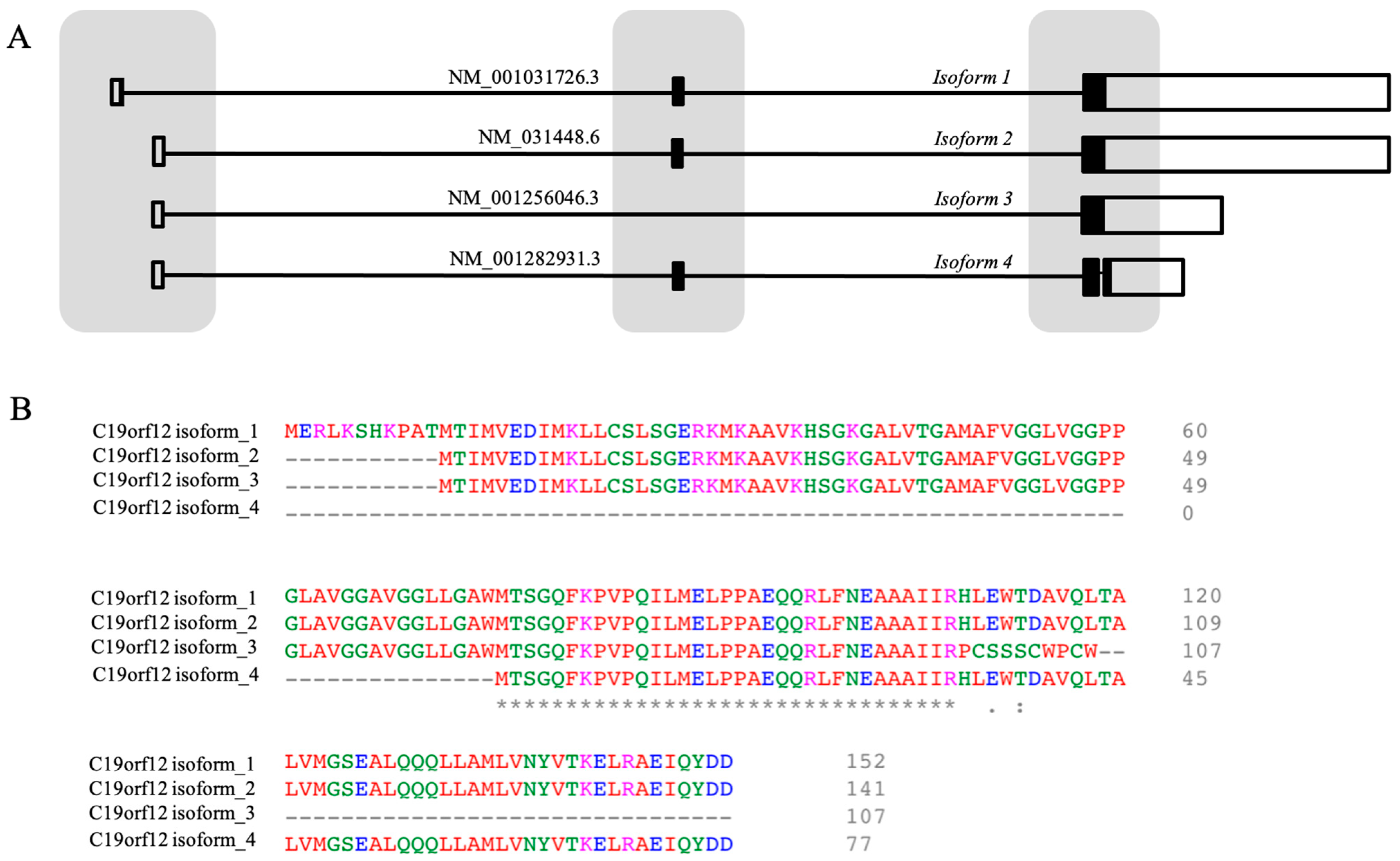
3.1. The Gene
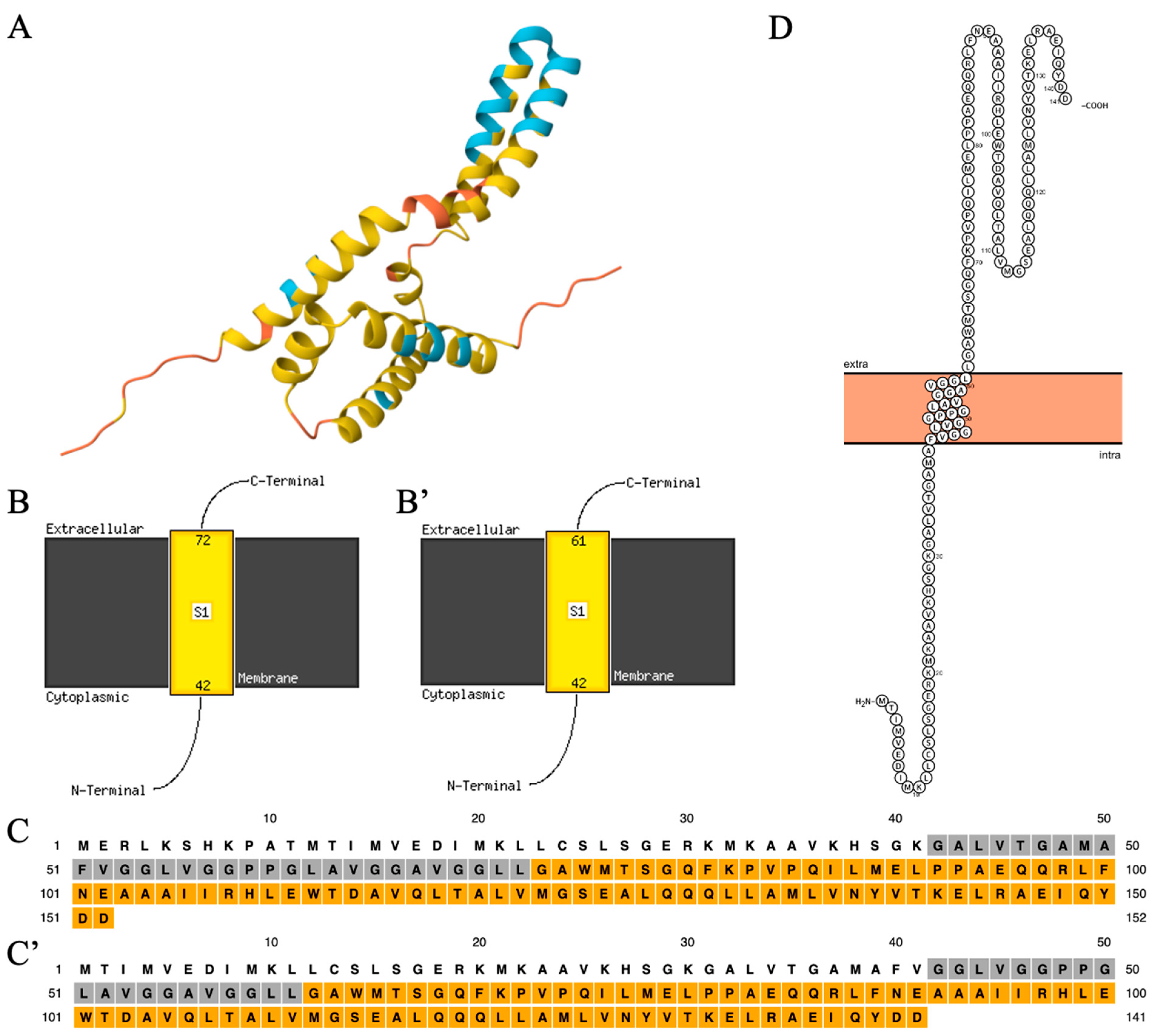
3.2. The Structure of the C19orf12 Protein
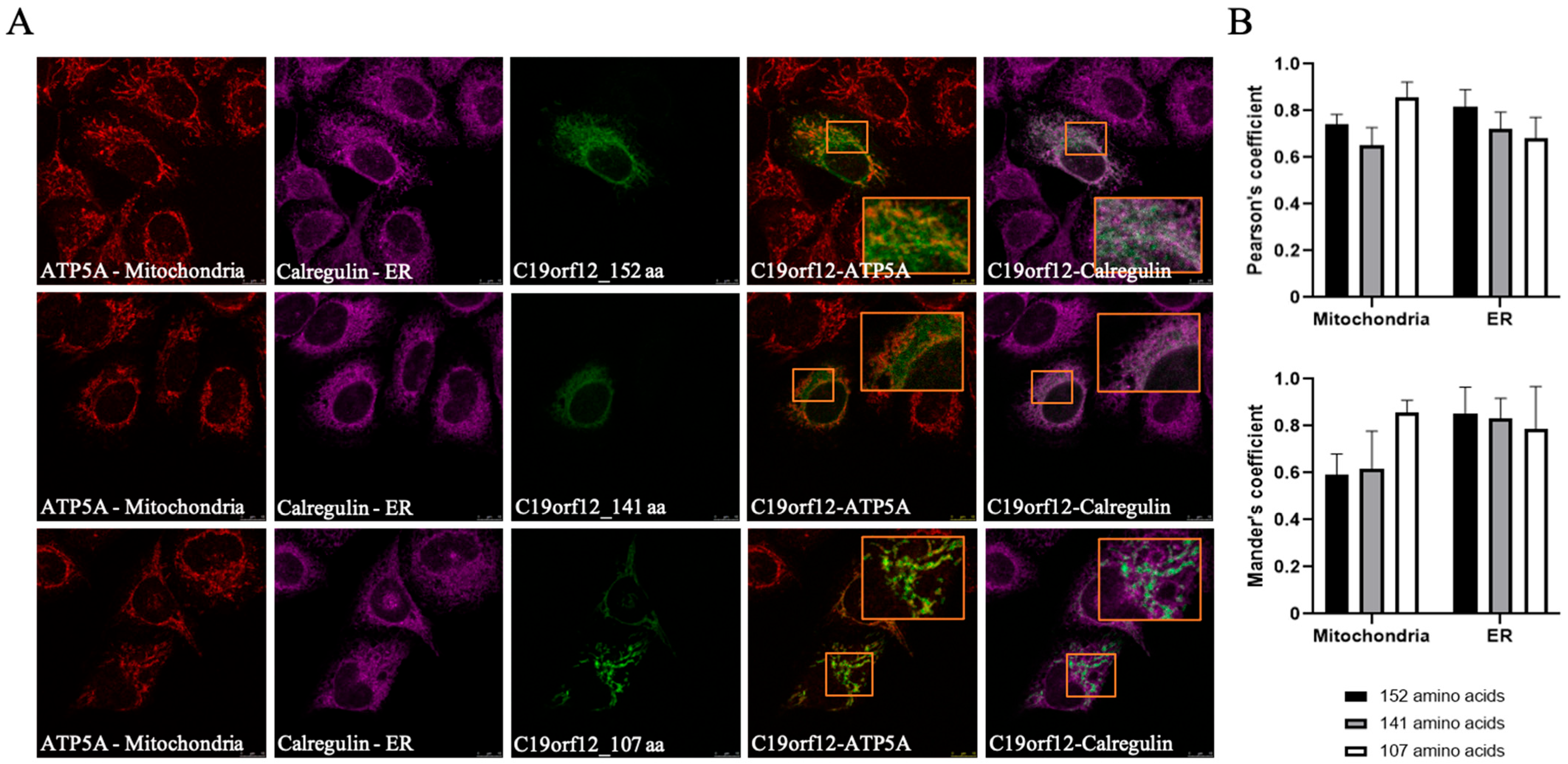
3.3. The Intracellular Localization of C19orf12 Protein
3.4. The Function of C19orf12
3.4.1. C19orf12 and Lipid Metabolism
3.4.2. C19orf12 and Autophagy
3.4.3. C19orf12 and Mitochondria/Oxidative Stress/Iron Homeostasis
3.5. Genotype–Phenotype Correlation
4. Animal Models of C19orf12 Deficiency
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Levi, S.; Finazzi, D. Neurodegeneration with brain iron accumulation: Update on pathogenic mechanisms. Front. Pharmacol. 2014, 5, 99. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Westaway, S.; Levinson, B.; Johnson, M.; Gitschier, J.; Hayflick, S. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat. Genet. 2001, 28, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Levi, S.; Tiranti, V. Neurodegeneration with Brain Iron Accumulation Disorders: Valuable Models Aimed at Understanding the Pathogenesis of Iron Deposition. Pharmaceuticals 2019, 12, 27. [Google Scholar] [CrossRef] [PubMed]
- Spaull, R.V.V.; Soo, A.K.S.; Hogarth, P.; Hayflick, S.J.; Kurian, M.A. Towards Precision Therapies for Inherited Disorders of Neurodegeneration with Brain Iron Accumulation. Tremor Other Hyperkinet. Mov. 2021, 11, 51. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, S.J.; Kurian, M.A.; Hogarth, P. Neurodegeneration with brain iron accumulation. Handb. Clin. Neurol. 2018, 147, 293–305. [Google Scholar] [PubMed]
- Iankova, V.; Karin, I.; Klopstock, T.; Schneider, S.A. Emerging Disease-Modifying Therapies in Neurodegeneration with Brain Iron Accumulation (NBIA) Disorders. Front. Neurol. 2021, 12, 629414. [Google Scholar] [CrossRef] [PubMed]
- Adam, M.P.; Feldman, J.; Mirzaa, G.M.; Pagon, R.A.; Wallace, S.E.; Amemiya, A. (Eds.) GeneReviews; University of Washington: Seattle, WA, USA, 1993.
- Kolarova, H.; Tan, J.; Strom, T.M.; Meitinger, T.; Wagner, M.; Klopstock, T. Lifetime risk of autosomal recessive neurodegeneration with brain iron accumulation (NBIA) disorders calculated from genetic databases. EBioMedicine 2022, 77, 103869. [Google Scholar] [CrossRef] [PubMed]
- Angelini, C.; Durand, C.M.; Fergelot, P.; Deforges, J.; Vital, A.; Menegon, P.; Sarrazin, E.; Bellance, R.; Mathis, S.; Gonzalez, V.; et al. Autosomal Dominant MPAN: Mosaicism Expands the Clinical Spectrum to Atypical Late-Onset Phenotypes. Mov. Disord. 2023, 38, 2103–2115. [Google Scholar] [CrossRef] [PubMed]
- Dogu, O.; Krebs, C.E.; Kaleagasi, H.; Demirtas, Z.; Oksuz, N.; Walker, R.H.; Paisán-Ruiz, C. Rapid disease progression in adult-onset mitochondrial membrane protein-associated neurodegeneration. Clin. Genet. 2013, 84, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Hogarth, P.; Gregory, A.; Kruer, M.C.; Sanford, L.; Wagoner, W.; Natowicz, M.R.; Egel, R.T.; Subramony, S.H.; Goldman, J.G.; Berry-Kravis, E.; et al. New NBIA subtype: Genetic, clinical, pathologic, and radiographic features of MPAN. Neurology 2013, 80, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Hartig, M.; Prokisch, H.; Meitinger, T.; Klopstock, T. Mitochondrial membrane protein-associated neurodegeneration (MPAN). Int. Rev. Neurobiol. 2013, 110, 73–84. [Google Scholar] [PubMed]
- Iankova, V.; Sparber, P.; Rohani, M.; Dusek, P.; Büchner, B.; Karin, I.; Schneider, S.A.; Gorriz, J.M.; Kmiec, T.; Klopstock, T. Phenotype and natural history of mitochondrial membrane protein-associated neurodegeneration. Brain 2024, 147, 1389–1398. [Google Scholar] [CrossRef] [PubMed]
- Kleffner, I.; Wessling, C.; Gess, B.; Korsukewitz, C.; Allkemper, T.; Schirmacher, A.; Young, P.; Senderek, J.; Husstedt, I.W. Behr syndrome with homozygous C19ORF12 mutation. J. Neurol. Sci. 2015, 357, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Kruer, M.C.; Salih, M.A.; Mooney, C.; Alzahrani, J.; Elmalik, S.A.; Kabiraj, M.M.; Khan, A.O.; Paudel, R.; Houlden, H.; Azzedine, H.; et al. C19orf12 mutation leads to a pallido-pyramidal syndrome. Gene 2014, 537, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Deschauer, M.; Gaul, C.; Behrmann, C.; Prokisch, H.; Zierz, S.; Haack, T.B. C19orf12 mutations in neurodegeneration with brain iron accumulation mimicking juvenile amyotrophic lateral sclerosis. J. Neurol. 2012, 259, 2434–2439. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Liao, Y.H.; Ionita, C.; Bale, A.E.; Darras, B.; Acsadi, G. Mitochondrial Membrane Protein-Associated Neurodegeneration Mimicking Juvenile Amyotrophic Lateral Sclerosis. Pediatr. Neurol. 2016, 64, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Remiche, G.; Vandernoot, I.; Sadeghi-Meibodi, N.; Desmyter, L. SPG43 and ALS-like syndrome in the same family due to compound heterozygous mutations of the C19orf12 gene: A case description and brief review. Neurogenetics 2021, 22, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Landouré, G.; Zhu, P.P.; Lourenço, C.M.; Johnson, J.O.; Toro, C.; Bricceno, K.V.; Rinaldi, C.; Meilleur, K.G.; Sangaré, M.; Diallo, O.; et al. Hereditary spastic paraplegia type 43 (SPG43) is caused by mutation in C19orf12. Hum. Mutat. 2013, 34, 1357–1360. [Google Scholar] [CrossRef] [PubMed]
- Akçakaya, N.H.; Haryanyan, G.; Mercan, S.; Sozer, N.; Ali, A.; Tombul, T.; Ozbek, U.; Uğur İşeri, S.A.; Yapıcı, Z. Clinical and genetic spectrum of an orphan disease MPAN: A series with new variants and a novel phenotype. Neurol. Neurochir. Pol. 2019, 53, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, R.; Deenadayalu, A.; Bhattacharjee, S.; Paramanandam, V. C19orf12 mutation causing mitochondrial membrane-protein Associated Neurodegeneration masquerading as spastic paraplegia. Parkinsonism Relat. Disord. 2021, 89, 146–147. [Google Scholar] [CrossRef] [PubMed]
- Kola, S.; Meka, S.S.L.; Fathima, S.T.; Wahed, A.; Kandadai, R.M.; Borgohain, R. Mitochondrial Membrane Protein-Associated Neurodegeneration (MPAN): Two Phenotypes-Dystonia and Spastic Paraparesis. Ann. Indian Acad. Neurol. 2022, 25, 1200–1202. [Google Scholar] [CrossRef] [PubMed]
- Saibaba, J.; Sibi, S.; Amalnath, D.; Subrahmanyam, D.K.S. Novel Variant in C19orf12 Gene Causing Mitochondrial Membrane Protein-Associated Neurodegeneration (MPAN)—Case Report and a Brief Review of Indian Literature on MPAN. Ann. Indian Acad. Neurol. 2025, 28, 450–452. [Google Scholar] [CrossRef] [PubMed]
- Skowronska, M.; Buksinska-Lisik, M.; Kmiec, T.; Litwin, T.; Kurkowska-Jastrzębska, I.; Czlonkowska, A. Is there heart disease in cases of neurodegeneration associated with mutations in C19orf12? Parkinsonism Relat. Disord. 2020, 80, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Olgiati, S.; Doğu, O.; Tufekcioglu, Z.; Diler, Y.; Saka, E.; Gultekin, M.; Kaleagasi, H.; Kuipers, D.; Graafland, J.; Breedveld, G.J.; et al. The p.Thr11Met mutation in c19orf12 is frequent among adult Turkish patients with MPAN. Parkinsonism Relat. Disord. 2017, 39, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Skowronska, M.; Kmiec, T.; Jurkiewicz, E.; Malczyk, K.; Kurkowska-Jastrzębska, I.; Czlonkowska, A. Evolution and novel radiological changes of neurodegeneration associated with mutations in C19orf12. Parkinsonism Relat. Disord. 2017, 39, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Yoganathan, S.; Sudhakar, S.V.; Thomas, M.; Dutta, A.K.; Danda, S. “Eye of tiger sign” mimic in an adolescent boy with mitochondrial membrane protein associated neurodegeneration (MPAN). Brain Dev. 2016, 38, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Hartig, M.B.; Iuso, A.; Haack, T.; Kmiec, T.; Jurkiewicz, E.; Heim, K.; Roeber, S.; Tarabin, V.; Dusi, S.; Krajewska-Walasek, M.; et al. Absence of an orphan mitochondrial protein, c19orf12, causes a distinct clinical subtype of neurodegeneration with brain iron accumulation. Am. J. Hum. Genet. 2011, 89, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Skowronska, M.; Kmiec, T.; Kurkowska-Jastrzębska, I.; Czlonkowska, A. Eye of the tiger sign in a 23 year patient with mitochondrial membrane protein associated neurodegeneration. J. Neurol. Sci. 2015, 352, 110–111. [Google Scholar] [CrossRef] [PubMed]
- Dehghan Manshadi, M.; Rohani, M.; Rezaei, A.; Aryani, O. A Case of MPAN with “Eye of the Tiger Sign,” Mimicking PKAN. Mov. Disord. Clin. Pract. 2022, 9, 693–695. [Google Scholar] [CrossRef] [PubMed]
- Garg, D.; Agarwal, A.; Garg, A.; Rajan, R.; Srivastava, A.K. ‘Comb Sign’: A Novel Appearance of Substantia Nigra in Mitochondrial Membrane Protein-Associated Neurodegeneration. Ann. Indian Acad. Neurol. 2023, 26, 1004–1005. [Google Scholar] [CrossRef] [PubMed]
- Gore, E.; Appleby, B.S.; Cohen, M.L.; DeBrosse, S.D.; Leverenz, J.B.; Miller, B.L.; Siedlak, S.L.; Zhu, X.; Lerner, A.J. Clinical and imaging characteristics of late onset mitochondrial membrane protein-associated neurodegeneration (MPAN). Neurocase 2016, 22, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Selikhova, M.; Fedotova, E.; Wiethoff, S.; Schottlaender, L.V.; Klyushnikov, S.; Illarioshkin, S.N.; Houlden, H. A 30-year history of MPAN case from Russia. Clin. Neurol. Neurosurg. 2017, 159, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Lehéricy, S.; Roze, E.; Goizet, C.; Mochel, F. MRI of neurodegeneration with brain iron accumulation. Curr. Opin. Neurol. 2020, 33, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Dusek, P.; Mekle, R.; Skowronska, M.; Acosta-Cabronero, J.; Huelnhagen, T.; Robinson, S.D.; Schubert, F.; Deschauer, M.; Els, A.; Ittermann, B.; et al. Brain iron and metabolic abnormalities in C19orf12 mutation carriers: A 7.0 tesla MRI study in mitochondrial membrane protein-associated neurodegeneration. Mov. Disord. 2020, 35, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.; Lotia, M.; Jeong, S.Y.; Fox, R.; Zhen, D.; Sanford, L.; Hamada, J.; Jahic, A.; Beetz, C.; Freed, A.; et al. Autosomal dominant mitochondrial membrane protein-associated neurodegeneration (MPAN). Mol. Genet. Genomic Med. 2019, 7, e00736. [Google Scholar] [CrossRef] [PubMed]
- Sparber, P.; Marakhonov, A.; Filatova, A.; Sharkova, I.; Skoblov, M. Novel case of neurodegeneration with brain iron accumulation 4 (NBIA4) caused by a pathogenic variant affecting splicing. Neurogenetics 2018, 19, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Mercan, S.; Ugur Iseri, S.A.; Yigiter, R.; Akcakaya, N.H.; Saka, E.; Yapici, Z. Two cases with mitochondrial membrane protein-associated neurodegeneration: Genetic features and long-term clinical follow-up. Neurocase 2022, 28, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Holla, V.V.; Sriram, N.; Kamble, N.; Asranna, A.; Saini, J.; Arunachal, G.; Yadav, R.; Pandey, A.; Pal, P.K.; et al. C19orf12 gene variants causing mitochondrial membrane protein-associated neurodegeneration (MPAN). Eur. J. Hum. Genet. 2025, 33, 878–886. [Google Scholar] [CrossRef] [PubMed]
- Al Macki, N.; Al Rashdi, I. A Novel Deletion Mutation of Exon 2 of the C19orf12 Gene in an Omani Family with Mitochondrial Membrane Protein-Associated Neurodegeneration (MPAN). Oman Med. J. 2017, 32, 66–68. [Google Scholar] [CrossRef] [PubMed]
- Mahesan, A.; Kamila, G.; Kumar, A.; Jauhari, P.; Chakrabarty, B.; Gulati, S. Teaching NeuroImage: Mitochondrial Membrane Protein-Associated Neurodegeneration: An MRI Pattern Recognition. Neurology 2024, 102, e209420. [Google Scholar] [CrossRef] [PubMed]
- Panteghini, C.; Zorzi, G.; Venco, P.; Dusi, S.; Reale, C.; Brunetti, D.; Chiapparini, L.; Zibordi, F.; Siegel, B.; Garavaglia, B.; et al. C19orf12 and FA2H mutations are rare in Italian patients with neurodegeneration with brain iron accumulation. Semin. Pediatr. Neurol. 2012, 19, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Monfrini, E.; Melzi, V.; Buongarzone, G.; Franco, G.; Ronchi, D.; Dilena, R.; Scola, E.; Vizziello, P.; Bordoni, A.; Bresolin, N.; et al. A de novo C19orf12 heterozygous mutation in a patient with MPAN. Parkinsonism Relat. Disord. 2018, 48, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Fraser, S.; Koenig, M.; Farach, L.; Mancias, P.; Mowrey, K. A De Novo case of autosomal dominant mitochondrial membrane protein-associated neurodegeneration. Mol. Genet. Genomic Med. 2021, 9, e1706. [Google Scholar] [CrossRef] [PubMed]
- Rickman, O.J.; Salter, C.G.; Gunning, A.C.; Fasham, J.; Voutsina, N.; Leslie, J.S.; McGavin, L.; Cross, H.E.; Posey, J.E.; Akdemir, Z.C.; et al. Dominant mitochondrial membrane protein-associated neurodegeneration (MPAN) variants cluster within a specific C19orf12 isoform. Parkinsonism Relat. Disord. 2021, 82, 84–86. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, S.; Yang, W.; Wei, T.; Hao, W.; Cheng, T.; Wang, J.; Dong, W.; Qian, N. Case Report: Identification of a De novo C19orf12 Variant in a Patient With Mitochondrial Membrane Protein–Associated Neurodegeneration. Front. Genet. 2022, 13, 852374. [Google Scholar] [PubMed]
- Chen, H.Y.; Lin, H.I.; Hsu, C.L.; Chen, P.L.; Huang, C.Y.; Teng, S.C.; Lin, C.H. A novel C19orf12 frameshift mutation in a MPAN pedigree impairs mitochondrial function and connectivity leading to neurodegeneration. Parkinsonism Relat. Disord. 2023, 109, 105353. [Google Scholar] [CrossRef] [PubMed]
- Schulte, E.C.; Claussen, M.C.; Jochim, A.; Haack, T.; Hartig, M.; Hempel, M.; Prokisch, H.; Haun-Jünger, U.; Winkelmann, J.; Hemmer, B.; et al. Mitochondrial membrane protein associated neurodegenration: A novel variant of neurodegeneration with brain iron accumulation. Mov. Disord. 2013, 28, 224–227. [Google Scholar] [CrossRef] [PubMed]
- Li, S.J.; Wang, L.L.; Qin, L.Z.; Wang, X.J.; Zhang, J.W.; Li, W. Pedigree analysis of C19ORF12 p.Asp18Tyr mutation in a family with mitochondrial membrane protein associated neurodegeneration. Zhonghua Yi Xue Za Zhi 2019, 99, 2926–2931. [Google Scholar] [PubMed]
- Nagarjunakonda, S.; Daggumati, R.; Uppala, V.; Gajula, R.; Amalakanti, S. A Novel Mutation in Neurodegeneration with Brain Iron Accumulation—A Case Report. Neurol. India 2019, 67, 1341–1343. [Google Scholar] [PubMed]
- Wydrych, A.; Pakuła, B.; Jakubek-Olszewska, P.; Janikiewicz, J.; Dobosz, A.M.; Cudna, A.; Rydzewski, M.; Pierzynowska, K.; Gaffke, L.; Cyske, Z.; et al. Metabolic alterations in fibroblasts of patients presenting with the MPAN subtype of neurodegeneration with brain iron accumulation (NBIA). Biochim. Biophys. Acta Mol. Basis Dis. 2025, 1871, 167541. [Google Scholar] [CrossRef] [PubMed]
- Imura, M.; Nakahara, K.; Hara, K.; Yoshino, H.; Nishioka, K.; Hattori, N.; Ueda, M. Rapidly Progressive Gait Disturbance and Extensive Iron Deposits in Late-Onset MPAN with a Pathogenic. Neurology 2024, 103, e209724. [Google Scholar] [CrossRef] [PubMed]
- de Vries, R.J.; Jaeger, B.; Hellebrekers, D.M.E.I.; Reneman, L.; Verhamme, C.; Smeets, H.J.M.; van Maarle, M.C.; de Visser, M.; Bleeker, F.E. Distal muscle weakness and optic atrophy without central nervous system involvement in a patient with a homozygous missense mutation in the C19ORF12-gene. Clin. Neurol. Neurosurg. 2021, 206, 106637. [Google Scholar] [CrossRef] [PubMed]
- Gowda, V.K.; Patil, A.; Srinivasan, V.M.; Kathrani, N. Mitochondrial Membrane Protein Associated Neurodegeneration (MPAN) with a Novel C19orf12 Mutation in the First Decade of Life. Indian J. Pediatr. 2019, 86, 746–748. [Google Scholar] [CrossRef] [PubMed]
- Sharawat, I.K.; Panda, P.K.; Sherwani, P.; Moirangthem, V. Mitochondrial Membrane Protein-associated Neurodegeneration due to Novel Homozygous Mutation in the C19orf12 Gene. Ann. Indian Acad. Neurol. 2021, 24, 417–418. [Google Scholar] [CrossRef] [PubMed]
- Gagliardi, M.; Annesi, G.; Lesca, G.; Broussolle, E.; Iannello, G.; Vaiti, V.; Gambardella, A.; Quattrone, A. C19orf12 gene mutations in patients with neurodegeneration with brain iron accumulation. Parkinsonism Relat. Disord. 2015, 21, 813–816. [Google Scholar] [CrossRef] [PubMed]
- Tariq, H.; Butt, J.U.R.; Houlden, H.; Naz, S. Are some C19orf12 variants monoallelic for neurological disorders? Parkinsonism Relat. Disord. 2019, 65, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Incecik, F.; Herguner, O.M.; Bisgin, A. Mitochondrial Membrane Protein-Associated Neurodegeneration: A Case Series of Six Children. Ann. Indian Acad. Neurol. 2020, 23, 802–804. [Google Scholar] [CrossRef] [PubMed]
- Schottmann, G.; Stenzel, W.; Lützkendorf, S.; Schuelke, M.; Knierim, E. A novel frameshift mutation of C19ORF12 causes NBIA4 with cerebellar atrophy and manifests with severe peripheral motor axonal neuropathy. Clin. Genet. 2014, 85, 290–292. [Google Scholar] [CrossRef] [PubMed]
- Lefter, A.; Mitrea, I.; Mitrea, D.; Plaiasu, V.; Bertoli-Avella, A.; Beetz, C.; Cozma, L.; Tulbă, D.; Mitu, C.E.; Popescu, B.O. Novel C19orf12 loss-of-function variant leading to neurodegeneration with brain iron accumulation. Neurocase 2021, 27, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Horvath, R.; Holinski-Feder, E.; Neeve, V.C.; Pyle, A.; Griffin, H.; Ashok, D.; Foley, C.; Hudson, G.; Rautenstrauss, B.; Nürnberg, G.; et al. A new phenotype of brain iron accumulation with dystonia, optic atrophy, and peripheral neuropathy. Mov. Disord. 2012, 27, 789–793. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Lai, X.; Fu, J.; Yang, J.; Zhao, B.; Shang, H.; Huang, R.; Chen, X. A novel C19ORF12 mutation in two MPAN sisters treated with deferiprone. BMC Neurol. 2023, 23, 134. [Google Scholar] [CrossRef] [PubMed]
- Balicza, P.; Bencsik, R.; Lengyel, A.; Gal, A.; Grosz, Z.; Csaban, D.; Rudas, G.; Danics, K.; Kovacs, G.G.; Molnar, M.J. Novel dominant MPAN family with a complex genetic architecture as a basis for phenotypic variability. Neurol. Genet. 2020, 6, e515. [Google Scholar] [CrossRef] [PubMed]
- Löbel, U.; Schweser, F.; Nickel, M.; Deistung, A.; Grosse, R.; Hagel, C.; Fiehler, J.; Schulz, A.; Hartig, M.; Reichenbach, J.R.; et al. Brain iron quantification by MRI in mitochondrial membrane protein-associated neurodegeneration under iron-chelating therapy. Ann. Clin. Transl. Neurol. 2014, 1, 1041–1046. [Google Scholar] [CrossRef] [PubMed]
- Zanuttigh, E.; Derderian, K.; Güra, M.A.; Geerlof, A.; Di Meo, I.; Cavestro, C.; Hempfling, S.; Ortiz-Collazos, S.; Mauthe, M.; Kmieć, T.; et al. Identification of Autophagy as a Functional Target Suitable for the Pharmacological Treatment of Mitochondrial Membrane Protein-Associated Neurodegeneration (MPAN) In Vitro. Pharmaceutics 2023, 15, 267. [Google Scholar] [CrossRef] [PubMed]
- Venco, P.; Bonora, M.; Giorgi, C.; Papaleo, E.; Iuso, A.; Prokisch, H.; Pinton, P.; Tiranti, V. Mutations of C19orf12, coding for a transmembrane glycine zipper containing mitochondrial protein, cause mis-localization of the protein, inability to respond to oxidative stress and increased mitochondrial Ca2+. Front. Genet. 2015, 6, 185. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T. Improving the accuracy of transmembrane protein topology prediction using evolutionary information. Bioinformatics 2007, 23, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Bertoni, D.; Magana, P.; Paramval, U.; Pidruchna, I.; Radhakrishnan, M.; Tsenkov, M.; Nair, S.; Mirdita, M.; Yeo, J.; et al. AlphaFold Protein Structure Database in 2024: Providing structure coverage for over 214 million protein sequences. Nucleic Acids Res. 2024, 52, D368–D375. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Jeon, T.J.; Oberai, A.; Yang, D.; Schmidt, J.J.; Bowie, J.U. Transmembrane glycine zippers: Physiological and pathological roles in membrane proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 14278–14283. [Google Scholar] [CrossRef] [PubMed]
- Mori, A.; Hatano, T.; Inoshita, T.; Shiba-Fukushima, K.; Koinuma, T.; Meng, H.; Kubo, S.I.; Spratt, S.; Cui, C.; Yamashita, C.; et al. Parkinson’s disease-associated iPLA2-VIA/PLA2G6 regulates neuronal functions and α-synuclein stability through membrane remodeling. Proc. Natl. Acad. Sci. USA 2019, 116, 20689–20699. [Google Scholar] [CrossRef] [PubMed]
- Klingelhuber, F.; Frendo-Cumbo, S.; Omar-Hmeadi, M.; Massier, L.; Kakimoto, P.; Taylor, A.J.; Couchet, M.; Ribicic, S.; Wabitsch, M.; Messias, A.C.; et al. A spatiotemporal proteomic map of human adipogenesis. Nat. Metab. 2024, 6, 861–879. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, M.; Zhang, C.; Méar, L.; Zhong, W.; Digre, A.; Katona, B.; Sjöstedt, E.; Butler, L.; Odeberg, J.; Dusart, P.; et al. A single-cell type transcriptomics map of human tissues. Sci. Adv. 2021, 7, eabh2169. [Google Scholar] [CrossRef] [PubMed]
- Sreejith, P.; Lolo, S.; Patten, K.R.; Gunasinghe, M.; More, N.; Pallanck, L.J.; Bharadwaj, R. Nazo, the Drosophila homolog of the NBIA-mutated protein-c19orf12, is required for triglyceride homeostasis. PLoS Genet. 2024, 20, e1011137. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.E.; Dennis, E.A. Phospholipase A2 structure/function, mechanism, and signaling. J. Lipid Res. 2009, 50, S237–S242. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Nakatani, Y.; Atsumi, G.I.; Inoue, K.; Kudo, I. Regulatory Functions of Phospholipase A2. Crit. Rev. Immunol. 2017, 37, 127–195. [Google Scholar] [CrossRef] [PubMed]
- Hanna, M.; Guillén-Samander, A.; De Camilli, P. RBG Motif Bridge-Like Lipid Transport Proteins: Structure, Functions, and Open Questions. Annu. Rev. Cell Dev. Biol. 2023, 39, 409–434. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Zhu, J.; Ma, X.; Siedlak, S.L.; Cohen, M.L.; Lerner, A.; Wang, W. C19orf12 ablation causes ferroptosis in mitochondrial membrane protein-associated with neurodegeneration. Free Radic. Biol. Med. 2022, 182, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Iuso, A.; Sibon, O.C.; Gorza, M.; Heim, K.; Organisti, C.; Meitinger, T.; Prokisch, H. Impairment of Drosophila orthologs of the human orphan protein C19orf12 induces bang sensitivity and neurodegeneration. PLoS ONE 2014, 9, e89439. [Google Scholar] [CrossRef] [PubMed]
- Goto, A.; Okado, K.; Martins, N.; Cai, H.; Barbier, V.; Lamiable, O.; Troxler, L.; Santiago, E.; Kuhn, L.; Paik, D.; et al. The Kinase IKKβ Regulates a STING- and NF-κB-Dependent Antiviral Response Pathway in Drosophila. Immunity 2018, 49, 225–234.e4. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Harding, A.T.; Sweeney, C.; Miao, D.; Swan, G.; Zhou, C.; Jiang, Z.; Fitzgerald, K.A.; Hammer, G.; Bergo, M.O.; et al. Control of antiviral innate immune response by protein geranylgeranylation. Sci. Adv. 2019, 5, eaav7999. [Google Scholar] [CrossRef] [PubMed]
- Foley, J.F. Making MAVS better. Sci. Signal. 2024, 17, eadt5916. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, M.; Lian, G.; Yang, S.; Cai, J.; Cai, Z.; Wu, Y.; Cui, J. Palmitoylation acts as a checkpoint for MAVS aggregation to promote antiviral innate immune responses. J. Clin. Investig. 2024, 134, e177924. [Google Scholar] [CrossRef] [PubMed]
- Mignani, L.; Zizioli, D.; Borsani, G.; Monti, E.; Finazzi, D. The Downregulation of c19orf12 Negatively Affects Neuronal and Musculature Development in Zebrafish Embryos. Front. Cell Dev. Biol. 2020, 8, 596069. [Google Scholar] [CrossRef] [PubMed]
- Deutschländer, A.; Konno, T.; Ross, O.A. Mitochondrial membrane protein-associated neurodegeneration. Park. Relat. Disord. 2017, 39, 1–3. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | NBIA Type | Protein Function |
|---|---|---|
| Pantothenate kinase 2 (PANK2) | Pantothenate Kinase-Associated Neurodegeneration (PKAN) | Coenzyme A biosynthesis |
| WD repeat domain 45 (WDR45) | β-propeller protein-associated neurodegeneration (BPAN) | Autophagy |
| Phospholipase A2 group IV (PLA2G6) | PLA2G6-Associated Neurodegeneration (PLAN) | Hydrolysis of membrane phospholipids |
| C19orf12 | Mitochondrial Membrane Protein-associated Neurodegeneration (MPAN) | Lipid metabolism? |
| Coenzyme A Synthase (COASY) | COASY Protein-Associated Neurodegeneration (CoPAN) | Coenzyme A biosynthesis |
| Ceruloplasmin (CP) | Aceruloplasminemia | Oxidation of Fe2+ to Fe3+; trafficking of iron |
| Ferritin light chain (FTL) | Neuroferritinopathy | Iron storage |
| Fatty acid 2-hydroxylase (FA2H) | Fatty Acid Hydroxylase-associated Neurodegeneration (FAHN) | Generation of 2-hydroxylated phospholipids |
| ATPase cation transporting 13A2 (ATP13A2) | Kufor–Rakeb Syndrome (KRS) | Translocation of ions and polyamines across the lysosomal membrane |
| DDB1- and CUL4-associated factor 17 (DCAF17) | Woodhouse–Sakati Syndrome (WSS) | Protein ubiquitination |
| Other genes potentially involved in NBIA: Sterol Carrier Protein 2 (SCP2), Carnitine O-acetyltransferase (CRAT), Adaptor-related Protein complex-4, M1 Subunit (AP4M1), RALBP1-Associated Eps Domain Containing 1 (REPS1), GTP Binding Protein 2 (GTPBP2), Ferritin Heavy Chain 1 (FTH1). | ||
| Category | Common Clinical Features |
|---|---|
| Prevalence and genetics | Ultra-rare disease (approx. 1 in 1,000,000); autosomal recessive (AR-MPAN) or dominant (AD-MPAN) forms; biallelic or heterozygous mutations in C19orf12. |
| Age of onset | Typically, between 3 and 16 years (juvenile form), but can also occur in early adulthood. |
| Initial symptoms | Gait disturbances Lower limb spasticity (toe walking, spastic gait, frequent falls) Psychiatric abnormalities (behavioral changes, anxiety, inattention, hyperactivity) Vision impairment due to optic atrophy |
| Movement disorders | Spasticity Dystonia (localized or generalized) Parkinsonism (bradykinesia, rigidity, tremor) Dysarthria, dysphagia Amyotrophy, fasciculations due to motor axonal neuropathy, visible on electroneuromyogram |
| Psychiatric symptoms | Anxiety Inattention and hyperactivity Behavioral changes |
| Cognitive symptoms | Progressive cognitive decline Severe dementia in later stages |
| Visual disturbances | Optic atrophy Progressive vision loss |
| Multisystem involvement | Cardiac disease (cardiomyopathy, arrhythmias, and autonomic dysfunction), muscle weakness, bowel and bladder incontinence. |
| Advanced disease | Wheelchair dependence after 15 years of evolution Marked weight loss Death due to complications (e.g., aspiration pneumonia) |
| Brain MRI findings | Symmetric hypointensity in globus pallidus and substantia nigra on T2 sequences Internal medullary lamina (hyperintense band in globus pallidus) Possible eye of the tiger sign Iron accumulation in caudate, putamen, and cortical/cerebellar atrophy White matter hyperintensities in periventricular areas |
| Progression | Slow progression over 15 years, followed by plateau phase High variability in symptoms among patients |
| AR-MPAN | |||
|---|---|---|---|
| NM_001031726.3 (Isoform 1) | NM_031448.6 (Isoform 2) | ||
| cDNA | Protein | cDNA | Protein |
| c.193+5G>A | p.? [37] | c.160+5G>A | p.? |
| c.194-2A>G | p.? [36,39] | c.161-2A>G | p.? |
| c.24G>C | p.Lys8Asn* [20] | c.-10G>C | p.? |
| c.32 C>T | p.Thr11Met [16,28,48,49] | c.-2C>T | p.? |
| c.52G>T | p.Asp18Tyr [49] | c.19G>T | p.Asp7Tyr |
| c.53A>G | p.Asp18Gly [48] | c.20A>G | p.Asp7Gly |
| c.94del | p.Met32fs* [36] | c.61del | p.Met21fs* |
| c.116C>T | p.Ser39Phe [11,36] | c.83C>T | p.Ser28Phe |
| c.142G>C | p.Ala48Pro [11,36] | c.109G>T | p.Ala37Ser |
| c.151T>G | p.Phe51Val [21,22,50] | c.118T>G | p.Phe40Val |
| c.157G>A | p.Gly53Arg [28,36] | c.124G>A | p.Gly42Arg |
| c.161del | p.Gly54Valfs*19 [51] | c.128del | p.Gly43Valfs*19 |
| c.161G>A | p.Gly54Glu [28] | c.128G>A | p.Gly43Asp |
| c.171_181del | p.Gly58Argfs*21 [36] | c.138_148del | p.Gly47Argfs*21 |
| c.172G>A | p.Gly58Ser [42] | c.139G>A | p.Gly47Ser |
| c.179C>T | p.Pro60Leu [11,36,52] | c.146C>T | p.Pro49Leu |
| c.187G>C | p.Ala63Pro [19,53] | c.154G>C | p.Ala52Pro |
| c.194G>T | p.Gly65Val [11,36] | c.161G>T | p.Gly54Val |
| c.194G>A | p.Gly65Glu [20,28,36] | c.161G>A | p.Gly54Glu |
| c.194-2del | p.Gly66_Gly73del [39] | c.161-2del | p.Gly55_Gly62del |
| c.196G>T | p.Gly66Trp [31,54,55] | c.163G>T | p.Gly55Trp |
| c.197_199del | p.Gly66del [16,19,48] | c.164_166del | p.Gly55del |
| c.199del | p.Ala67Leufs*6 [31,36,56,57] | c.166del | p.Ala56Leufs*6 |
| c.199dup | p.Ala67Glyfs*16 [58] | c.166dup | p.Ala56Glyfs*16 |
| c.204_214del | p.Gly69Argfs*10 [16,26,28,37,48] | c.171_181del | p.Gly58Argfs*10 |
| c.205G>A | p.Gly69Arg [28,36,51] | c.172G>A | p.Gly58Arg |
| c.210dup | p.Leu71Alafs10* [59] | c.177dup | p.Leu60Alafs10* |
| c.215T>G | p.Leu72* [60] | c.182T>G | p.Leu61* |
| c.248C>T | p.Pro83Leu [11,28,56] | c.215C>T | p.Pro72Leu |
| c.287A>C | p.Gln96Pro [42,56] | c.254A>C | p.Gln85Pro |
| c.294G>C | p.Arg98Ser [11] | c.261G>C | p.Arg87Ser |
| c.362T>A | p.Leu121Gln [61] | c.329T>A | p.Leu110Gln |
| c.376_388del | p.Glu126Serfs*8 [26] | c.343_355del | p.Glu115Serfs*8 |
| c.395 T>A | p.Leu132Gln [48,49] | c.362T>A | p.Leu121Gln |
| c.400G>C | p.Ala134Pro [11,56] | c.367G>C | p.Ala123Pro |
| c.404T>G | p.Met135Arg [62] | c.371T>G | p.Met124Arg |
| c.404dup | p.Met135Ilefs*17 [58] | c.371dup | p.Met124Ilefs*17 |
| c.405del | p.Met135Ilefs*3 [45] | c.372del | p.Met124Ilefs*3 |
| c.416 A>G | p.Tyr139Cys [56] | c.383A>G | p.Tyr128Cys |
| c.424A>G | p.Lys142Glu [28,35] | c.391A>G | p.Lys131Glu |
| c.436dup | p.Ala146Glyfs*6 [56] | c.403dup | p.Ala135Glyfs*6 |
| AD-MPAN | |||
|---|---|---|---|
| NM_001031726.3 (Isoform 1) | NM_031448.6 (Isoform 2) | ||
| cDNA | Protein | cDNA | Protein |
| c.227_237del | p.Met76Thrfs*3 [36] | c.194_204del | p.Met65Thrfs*3 |
| c.238C>T | p.Gln80* [36] | c.205C>T | p.Gln69* |
| c.244A>T | p.Lys82* [9,56] | c.211A>T | p.Lys71* |
| c.256C>T | p.Gln86* [36,44] | c.223C>T | p.Gln75* |
| c.265_266del | p.Met89Glyfs*12 [43] | c.232_233del | p.Met78Glyfs*12 |
| c.268G>T | p.Glu90* [36] | c.235G>T | p.Glu79* |
| c.273_274insA | p.Pro92Thrfs*10 [47] | c.240_241insA | p.Pro81Thrfs*10 |
| c.278del | p.Pro93Leufs*26 [36,45] | c.245del | p.Pro82Leufs*26 |
| c.278dup | p.Ala94Cysfs*8 [36] | c.245dup | p.Ala83Cysfs*8 |
| c.279_282del | p.Ala94Serfs*24 [36] | c.246_249del | p.Ala83Serfs*24 |
| c.279delT | p.Ala94Profs*25 [36] | c.246delT | p.Ala83Profs*25 |
| c.283G > T | p.Glu95* [9] | c.250G>T | p.Glu84* |
| c.297insGCTC | p.Leu99fs*102 [42] | c.264insGCTC | p.Leu88fs*102 |
| c.300del | p.Phe100Leufs*19 [36] | c.267del | p.Phe89Leufs*19 |
| c.304G > T | p.Glu102* [9] | c.271G>T | p.Glu91* |
| c.335G>A | p.Trp112* [36,63] | c.302G>A | p.Trp101* |
| c.336_338delGACinsCACA | p.Trp112Cysfs*40 [46] | c.303_305delGACinsCACA | p.Trp101Cysfs*40 |
| c.349C>T | p.Gln117* [36] | c.316C>T | p.GIn106* |
| c.357dup | p.Ala120Glyfs*32 [36] | c.324dup | p.Ala109Glyfs*32 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gnutti, B.; Iuso, A.; Angelini, C.; Finazzi, D. An Update and Perspectives on Mitochondrial Membrane Protein-Associated Neurodegeneration and C19orf12 Research. Brain Sci. 2025, 15, 777. https://doi.org/10.3390/brainsci15080777
Gnutti B, Iuso A, Angelini C, Finazzi D. An Update and Perspectives on Mitochondrial Membrane Protein-Associated Neurodegeneration and C19orf12 Research. Brain Sciences. 2025; 15(8):777. https://doi.org/10.3390/brainsci15080777
Chicago/Turabian StyleGnutti, Barbara, Arcangela Iuso, Chloé Angelini, and Dario Finazzi. 2025. "An Update and Perspectives on Mitochondrial Membrane Protein-Associated Neurodegeneration and C19orf12 Research" Brain Sciences 15, no. 8: 777. https://doi.org/10.3390/brainsci15080777
APA StyleGnutti, B., Iuso, A., Angelini, C., & Finazzi, D. (2025). An Update and Perspectives on Mitochondrial Membrane Protein-Associated Neurodegeneration and C19orf12 Research. Brain Sciences, 15(8), 777. https://doi.org/10.3390/brainsci15080777