
Looking into Abnormal Co-Expressions of Tau and TDP-43 in the Realm of Mixed Dementia Types: A Double-Punch Scenario




Abstract
1. Introduction
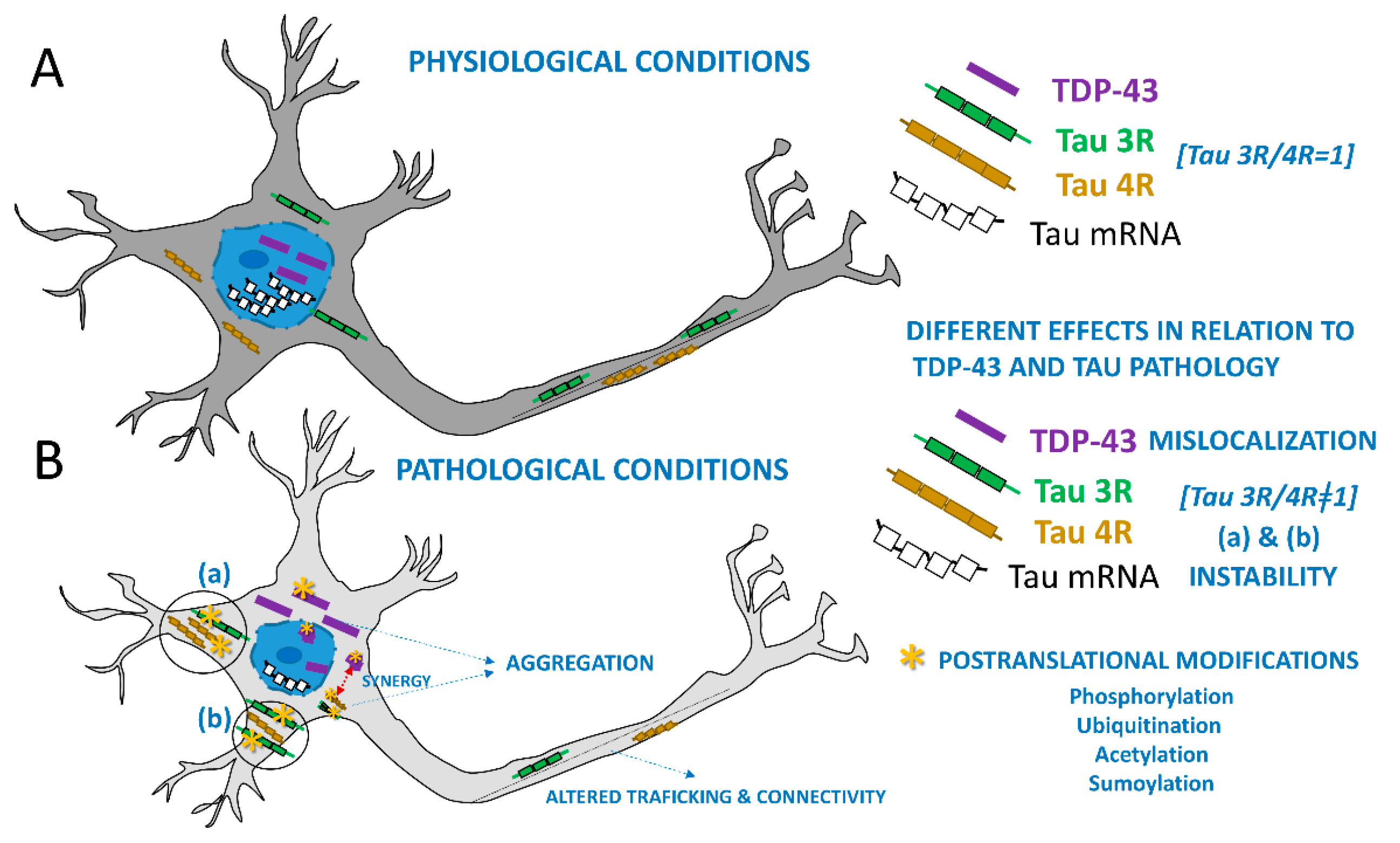
2. TDP-43 and Tau: Molecular Perspectives
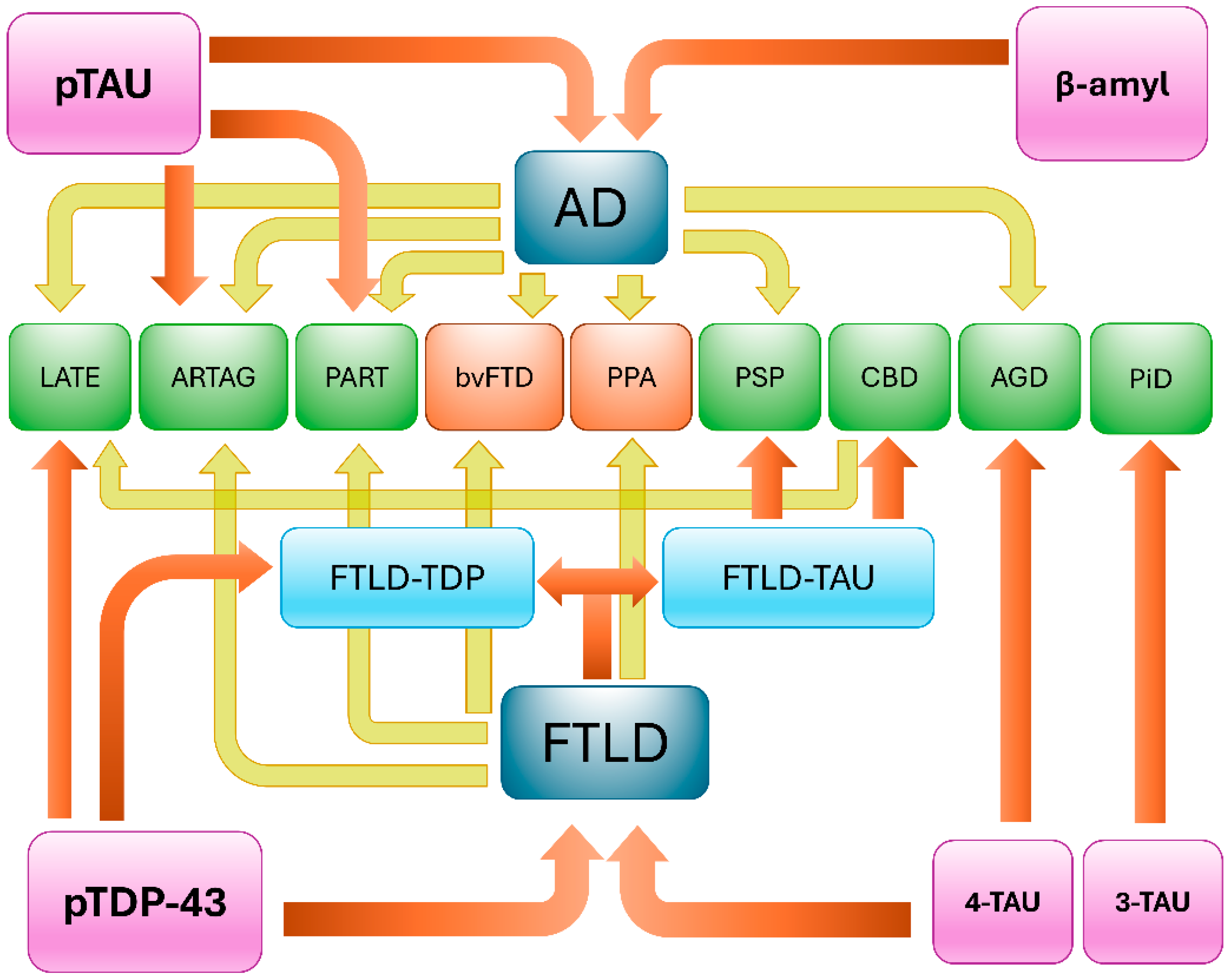
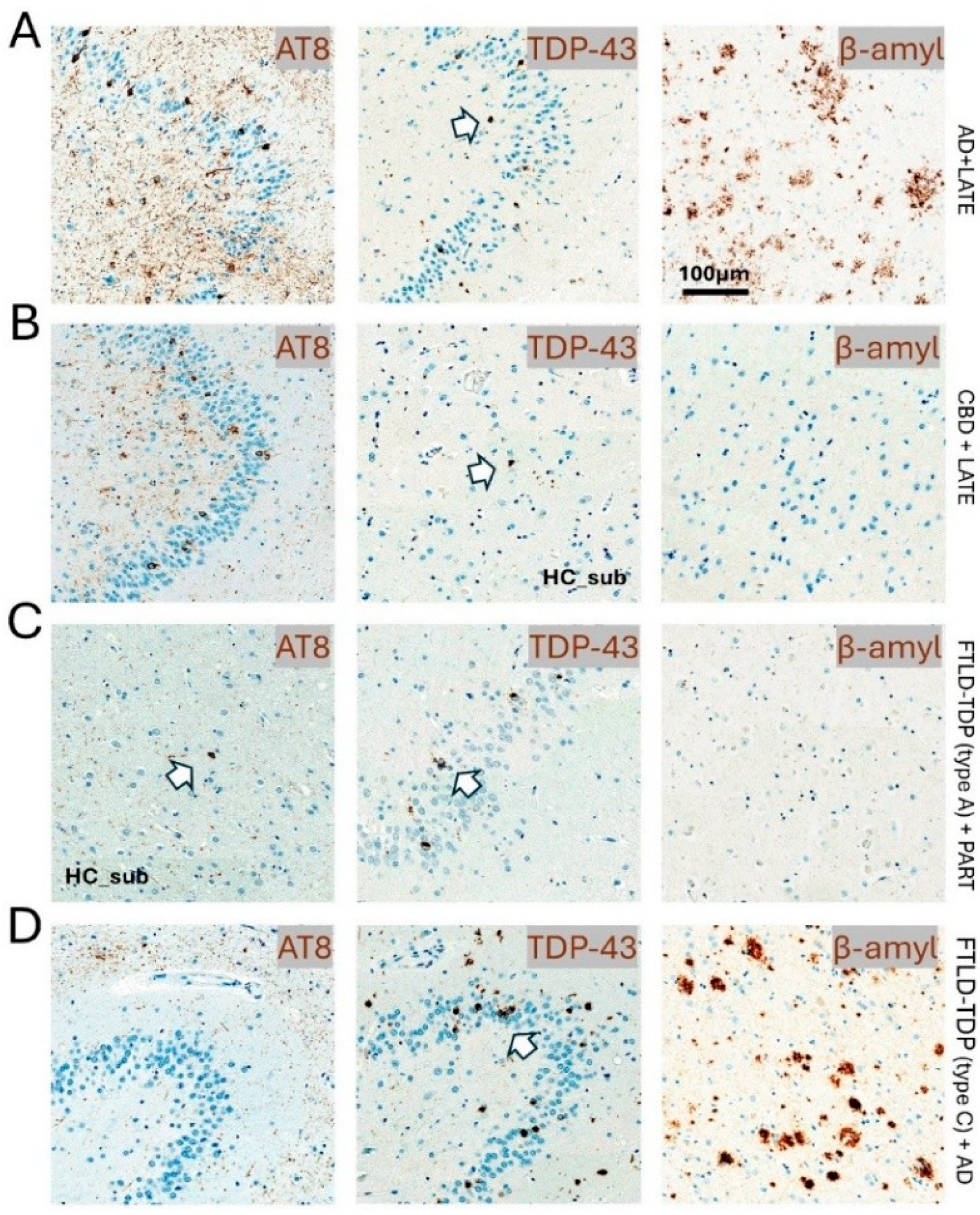
3. Clinical Presentations and Neuropathological Findings
3.1. Frontotemporal Lobar Degeneration

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Institution | Study Criteria | Total Cases | Tau Cases | TDP Cases | FUS Cases | Other Cases | Neuropathological and Clinical Findings |
|---|---|---|---|---|---|---|---|---|
| Hodges et al., 2004 [71] | Sydney, Australia, and Cambridge, UK | Pathologically confirmed FTLD or CBD from dementia clinics. No PSP | 61 | 31 | 30 | 0 | 0 | One out of nine CBD had ubiquitin-positive. CBD pathology was present in seven of nine cases (78%), with one each having FTLD-MND and DLDH. |
| Kertez et al., 2005 [72] | London, Ontario, Canada | Clinical diagnosis of FTD, CBS, or PSP that went to autopsy | 60 | 21 | 24 | 0 | 15 | A case exhibited features of both CBD and MNDI, suggesting transitional patterns between tau-positive and tau-negative types. Clinical CBS with non-CBD pathologies highlights this overlap, making it difficult to predict pathological variety from clinical phenotype. This indicates that initially distinct syndromes may converge in a patient, representing different histological varieties. |
| Forman et al., 2006 [73] | Philadelphia, PA, USA | Clinical diagnosis of dementia, as well as FTLD, CBD, or PSP pathologic diagnosis | 90 | 53 | 37 | 0 | 0 | Progressive aphasia is linked to tau-negative pathologies like FTLD-U, while progressive nonfluent aphasia is associated with PiD or CBD. No specific clinical association between language disorder and pathological subgroups was found. Tauopathy patients often show extrapyramidal features indicative of CBD and PSP, aligning with smaller studies that connect rigidity to CBD and pyramidal features of clinical MND to FTLD-U. |
| Snowden et al., 2007 [74] | Manchester, UK | Pathologic diagnosis of FTLD or CBD from a cerebral function unit | 65 | 25 | 40 | 0 | 0 | One case of tauopathy exhibited CBD-related immunohistochemical changes, despite presenting typically as bvFTD without Parkinsonism or asymmetric apraxia. This highlights that predictable patterns do not guarantee a direct link between clinical and pathological phenotypes. |
| Robinson et al., 2014 [61] | Manchester, UK | Pathological diagnosis of FTLD-tau, FTLD-TDP, and MND | 78 | 33 | 45 | 0 | 23 | In 33 FTLD-tau cases, IHC for non-pTDP-43 showed normal nuclear staining in all but two cases with pathological TDP-43 (NCI, DN, NII). One case had a MAPT+13 mutation with TDP-43 in a few surviving cells and extensive involvement of the entorhinal cortex and fusiform gyrus, alongside Aβ deposition. The CBD case had few NCI and DN in similar regions. Tau immunostaining revealed AT8-positive neurons in 60% of FTLD-TDP patients, 30% of MND patients, and 81% of controls. TDP-43 pathology resembled FTLD-TDP type A but was limited to medial temporal lobe structures, suggesting a secondary pathology akin to changes in elderly AD patients. |
| Kim et al., 2018 [60] | University of California, LA, USA Pusan national university, Busan, Republic of Korea | Neuropathologic diagnosis of FTLD-TDP and FTLD-tau | 9 | 4 | 5 | 0 | 0 | Primary FTLD-TDP presents with unclassifiable FTLD-tau inclusions and conditions like PSP. Mixed cases of FTLD-TDP and FTLD-tau exhibited widespread tau pathologies distinct from Alzheimer’s disease. Among five mixed cases, three showed unclassifiable tauopathy, while two with FTLD-TDP type C were associated with FTLD-tau and PSP. Additionally, TDP-43 pathology was found in four FTLD-tau, CBD cases, with no significant clinical differences between mixed and pure FTLD-tau groups. FTLD-CBD emerged as the predominant form with TDP-43 pathology, primarily affecting regions beyond limbic areas, correlating with clinical symptoms such as abnormal behaviors and nonfluent aphasia. |
| Pennington et al., 2020 [5] | Bristol & Edinburgh | Neuropathologic diagnosis of FTLD-TDP and FTLD-tau | 515 | 139 | 359 | 7 | 10 | In nine cases, primary tauopathy and secondary TDP-43 deposition were noted; four cases had a primary FTLD-TDP diagnosis with additional tauopathy. Mixed neuropathology occurred more frequently in FTLD-TDP (49%) than in FTLD-tau (24%). |
| Koga et al., 2021 [59] | Jacksonville, Florida, USA | Pathologically confirmed cases of FTLD-TDP with and without MND | 201 | 0 | 146 | 0 | 55 | In the FTLD-TDP cases (n = 146), PART was found in 34% (50), ARTAG in 44% (64), AGD in 23% (33), and CBD in 1% (2). |
3.2. Argyrophilic Grain Disease and TDP-43
3.3. Corticobasal Degeneration and TDP-43
3.4. TDP-43 in Alzheimer’s Disease and Primary Age-Related Tauopathy
3.5. Tau in Limbic-Predominant Age-Related TDP-43 Encephalopathy
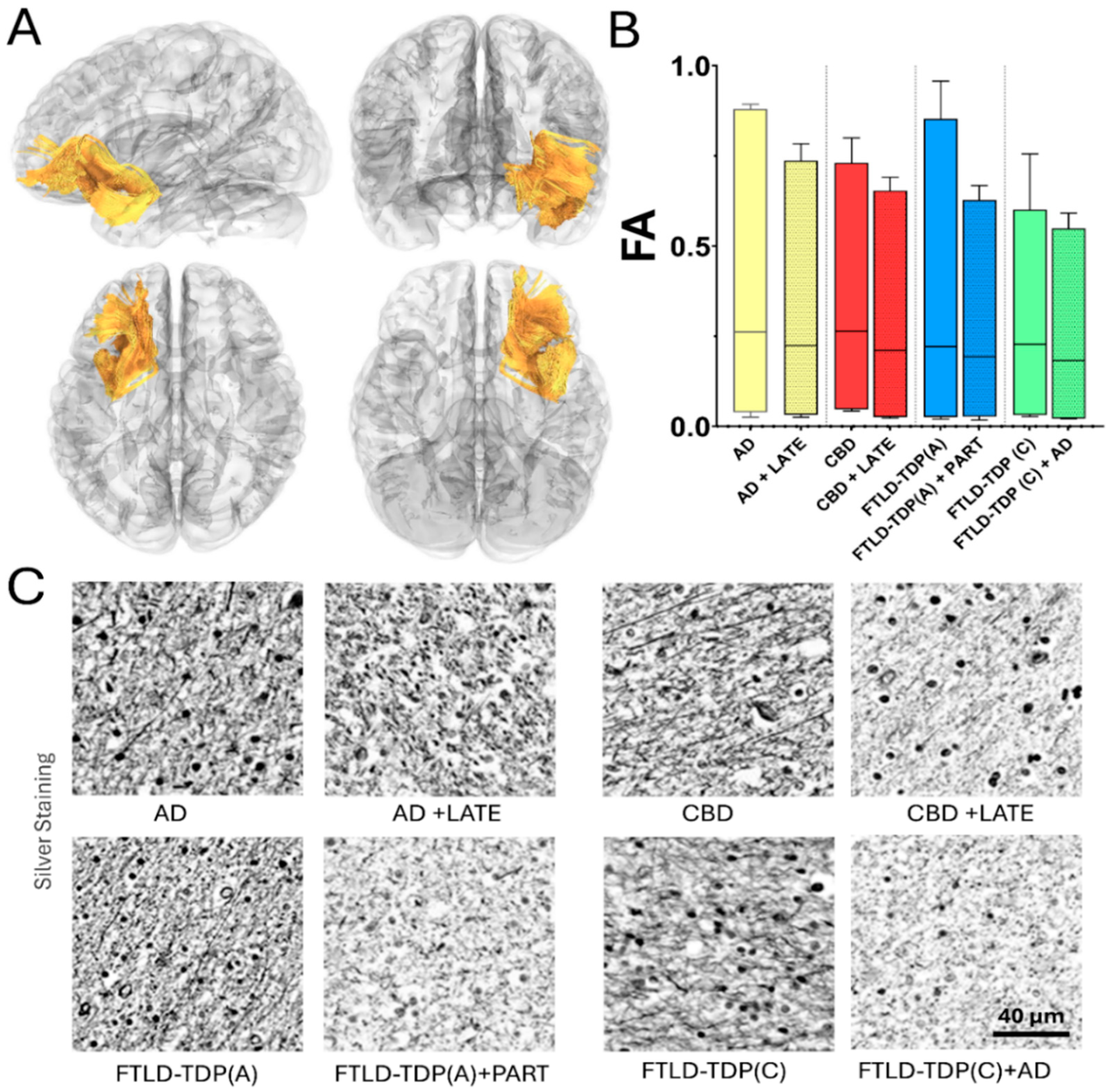
4. Macro- and Microstructural MRI Findings in TDP and Tau Co-Pathologies
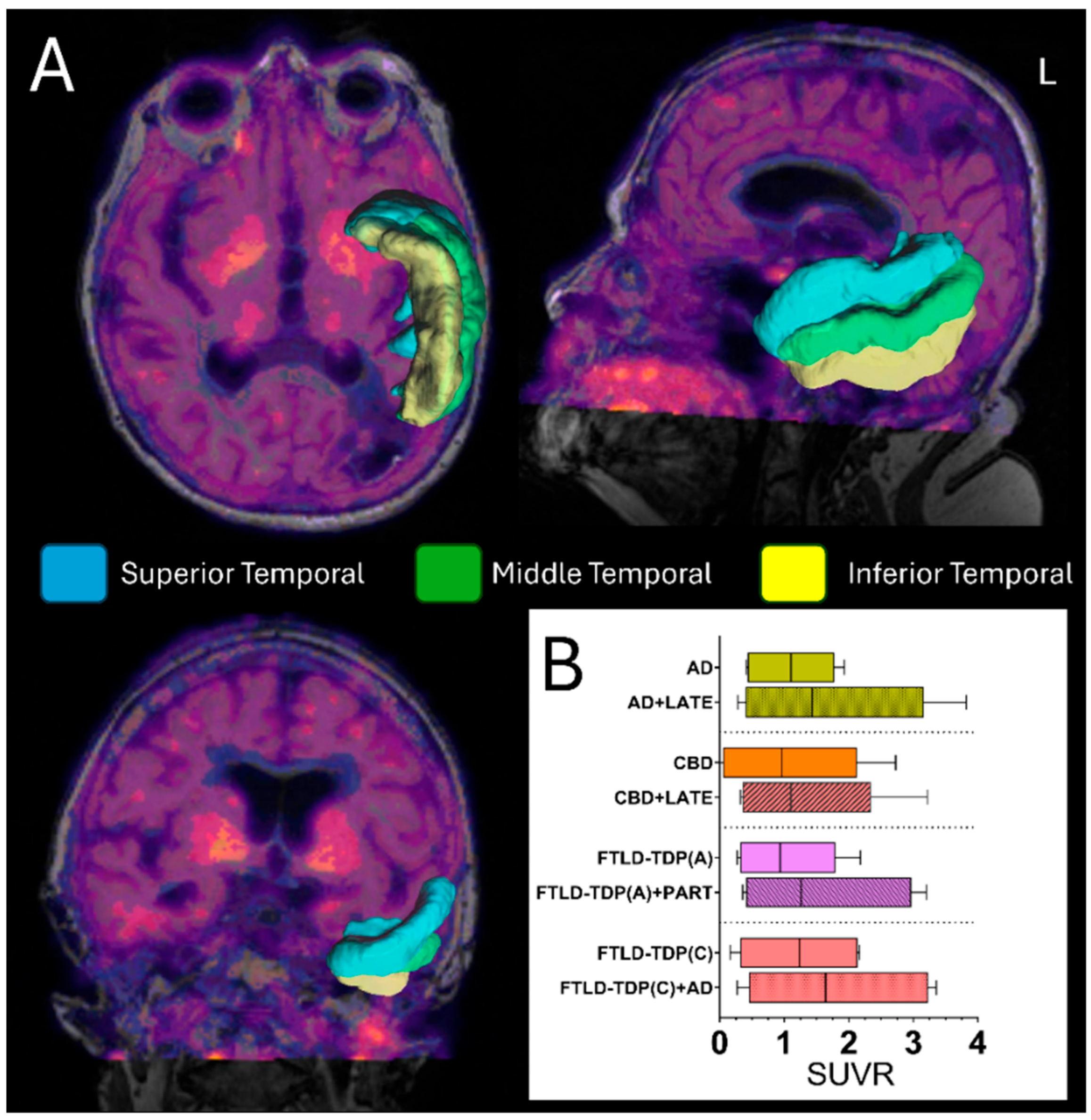
5. Positron Emission Tomography (PET) Scan
5.1. Tau PET Scan and TDP-43 Detection
5.2. Fluorodeoxyglucose (FDG) PET Scan Significance in Detecting Tau and TDP-43
5.3. Neuroimaging and Neuromodulation in Untangling Mixed Pathology Networks
6. Summary
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gefen, T.; Ahmadian, S.S.; Mao, Q.; Kim, G.; Seckin, M.; Bonakdarpour, B.; Ramos, E.M.; Coppola, G.; Rademakers, R.; Rogalski, E.; et al. Combined Pathologies in FTLD-TDP Types A and C. J. Neuropathol. Exp. Neurol. 2018, 77, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Latimer, C.S.; Liachko, N.F. Tau and TDP-43 synergy: A novel therapeutic target for sporadic late-onset Alzheimer’s disease. Geroscience 2021, 43, 1627–1634. [Google Scholar] [CrossRef] [PubMed]
- Meneses, A.; Koga, S.; O’Leary, J.; Dickson, D.W.; Bu, G.; Zhao, N. TDP-43 Pathology in Alzheimer’s Disease. Mol. Neurodegener. 2021, 16, 84. [Google Scholar] [CrossRef]
- Nelson, P.T.; Fardo, D.W.; Wu, X.; Aung, K.Z.; Cykowski, M.D.; Katsumata, Y. Limbic-predominant age-related TDP-43 encephalopathy (LATE-NC): Co-pathologies and genetic risk factors provide clues about pathogenesis. J. Neuropathol. Exp. Neurol. 2024, 83, 396–415. [Google Scholar] [CrossRef] [PubMed]
- Pennington, C.; Marini, L.; Coulthard, E.; Love, S. Mixed neuropathology in frontotemporal lobar degeneration. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 301–308. [Google Scholar] [CrossRef]
- Tome, S.O.; Tsaka, G.; Ronisz, A.; Ospitalieri, S.; Gawor, K.; Gomes, L.A.; Otto, M.; von Arnim, C.A.F.; Van Damme, P.; Van Den Bosch, L.; et al. TDP-43 pathology is associated with increased tau burdens and seeding. Mol. Neurodegener. 2023, 18, 71. [Google Scholar] [CrossRef]
- Tome, S.O.; Vandenberghe, R.; Ospitalieri, S.; Van Schoor, E.; Tousseyn, T.; Otto, M.; von Arnim, C.A.F.; Thal, D.R. Distinct molecular patterns of TDP-43 pathology in Alzheimer’s disease: Relationship with clinical phenotypes. Acta Neuropathol. Commun. 2020, 8, 61. [Google Scholar] [CrossRef]
- Josephs, K.A.; Murray, M.E.; Tosakulwong, N.; Weigand, S.D.; Knopman, D.S.; Petersen, R.C.; Jack, C.R., Jr.; Whitwell, J.L.; Dickson, D.W. Brain atrophy in primary age-related tauopathy is linked to transactive response DNA-binding protein of 43 kDa. Alzheimers Dement. 2019, 15, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Riku, Y.; Yoshida, M.; Iwasaki, Y.; Sobue, G.; Katsuno, M.; Ishigaki, S. TDP-43 Proteinopathy and Tauopathy: Do They Have Pathomechanistic Links? Int. J. Mol. Sci. 2022, 23, 15755. [Google Scholar] [CrossRef]
- Youssef, H.; Gatto, R.G.; Pham, N.T.T.; Petersen, R.C.; Machulda, M.M.; Reichard, R.R.; Dickson, D.W.; Jack, C.R.; Whitwell, J.L.; Josephs, K.A. TDP-43 Is Associated with Subiculum and Cornu Ammonis 1 Hippocampal Subfield Atrophy in Primary Age-Related Tauopathy. J. Alzheimers Dis. 2024, 99, 1023–1032. [Google Scholar] [CrossRef]
- McAleese, K.E.; Walker, L.; Erskine, D.; Johnson, M.; Koss, D.; Thomas, A.J.; Attems, J. Concomitant LATE-NC in Alzheimer’s disease is not associated with increased tau or amyloid-beta pathological burden. Neuropathol. Appl. Neurobiol. 2020, 46, 722–734. [Google Scholar] [CrossRef]
- Riku, Y.; Iwasaki, Y.; Ishigaki, S.; Akagi, A.; Hasegawa, M.; Nishioka, K.; Li, Y.; Riku, M.; Ikeuchi, T.; Fujioka, Y.; et al. Motor neuron TDP-43 proteinopathy in progressive supranuclear palsy and corticobasal degeneration. Brain 2022, 145, 2769–2784. [Google Scholar] [CrossRef]
- Tome, S.O.; Gawor, K.; Thal, D.R. LATE-NC in Alzheimer’s disease: Molecular aspects and synergies. Brain Pathol. 2024, 34, e13213. [Google Scholar] [CrossRef] [PubMed]
- Josephs, K.A.; Murray, M.E.; Tosakulwong, N.; Whitwell, J.L.; Knopman, D.S.; Machulda, M.M.; Weigand, S.D.; Boeve, B.F.; Kantarci, K.; Petrucelli, L.; et al. Tau aggregation influences cognition and hippocampal atrophy in the absence of beta-amyloid: A clinico-imaging-pathological study of primary age-related tauopathy (PART). Acta Neuropathol. 2017, 133, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Josephs, K.A.; Whitwell, J.L.; Knopman, D.S.; Hu, W.T.; Stroh, D.A.; Baker, M.; Rademakers, R.; Boeve, B.F.; Parisi, J.E.; Smith, G.E.; et al. Abnormal TDP-43 immunoreactivity in AD modifies clinicopathologic and radiologic phenotype. Neurology 2008, 70, 1850–1857. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Schneider, J.A.; Jicha, G.A.; Duong, M.T.; Wolk, D.A. When Alzheimer’s is LATE: Why Does it Matter? Ann. Neurol. 2023, 94, 211–222. [Google Scholar] [CrossRef]
- Jiang, L.L.; Zhang, X.L.; Hu, H.Y. Co-Aggregation of TDP-43 with Other Pathogenic Proteins and Their Co-Pathologies in Neurodegenerative Diseases. Int. J. Mol. Sci. 2024, 25, 12380. [Google Scholar] [CrossRef]
- Youssef, H.; Gatto, R.G.; Pham, N.T.T.; Jones, D.; Petersen, R.C.; Machulda, M.M.; Whitwell, J.L.; Josephs, K.A. Multiple Neuropathologies Underly Hippocampal Subfield Atrophy in a Case With a Slowly Progressive Amnestic Syndrome: Challenging the Notion of Pure LATE-NC. Neuropathology 2025. [Google Scholar] [CrossRef]
- Dulski, J.; Cerquera-Cleves, C.; Milanowski, L.; Kidd, A.; Sitek, E.J.; Strongosky, A.; Vanegas Monroy, A.M.; Dickson, D.W.; Ross, O.A.; Pentela-Nowicka, J.; et al. Clinical, pathological and genetic characteristics of Perry disease-new cases and literature review. Eur. J. Neurol. 2021, 28, 4010–4021. [Google Scholar] [CrossRef]
- Mishima, T.; Fujioka, S.; Tomiyama, H.; Yabe, I.; Kurisaki, R.; Fujii, N.; Neshige, R.; Ross, O.A.; Farrer, M.J.; Dickson, D.W.; et al. Establishing diagnostic criteria for Perry syndrome. J. Neurol. Neurosurg. Psychiatry 2018, 89, 482–487. [Google Scholar] [CrossRef]
- Baloh, R.H. TDP-43: The relationship between protein aggregation and neurodegeneration in amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Febs J. 2011, 278, 3539–3549. [Google Scholar] [CrossRef] [PubMed]
- Tollervey, J.R.; Curk, T.; Rogelj, B.; Briese, M.; Cereda, M.; Kayikci, M.; Konig, J.; Hortobagyi, T.; Nishimura, A.L.; Zupunski, V.; et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 2011, 14, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; Baralle, F.E. The multiple roles of TDP-43 in pre-mRNA processing and gene expression regulation. RNA Biol. 2010, 7, 420–429. [Google Scholar] [CrossRef]
- Ishiguro, T.; Sato, N.; Ueyama, M.; Fujikake, N.; Sellier, C.; Kanegami, A.; Tokuda, E.; Zamiri, B.; Gall-Duncan, T.; Mirceta, M.; et al. Regulatory Role of RNA Chaperone TDP-43 for RNA Misfolding and Repeat-Associated Translation in SCA31. Neuron 2017, 94, 108–124.E7. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Ying, Y.; Xie, H.; Liu, X.; Wang, X.; Li, J. The Regulatory Role of RNA Metabolism Regulator TDP-43 in Human Cancer. Front. Oncol. 2021, 11, 755096. [Google Scholar] [CrossRef]
- Freibaum, B.D.; Chitta, R.K.; High, A.A.; Taylor, J.P. Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J. Proteome Res. 2010, 9, 1104–1120. [Google Scholar] [CrossRef]
- Lalmansingh, A.S.; Urekar, C.J.; Reddi, P.P. TDP-43 is a transcriptional repressor: The testis-specific mouse acrv1 gene is a TDP-43 target in vivo. J. Biol. Chem. 2011, 286, 10970–10982. [Google Scholar] [CrossRef]
- Dutta, K.; Thammisetty, S.S.; Boutej, H.; Bareil, C.; Julien, J.P. Mitigation of ALS Pathology by Neuron-Specific Inhibition of Nuclear Factor Kappa B Signaling. J. Neurosci. 2020, 40, 5137–5154. [Google Scholar] [CrossRef]
- Chhangani, D.; Martin-Pena, A.; Rincon-Limas, D.E. Molecular, functional, and pathological aspects of TDP-43 fragmentation. iScience 2021, 24, 102459. [Google Scholar] [CrossRef]
- Fang, Y.S.; Tsai, K.J.; Chang, Y.J.; Kao, P.; Woods, R.; Kuo, P.H.; Wu, C.C.; Liao, J.Y.; Chou, S.C.; Lin, V.; et al. Full-length TDP-43 forms toxic amyloid oligomers that are present in frontotemporal lobar dementia-TDP patients. Nat. Commun. 2014, 5, 4824. [Google Scholar] [CrossRef]
- Liachko, N.F.; Guthrie, C.R.; Kraemer, B.C. Phosphorylation promotes neurotoxicity in a Caenorhabditis elegans model of TDP-43 proteinopathy. J. Neurosci. 2010, 30, 16208–16219. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Lee, E.B.; Mackenzie, I.R. Frontotemporal Lobar Degeneration TDP-43-Immunoreactive Pathological Subtypes: Clinical and Mechanistic Significance. Adv. Exp. Med. Biol. 2021, 1281, 201–217. [Google Scholar] [CrossRef]
- Kawakami, I.; Arai, T.; Hasegawa, M. The basis of clinicopathological heterogeneity in TDP-43 proteinopathy. Acta Neuropathol. 2019, 138, 751–770. [Google Scholar] [CrossRef]
- Barbier, P.; Zejneli, O.; Martinho, M.; Lasorsa, A.; Belle, V.; Smet-Nocca, C.; Tsvetkov, P.O.; Devred, F.; Landrieu, I. Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Front. Aging Neurosci. 2019, 11, 204. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, D. Tau Protein and Tauopathies: Exploring Tau Protein–Protein and Microtubule Interactions, Cross-Interactions and Therapeutic Strategies. ChemMedChem 2024, 19, e202400180. [Google Scholar] [CrossRef] [PubMed]
- Avila, J.; Lucas, J.J.; Perez, M.; Hernandez, F. Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 2004, 84, 361–384. [Google Scholar] [CrossRef]
- Regan, P.; Mitchell, S.J.; Kim, S.C.; Lee, Y.; Yi, J.H.; Barbati, S.A.; Shaw, C.; Cho, K. Regulation of Synapse Weakening through Interactions of the Microtubule Associated Protein Tau with PACSIN1. J. Neurosci. 2021, 41, 7162–7170. [Google Scholar] [CrossRef]
- Robbins, M.; Clayton, E.; Kaminski Schierle, G.S. Synaptic tau: A pathological or physiological phenomenon? Acta Neuropathol. Commun. 2021, 9, 149. [Google Scholar] [CrossRef]
- Kanaan, N.M. Tau here, tau there, tau almost everywhere: Clarifying the distribution of tau in the adult CNS. Cytoskelet. 2024, 81, 107–115. [Google Scholar] [CrossRef]
- Younas, N.; Saleem, T.; Younas, A.; Zerr, I. Nuclear face of Tau: An inside player in neurodegeneration. Acta Neuropathol. Commun. 2023, 11, 196. [Google Scholar] [CrossRef]
- Gil, L.; Federico, C.; Pinedo, F.; Bruno, F.; Rebolledo, A.B.; Montoya, J.J.; Olazabal, I.M.; Ferrer, I.; Saccone, S. Aging dependent effect of nuclear tau. Brain Res. 2017, 1677, 129–137. [Google Scholar] [CrossRef]
- Tang, X.; Jiao, L.; Zheng, M.; Yan, Y.; Nie, Q.; Wu, T.; Wan, X.; Zhang, G.; Li, Y.; Wu, S.; et al. Tau Deficiency Down-Regulated Transcription Factor Orthodenticle Homeobox 2 Expression in the Dopaminergic Neurons in Ventral Tegmental Area and Caused No Obvious Motor Deficits in Mice. Neuroscience 2018, 373, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Bukar Maina, M.; Al-Hilaly, Y.K.; Serpell, L.C. Nuclear Tau and Its Potential Role in Alzheimer’s Disease. Biomolecules 2016, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Grundke-Iqbal, I.; Iqbal, K.; Quinlan, M.; Tung, Y.C.; Zaidi, M.S.; Wisniewski, H.M. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem. 1986, 261, 6084–6089. [Google Scholar] [CrossRef] [PubMed]
- Corsi, A.; Bombieri, C.; Valenti, M.T.; Romanelli, M.G. Tau Isoforms: Gaining Insight into MAPT Alternative Splicing. Int. J. Mol. Sci. 2022, 23, 15383. [Google Scholar] [CrossRef]
- Liu, F.; Gong, C.X. Tau exon 10 alternative splicing and tauopathies. Mol. Neurodegener. 2008, 3, 8. [Google Scholar] [CrossRef]
- Strang, K.H.; Golde, T.E.; Giasson, B.I. MAPT mutations, tauopathy, and mechanisms of neurodegeneration. Lab. Investig. 2019, 99, 912–928. [Google Scholar] [CrossRef]
- Andreadis, A.; Brown, W.M.; Kosik, K.S. Structure and novel exons of the human tau gene. Biochemistry 1992, 31, 10626–10633. [Google Scholar] [CrossRef]
- Ferrer, I.; López-González, I.; Carmona, M.; Arregui, L.; Dalfó, E.; Torrejón-Escribano, B.; Diehl, R.; Kovacs, G.G. Glial and neuronal tau pathology in tauopathies: Characterization of disease-specific phenotypes and tau pathology progression. J. Neuropathol. Exp. Neurol. 2014, 73, 81–97. [Google Scholar] [CrossRef]
- Kahlson, M.A.; Colodner, K.J. Glial Tau Pathology in Tauopathies: Functional Consequences. J. Exp. Neurosci. 2015, 9, 43–50. [Google Scholar] [CrossRef]
- Eltom, K.; Mothes, T.; Libard, S.; Ingelsson, M.; Erlandsson, A. Astrocytic accumulation of tau fibrils isolated from Alzheimer’s disease brains induces inflammation, cell-to-cell propagation and neuronal impairment. Acta Neuropathol. Commun. 2024, 12, 34. [Google Scholar] [CrossRef]
- Dickson, D.W.; Kouri, N.; Murray, M.E.; Josephs, K.A. Neuropathology of frontotemporal lobar degeneration-tau (FTLD-tau). J. Mol. Neurosci. 2011, 45, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Gatto, R.G.; Carlos, A.F.; Reichard, R.R.; Lowe, V.J.; Whitwell, J.L.; Josephs, K.A. Comparative assessment of regional tau distribution by Tau-PET and Post-mortem neuropathology in a representative set of Alzheimer’s & frontotemporal lobar degeneration patients. PLoS ONE 2023, 18, e0284182. [Google Scholar] [CrossRef]
- Gatto, R.G.; Hossam, Y.; Reichard, R.R.; Lowe, V.J.; Whitwell, J.L.; Josephs, K.A. Microscopy assessment of a fluorescence [(18)F] flortaucipir analog (T726) shows neuropathological overlap with 3R and 4R tau lesions. Alzheimers Dement. 2024, 20, 8758–8768. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Ozdemir, S.; Fritz, C.; Mobius, W.; Kleineidam, L.; Mandelkow, E.; Biernat, J.; Dogdu, C.; Peters, O.; Cosma, N.C.; et al. Plasma extracellular vesicle tau and TDP-43 as diagnostic biomarkers in FTD and ALS. Nat. Med. 2024, 30, 1771–1783. [Google Scholar] [CrossRef]
- Chornenkyy, Y.; Fardo, D.W.; Nelson, P.T. Tau and TDP-43 proteinopathies: Kindred pathologic cascades and genetic pleiotropy. Lab. Invest. 2019, 99, 993–1007. [Google Scholar] [CrossRef]
- Taylor, L.M.; McMillan, P.J.; Liachko, N.F.; Strovas, T.J.; Ghetti, B.; Bird, T.D.; Keene, C.D.; Kraemer, B.C. Pathological phosphorylation of tau and TDP-43 by TTBK1 and TTBK2 drives neurodegeneration. Mol. Neurodegener. 2018, 13, 7. [Google Scholar] [CrossRef]
- Liachko, N.F.; McMillan, P.J.; Strovas, T.J.; Loomis, E.; Greenup, L.; Murrell, J.R.; Ghetti, B.; Raskind, M.A.; Montine, T.J.; Bird, T.D.; et al. The tau tubulin kinases TTBK1/2 promote accumulation of pathological TDP-43. PLoS Genet. 2014, 10, e1004803. [Google Scholar] [CrossRef]
- Koga, S.; Zhou, X.; Murakami, A.; Fernandez De Castro, C.; Baker, M.C.; Rademakers, R.; Dickson, D.W. Concurrent tau pathologies in frontotemporal lobar degeneration with TDP-43 pathology. Neuropathol. Appl. Neurobiol. 2022, 48, e12778. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Brown, J.A.; Deng, J.; Hwang, J.L.; Spina, S.; Miller, Z.A.; DeMay, M.G.; Valcour, V.; Karydas, A.; Ramos, E.M.; et al. Mixed TDP-43 proteinopathy and tauopathy in frontotemporal lobar degeneration: Nine case series. J. Neurol. 2018, 265, 2960–2971. [Google Scholar] [CrossRef]
- Robinson, A.C.; Thompson, J.C.; Weedon, L.; Rollinson, S.; Pickering-Brown, S.; Snowden, J.S.; Davidson, Y.S.; Mann, D.M. No interaction between tau and TDP-43 pathologies in either frontotemporal lobar degeneration or motor neurone disease. Neuropathol. Appl. Neurobiol. 2014, 40, 844–854. [Google Scholar] [CrossRef]
- Jo, M.; Lee, S.; Jeon, Y.-M.; Kim, S.; Kwon, Y.; Kim, H.-J. The role of TDP-43 propagation in neurodegenerative diseases: Integrating insights from clinical and experimental studies. Exp. Mol. Med. 2020, 52, 1652–1662. [Google Scholar] [CrossRef]
- Gu, J.; Wu, F.; Xu, W.; Shi, J.; Hu, W.; Jin, N.; Qian, W.; Wang, X.; Iqbal, K.; Gong, C.X.; et al. TDP-43 suppresses tau expression via promoting its mRNA instability. Nucleic Acids Res. 2017, 45, 6177–6193. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Chen, F.; Iqbal, K.; Gong, C.X.; Wang, X.; Liu, F. Transactive response DNA-binding protein 43 (TDP-43) regulates alternative splicing of tau exon 10: Implications for the pathogenesis of tauopathies. J. Biol. Chem. 2017, 292, 10600–10612. [Google Scholar] [CrossRef] [PubMed]
- Dang, M.; Wu, L.; Zhang, X. Structural insights and milestones in TDP-43 research: A comprehensive review of its pathological and therapeutic advances. Int. J. Biol. Macromol. 2025, 306, 141677. [Google Scholar] [CrossRef]
- Montalbano, M.; McAllen, S.; Cascio, F.L.; Sengupta, U.; Garcia, S.; Bhatt, N.; Ellsworth, A.; Heidelman, E.A.; Johnson, O.D.; Doskocil, S.; et al. TDP-43 and Tau Oligomers in Alzheimer’s Disease, Amyotrophic Lateral Sclerosis, and Frontotemporal Dementia. Neurobiol. Dis. 2020, 146, 105130. [Google Scholar] [CrossRef]
- Nelson, P.T.; Abner, E.L.; Patel, E.; Anderson, S.; Wilcock, D.M.; Kryscio, R.J.; Van Eldik, L.J.; Jicha, G.A.; Gal, Z.; Nelson, R.S.; et al. The Amygdala as a Locus of Pathologic Misfolding in Neurodegenerative Diseases. J. Neuropathol. Exp. Neurol. 2018, 77, 2–20. [Google Scholar] [CrossRef]
- Kapasi, A.; DeCarli, C.; Schneider, J.A. Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathol. 2017, 134, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.X.; Bajaj, S.; McRae-McKee, K.; Hadjichrysanthou, C.; Anderson, R.M.; Collinge, J. Association of TDP-43 proteinopathy, cerebral amyloid angiopathy, and Lewy bodies with cognitive impairment in individuals with or without Alzheimer’s disease neuropathology. Sci. Rep. 2020, 10, 14579. [Google Scholar] [CrossRef] [PubMed]
- Kawas, C.H.; Kim, R.C.; Sonnen, J.A.; Bullain, S.S.; Trieu, T.; Corrada, M.M. Multiple pathologies are common and related to dementia in the oldest-old: The 90 + Study. Neurology 2015, 85, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Hodges, J.R.; Davies, R.R.; Xuereb, J.H.; Casey, B.; Broe, M.; Bak, T.H.; Kril, J.J.; Halliday, G.M. Clinicopathological correlates in frontotemporal dementia. Ann. Neurol. 2004, 56, 399–406. [Google Scholar] [CrossRef]
- Kertesz, A.; McMonagle, P.; Blair, M.; Davidson, W.; Munoz, D.G. The evolution and pathology of frontotemporal dementia. Brain 2005, 128, 1996–2005. [Google Scholar] [CrossRef]
- Forman, M.S.; Farmer, J.; Johnson, J.K.; Clark, C.M.; Arnold, S.E.; Coslett, H.B.; Chatterjee, A.; Hurtig, H.I.; Karlawish, J.H.; Rosen, H.J.; et al. Frontotemporal dementia: Clinicopathological correlations. Ann. Neurol. 2006, 59, 952–962. [Google Scholar] [CrossRef]
- Snowden, J.; Neary, D.; Mann, D. Frontotemporal lobar degeneration: Clinical and pathological relationships. Acta Neuropathol. 2007, 114, 31–38. [Google Scholar] [CrossRef]
- Torres, A.K.; Jara, C.; Olesen, M.A.; Tapia-Rojas, C. Pathologically phosphorylated tau at S396/404 (PHF-1) is accumulated inside of hippocampal synaptic mitochondria of aged Wild-type mice. Sci. Rep. 2021, 11, 4448. [Google Scholar] [CrossRef]
- Mammeri, N.E.; Dregni, A.J.; Duan, P.; Hong, M. Structures of AT8 and PHF1 phosphomimetic tau: Insights into the posttranslational modification code of tau aggregation. Proc. Natl. Acad. Sci. USA 2024, 121, e2316175121. [Google Scholar] [CrossRef]
- Uryu, K.; Nakashima-Yasuda, H.; Forman, M.S.; Kwong, L.K.; Clark, C.M.; Grossman, M.; Miller, B.L.; Kretzschmar, H.A.; Lee, V.M.; Trojanowski, J.Q.; et al. Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J. Neuropathol. Exp. Neurol. 2008, 67, 555–564. [Google Scholar] [CrossRef]
- Wisse, L.E.M.; Wuestefeld, A.; Murray, M.E.; Jagust, W.; La Joie, R. Role of tau versus TDP-43 pathology on medial temporal lobe atrophy in aging and Alzheimer’s disease. Alzheimer’s Dement. 2025, 21, e14582. [Google Scholar] [CrossRef] [PubMed]
- Brenowitz, W.D.; Hubbard, R.A.; Keene, C.D.; Hawes, S.E.; Longstreth, W.T., Jr.; Woltjer, R.L.; Kukull, W.A. Mixed neuropathologies and estimated rates of clinical progression in a large autopsy sample. Alzheimers Dement. 2017, 13, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, R.A. Survival in the pre-senile dementia frontotemporal lobar degeneration with TDP-43 proteinopathy: Effects of genetic, demographic and neuropathological variables. Folia Neuropathol. 2016, 54, 137–148. [Google Scholar] [CrossRef]
- Perry, D.C.; Brown, J.A.; Possin, K.L.; Datta, S.; Trujillo, A.; Radke, A.; Karydas, A.; Kornak, J.; Sias, A.C.; Rabinovici, G.D.; et al. Clinicopathological correlations in behavioural variant frontotemporal dementia. Brain 2017, 140, 3329–3345. [Google Scholar] [CrossRef]
- Koga, S.; Murakami, A.; Soto-Beasley, A.I.; Walton, R.L.; Baker, M.C.; Castanedes-Casey, M.; Josephs, K.A.; Ross, O.A.; Dickson, D.W. Publisher Correction to: Diffuse argyrophilic grain disease with TDP-43 proteinopathy and neuronal intermediate filament inclusion disease: FTLD with mixed tau, TDP-43 and FUS pathologies. Acta Neuropathol. Commun. 2023, 11, 117. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.J.; Dugger, B.N.; Beach, T.G. TDP-43 deposition in prospectively followed, cognitively normal elderly individuals: Correlation with argyrophilic grains but not other concomitant pathologies. Acta Neuropathol. 2013, 126, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Santpere, G.; van Leeuwen, F.W. Argyrophilic grain disease. Brain 2008, 131, 1416–1432. [Google Scholar] [CrossRef]
- Koga, S.; Murakami, A.; Martin, N.B.; Dickson, D.W. The frequency and distribution of TDP-43 pathology in argyrophilic grain disease. J. Neuropathol. Exp. Neurol. 2023, 82, 739–741. [Google Scholar] [CrossRef]
- Fujishiro, H.; Uchikado, H.; Arai, T.; Hasegawa, M.; Akiyama, H.; Yokota, O.; Tsuchiya, K.; Togo, T.; Iseki, E.; Hirayasu, Y. Accumulation of phosphorylated TDP-43 in brains of patients with argyrophilic grain disease. Acta Neuropathol. 2009, 117, 151–158. [Google Scholar] [CrossRef]
- Koga, S.; Kouri, N.; Walton, R.L.; Ebbert, M.T.W.; Josephs, K.A.; Litvan, I.; Graff-Radford, N.; Ahlskog, J.E.; Uitti, R.J.; van Gerpen, J.A.; et al. Corticobasal degeneration with TDP-43 pathology presenting with progressive supranuclear palsy syndrome: A distinct clinicopathologic subtype. Acta Neuropathol. 2018, 136, 389–404. [Google Scholar] [CrossRef]
- Sainouchi, M.; Tada, M.; Fitrah, Y.A.; Hara, N.; Tanaka, K.; Idezuka, J.; Aida, I.; Nakajima, T.; Miyashita, A.; Akazawa, K.; et al. Brain TDP-43 pathology in corticobasal degeneration: Topographical correlation with neuronal loss. Neuropathol. Appl. Neurobiol. 2022, 48, e12786. [Google Scholar] [CrossRef]
- Tomenaga, T.; Minatani, S.; Namba, H.; Takeda, A.; Yoshizaki, T.; Kawabe, J.; Keyoumu, N.; Morino, H.; Higuchi, M.; Matsubara, T.; et al. An autopsy case of type A FTLD-TDP with a GRN mutation presenting with the logopenic variant of primary progressive aphasia at onset and with corticobasal syndrome subsequently. Neuropathology 2025, 45, 38–47. [Google Scholar] [CrossRef]
- Tando, S.; Kasai, T.; Mizuta, I.; Takahashi, H.; Yaoi, T.; Saito, K.; Hojo, T.; Mizuno, T.; Hasegawa, M.; Itoh, K. An autopsy case of corticobasal syndrome due to asymmetric degeneration of the motor cortex and substantia nigra with TDP-43 proteinopathy, associated with Alzheimer’s disease pathology. Neuropathology 2021, 41, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Smirnov, D.S.; Salmon, D.P.; Galasko, D.; Edland, S.D.; Pizzo, D.P.; Goodwill, V.; Hiniker, A. TDP-43 Pathology Exacerbates Cognitive Decline in Primary Age-Related Tauopathy. Ann. Neurol. 2022, 92, 425–438. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, B.; Wang, X.; Lu, H.; Shao, F.; Rozemuller, A.J.M.; Liang, H.; Liu, C.; Chen, J.; Huang, M.; et al. Phosphorylated TDP-43 Staging of Primary Age-Related Tauopathy. Neurosci. Bull. 2019, 35, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Glashutter, M.; Wijesinghe, P.; Matsubara, J.A. TDP-43 as a potential retinal biomarker for neurodegenerative diseases. Front. Neurosci. 2025, 19, 1533045. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, H.S.; Choi, S.M.; Kim, B.C.; Lee, M.C.; Lee, K.H.; Lee, J.H. Primary Age-Related Tauopathy: An Elderly Brain Pathology Frequently Encountered during Autopsy. J. Pathol. Transl. Med. 2019, 53, 159–163. [Google Scholar] [CrossRef]
- Shih, Y.H.; Tu, L.H.; Chang, T.Y.; Ganesan, K.; Chang, W.W.; Chang, P.S.; Fang, Y.S.; Lin, Y.T.; Jin, L.W.; Chen, Y.R. TDP-43 interacts with amyloid-beta, inhibits fibrillization, and worsens pathology in a model of Alzheimer’s disease. Nat. Commun. 2020, 11, 5950. [Google Scholar] [CrossRef]
- Josephs, K.A.; Murray, M.E.; Whitwell, J.L.; Parisi, J.E.; Petrucelli, L.; Jack, C.R.; Petersen, R.C.; Dickson, D.W. Staging TDP-43 pathology in Alzheimer’s disease. Acta Neuropathol. 2014, 127, 441–450. [Google Scholar] [CrossRef]
- Carlos, A.F.; Koga, S.; Graff-Radford, N.R.; Baker, M.C.; Rademakers, R.; Ross, O.A.; Dickson, D.W.; Josephs, K.A. Senile plaque-associated transactive response DNA-binding protein 43 in Alzheimer’s disease: A case report spanning 16 years of memory loss. Neuropathology 2024, 44, 115–125. [Google Scholar] [CrossRef]
- Huang, W.; Zhou, Y.; Tu, L.; Ba, Z.; Huang, J.; Huang, N.; Luo, Y. TDP-43: From Alzheimer’s Disease to Limbic-Predominant Age-Related TDP-43 Encephalopathy. Front. Mol. Neurosci. 2020, 13, 20. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, Y.; Liu, M.; Wang, Y.; Peng, G. TDP-43 and Limbic-Predominant Age-Related TDP-43 Encephalopathy. Front. Aging Neurosci. 2020, 11, 376. [Google Scholar] [CrossRef]
- Wolk, D.A.; Nelson, P.T.; Apostolova, L.; Arfanakis, K.; Boyle, P.A.; Carlsson, C.M.; Corriveau-Lecavalier, N.; Dacks, P.; Dickerson, B.C.; Domoto-Reilly, K.; et al. Clinical criteria for limbic-predominant age-related TDP-43 encephalopathy. Alzheimers Dement. 2025, 21, e14202. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Yu, L.; Capuano, A.W.; Wilson, R.S.; Leurgans, S.E.; Bennett, D.A.; Schneider, J.A. Hippocampal sclerosis and TDP-43 pathology in aging and Alzheimer disease. Ann. Neurol. 2015, 77, 942–952. [Google Scholar] [CrossRef]
- Wilson, A.C.; Dugger, B.N.; Dickson, D.W.; Wang, D.S. TDP-43 in aging and Alzheimer’s disease—A review. Int. J. Clin. Exp. Pathol. 2011, 4, 147–155. [Google Scholar]
- Hokkanen, S.R.K.; Hunter, S.; Polvikoski, T.M.; Keage, H.A.D.; Minett, T.; Matthews, F.E.; Brayne, C.; Mrc, C.; Group, C.C.S. Hippocampal sclerosis, hippocampal neuron loss patterns and TDP-43 in the aged population. Brain Pathol. 2018, 28, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Josephs, K.A.; Mackenzie, I.; Frosch, M.P.; Bigio, E.H.; Neumann, M.; Arai, T.; Dugger, B.N.; Ghetti, B.; Grossman, M.; Hasegawa, M.; et al. LATE to the PART-y. Brain 2019, 142, e47. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Dickson, D.W.; Trojanowski, J.Q.; Jack, C.R.; Boyle, P.A.; Arfanakis, K.; Rademakers, R.; Alafuzoff, I.; Attems, J.; Brayne, C.; et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): Consensus working group report. Brain 2019, 142, 1503–1527. [Google Scholar] [CrossRef]
- Amador-Ortiz, C.; Lin, W.L.; Ahmed, Z.; Personett, D.; Davies, P.; Duara, R.; Graff-Radford, N.R.; Hutton, M.L.; Dickson, D.W. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease. Ann. Neurol. 2007, 61, 435–445. [Google Scholar] [CrossRef]
- Josephs, K.A.; Whitwell, J.L.; Weigand, S.D.; Murray, M.E.; Tosakulwong, N.; Liesinger, A.M.; Petrucelli, L.; Senjem, M.L.; Knopman, D.S.; Boeve, B.F.; et al. TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathol. 2014, 127, 811–824. [Google Scholar] [CrossRef]
- Llamas-Rodriguez, J.; Oltmer, J.; Marshall, M.; Champion, S.; Frosch, M.P.; Augustinack, J.C. TDP-43 and tau concurrence in the entorhinal subfields in primary age-related tauopathy and preclinical Alzheimer’s disease. Brain Pathol. 2023, 33, e13159. [Google Scholar] [CrossRef]
- Josephs, K.A.; Koga, S.; Tosakulwong, N.; Weigand, S.D.; Nha Pham, T.T.; Baker, M.; Whitwell, J.L.; Rademakers, R.; Petrucelli, L.; Dickson, D.W. Molecular fragment characteristics and distribution of tangle associated TDP-43 (TATs) and other TDP-43 lesions in Alzheimer’s disease. Free Neuropathol. 2023, 4, 22. [Google Scholar] [CrossRef]
- Tan, C.H.; Hilal, S.; Xu, X.; Vrooman, H.; Cheng, C.Y.; Wong, T.Y.; Venketasubramanian, N.; Chen, C. MRI Markers of Mixed Pathology and Cognitive Impairment in Multiethnic Asians. J. Alzheimers Dis. 2020, 73, 1501–1509. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.A.; Arvanitakis, Z.; Bang, W.; Bennett, D.A. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 2007, 69, 2197–2204. [Google Scholar] [CrossRef]
- Carlos, A.F.; Sekiya, H.; Koga, S.; Gatto, R.G.; Casey, M.C.; Pham, N.T.T.; Sintini, I.; Machulda, M.M.; Jack, C.R.; Lowe, V.J.; et al. Clinicopathologic features of a novel star-shaped transactive response DNA-binding protein 43 (TDP-43) pathology in the oldest old. J. Neuropathol. Exp. Neurol. 2023, 83, 36–52. [Google Scholar] [CrossRef]
- Hu, W.T.; Josephs, K.A.; Knopman, D.S.; Boeve, B.F.; Dickson, D.W.; Petersen, R.C.; Parisi, J.E. Temporal lobar predominance of TDP-43 neuronal cytoplasmic inclusions in Alzheimer disease. Acta Neuropathol. 2008, 116, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Kadokura, A.; Yamazaki, T.; Lemere, C.A.; Takatama, M.; Okamoto, K. Regional distribution of TDP-43 inclusions in Alzheimer disease (AD) brains: Their relation to AD common pathology. Neuropathology 2009, 29, 566–573. [Google Scholar] [CrossRef]
- Nelson, P.T.; Brayne, C.; Flanagan, M.E.; Abner, E.L.; Agrawal, S.; Attems, J.; Castellani, R.J.; Corrada, M.M.; Cykowski, M.D.; Di, J.; et al. Frequency of LATE neuropathologic change across the spectrum of Alzheimer’s disease neuropathology: Combined data from 13 community-based or population-based autopsy cohorts. Acta Neuropathol. 2022, 144, 27–44. [Google Scholar] [CrossRef]
- Hiya, S.; Maldonado-Díaz, C.; Walker, J.M.; Richardson, T.E. Cognitive symptoms progress with limbic-predominant age-related TDP-43 encephalopathy stage and co-occurrence with Alzheimer disease. J. Neuropathol. Exp. Neurol. 2023, 83, 2–10. [Google Scholar] [CrossRef]
- Butler Pagnotti, R.M.; Pudumjee, S.B.; Cross, C.L.; Miller, J.B. Cognitive and Clinical Characteristics of Patients With Limbic-Predominant Age-Related TDP-43 Encephalopathy. Neurology 2023, 100, e2027–e2035. [Google Scholar] [CrossRef]
- Josephs, K.A.; Martin, P.R.; Weigand, S.D.; Tosakulwong, N.; Buciuc, M.; Murray, M.E.; Petrucelli, L.; Senjem, M.L.; Spychalla, A.J.; Knopman, D.S.; et al. Protein contributions to brain atrophy acceleration in Alzheimer’s disease and primary age-related tauopathy. Brain 2020, 143, 3463–3476. [Google Scholar] [CrossRef]
- Buciuc, M.; Wennberg, A.M.; Weigand, S.D.; Murray, M.E.; Senjem, M.L.; Spychalla, A.J.; Boeve, B.F.; Knopman, D.S.; Jack, C.R.; Kantarci, K.; et al. Effect Modifiers of TDP-43-Associated Hippocampal Atrophy Rates in Patients with Alzheimer’s Disease Neuropathological Changes. J. Alzheimers Dis. 2020, 73, 1511–1523. [Google Scholar] [CrossRef] [PubMed]
- Josephs, K.A.; Dickson, D.W.; Tosakulwong, N.; Weigand, S.D.; Murray, M.E.; Petrucelli, L.; Liesinger, A.M.; Senjem, M.L.; Spychalla, A.J.; Knopman, D.S.; et al. Rates of hippocampal atrophy and presence of post-mortem TDP-43 in patients with Alzheimer’s disease: A longitudinal retrospective study. Lancet Neurol. 2017, 16, 917–924. [Google Scholar] [CrossRef]
- Boelmans, K.; Kaufmann, J.; Bodammer, N.; Ebersbach, G.; Behlau, G.; Heinze, H.J.; Niehaus, L. Involvement of motor pathways in corticobasal syndrome detected by diffusion tensor tractography. Mov. Disord. 2009, 24, 168–175. [Google Scholar] [CrossRef]
- Josephs, K.A.; Whitwell, J.L.; Boeve, B.F.; Knopman, D.S.; Petersen, R.C.; Hu, W.T.; Parisi, J.E.; Dickson, D.W.; Jack, C.R., Jr. Anatomical differences between CBS-corticobasal degeneration and CBS-Alzheimer’s disease. Mov. Disord. 2010, 25, 1246–1252. [Google Scholar] [CrossRef] [PubMed]
- Constantinides, V.C.; Paraskevas, G.P.; Paraskevas, P.G.; Stefanis, L.; Kapaki, E. Corticobasal degeneration and corticobasal syndrome: A review. Clin. Park. Relat. Disord. 2019, 1, 66–71. [Google Scholar] [CrossRef]
- Di Stasio, F.; Suppa, A.; Marsili, L.; Upadhyay, N.; Asci, F.; Bologna, M.; Colosimo, C.; Fabbrini, G.; Pantano, P.; Berardelli, A. Corticobasal syndrome: Neuroimaging and neurophysiological advances. Eur. J. Neurol. 2019, 26, 701-e52. [Google Scholar] [CrossRef]
- Whitwell, J.L.; Avula, R.; Senjem, M.L.; Kantarci, K.; Weigand, S.D.; Samikoglu, A.; Edmonson, H.A.; Vemuri, P.; Knopman, D.S.; Boeve, B.F.; et al. Gray and white matter water diffusion in the syndromic variants of frontotemporal dementia. Neurology 2010, 74, 1279–1287. [Google Scholar] [CrossRef]
- Kouri, N.; Oshima, K.; Takahashi, M.; Murray, M.E.; Ahmed, Z.; Parisi, J.E.; Yen, S.-H.C.; Dickson, D.W. Corticobasal degeneration with olivopontocerebellar atrophy and TDP-43 pathology: An unusual clinicopathologic variant of CBD. Acta Neuropathol. 2013, 125, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Jacob, C.; Espay, A.J.; Hagen, M.C.; Duker, A.P. Misleading Imaging and Clinical Features in Pathology-Proven Corticobasal Degeneration. Mov. Disord. Clin. Pract. 2016, 3, 315–317. [Google Scholar] [CrossRef]
- Koga, S.; Sanchez-Contreras, M.; Josephs, K.A.; Uitti, R.J.; Graff-Radford, N.; van Gerpen, J.A.; Cheshire, W.P.; Wszolek, Z.K.; Rademakers, R.; Dickson, D.W. Distribution and characteristics of transactive response DNA binding protein 43 kDa pathology in progressive supranuclear palsy. Mov. Disord. 2017, 32, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, E.P.; Duffy, J.R.; Whitwell, J.L.; Vemuri, P.; Dickson, D.W.; Josephs, K.A. Mixed tau and TDP-43 pathology in a patient with unclassifiable primary progressive aphasia. Neurocase 2016, 22, 55–59. [Google Scholar] [CrossRef]
- Tazwar, M.; Evia, A.M.; Ridwan, A.R.; Leurgans, S.E.; Bennett, D.A.; Schneider, J.A.; Arfanakis, K. Limbic-predominant age-related TDP-43 encephalopathy neuropathological change (LATE-NC) is associated with abnormalities in white matter structural integrity and connectivity: An ex-vivo diffusion MRI and pathology investigation. Neurobiol. Aging 2024, 140, 81–92. [Google Scholar] [CrossRef]
- Lavrova, A.; Pham, N.T.T.; Reid, R.I.; Boeve, B.F.; Knopman, D.S.; Petersen, R.C.; Nguyen, A.T.; Ross Reichard, R.; Dickson, D.W.; Jack, C.R., Jr.; et al. Relation of Alzheimer’s disease-related TDP-43 proteinopathy to metrics from diffusion tensor imaging (DTI) and neurite orientation dispersion and density imaging (NODDI). Neurobiol. Aging 2025, 150, 97–108. [Google Scholar] [CrossRef]
- Gatto, R.G.; Martin, P.R.; Utianski, R.L.; Duffy, J.R.; Clark, H.M.; Botha, H.; Machulda, M.M.; Josephs, K.A.; Whitwell, J.L. Diffusion tensor imaging-based multi-fiber tracking reconstructions can regionally differentiate phonetic versus prosodic subtypes of progressive apraxia of speech. Cortex 2024, 171, 272–286. [Google Scholar] [CrossRef]
- Badihian, N.; Gatto, R.G.; Satoh, R.; Ali, F.; Clark, H.M.; Pham, N.T.T.; Whitwell, J.L.; Josephs, K.A. Clinical and neuroimaging characteristics of primary lateral sclerosis with overlapping features of progressive supranuclear palsy. Eur. J. Neurol. 2024, 31, e16320. [Google Scholar] [CrossRef]
- Costa, F.; Gatto, R.G.; Pham, N.T.T.; Ali, F.; Clark, H.M.; Stierwalt, J.; Machulda, M.M.; Agosta, F.; Filippi, M.; Josephs, K.A.; et al. Longitudinal assessment of white matter alterations in progressive supranuclear palsy variants using diffusion tractography. Park. Relat. Disord. 2025, 132, 107272. [Google Scholar] [CrossRef]
- Chouliaras, L.; O’Brien, J.T. The use of neuroimaging techniques in the early and differential diagnosis of dementia. Mol. Psychiatry 2023, 28, 4084–4097. [Google Scholar] [CrossRef] [PubMed]
- Juengling, F.; Wuest, F.; Schirrmacher, R.; Abele, J.; Thiel, A.; Soucy, J.P.; Camicioli, R.; Garibotto, V. PET Imaging in Dementia: Mini-Review and Canadian Perspective for Clinical Use. Can. J. Neurol. Sci. 2025, 52, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Carbonero, J.I.; Garcia-Toledo, I.; Fernandez-Hernandez, L.; Bascunana, P.; Gil-Moreno, M.J.; Matias-Guiu, J.A.; Corrochano, S. In vivo diagnosis of TDP-43 proteinopathies: In search of biomarkers of clinical use. Transl. Neurodegener. 2024, 13, 29. [Google Scholar] [CrossRef]
- Minoshima, S.; Cross, D.; Thientunyakit, T.; Foster, N.L.; Drzezga, A. (18)F-FDG PET Imaging in Neurodegenerative Dementing Disorders: Insights into Subtype Classification, Emerging Disease Categories, and Mixed Dementia with Copathologies. J. Nucl. Med. 2022, 63, 2S–12S. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Xie, F.; Zuo, C.; Guan, Y.; Huang, Y.H. PET Neuroimaging of Alzheimer’s Disease: Radiotracers and Their Utility in Clinical Research. Front. Aging Neurosci. 2021, 13, 624330. [Google Scholar] [CrossRef]
- Conte, M.; De Feo, M.S.; Sidrak, M.M.A.; Corica, F.; Gorica, J.; Granese, G.M.; Filippi, L.; De Vincentis, G.; Frantellizzi, V. Imaging of Tauopathies with PET Ligands: State of the Art and Future Outlook. Diagnostics 2023, 13, 1682. [Google Scholar] [CrossRef]
- Jiao, F.; Wang, M.; Sun, X.; Ju, Z.; Lu, J.; Wang, L.; Jiang, J.; Zuo, C. Based on Tau PET Radiomics Analysis for the Classification of Alzheimer’s Disease and Mild Cognitive Impairment. Brain Sci. 2023, 13, 367. [Google Scholar] [CrossRef]
- Leuzy, A.; Chiotis, K.; Lemoine, L.; Gillberg, P.G.; Almkvist, O.; Rodriguez-Vieitez, E.; Nordberg, A. Tau PET imaging in neurodegenerative tauopathies-still a challenge. Mol. Psychiatry 2019, 24, 1112–1134. [Google Scholar] [CrossRef]
- Knight, A.C.; Morrone, C.D.; Varlow, C.; Yu, W.H.; McQuade, P.; Vasdev, N. Head-to-Head Comparison of Tau-PET Radioligands for Imaging TDP-43 in Post-Mortem ALS Brain. Mol. Imaging Biol. 2023, 25, 513–527. [Google Scholar] [CrossRef]
- Carlos, A.F.; Tosakulwong, N.; Weigand, S.D.; Senjem, M.L.; Schwarz, C.G.; Knopman, D.S.; Boeve, B.F.; Petersen, R.C.; Nguyen, A.T.; Reichard, R.R.; et al. TDP-43 pathology effect on volume and flortaucipir uptake in Alzheimer’s disease. Alzheimers Dement. 2023, 19, 2343–2354. [Google Scholar] [CrossRef]
- Bevan-Jones, W.R.; Cope, T.E.; Jones, P.S.; Passamonti, L.; Hong, Y.T.; Fryer, T.D.; Arnold, R.; Allinson, K.S.J.; Coles, J.P.; Aigbirhio, F.I.; et al. [(18)F]AV-1451 binding in vivo mirrors the expected distribution of TDP-43 pathology in the semantic variant of primary progressive aphasia. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1032–1037. [Google Scholar] [CrossRef] [PubMed]
- Matias-Guiu, J.A.; Pytel, V.; Cabrera-Martin, M.N.; Galan, L.; Valles-Salgado, M.; Guerrero, A.; Moreno-Ramos, T.; Matias-Guiu, J.; Carreras, J.L. Amyloid- and FDG-PET imaging in amyotrophic lateral sclerosis. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 2050–2060. [Google Scholar] [CrossRef] [PubMed]
- Sennfalt, S.; Pagani, M.; Fang, F.; Savitcheva, I.; Estenberg, U.; Ingre, C. FDG-PET shows weak correlation between focal motor weakness and brain metabolic alterations in ALS. Amyotroph. Lateral Scler. Front. Degener. 2023, 24, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Caminiti, S.P.; De Francesco, S.; Tondo, G.; Galli, A.; Redolfi, A.; Perani, D.; Alzheimer’s Disease Neuroimaging, I.; Interceptor, P. FDG-PET markers of heterogeneity and different risk of progression in amnestic MCI. Alzheimers Dement. 2024, 20, 159–172. [Google Scholar] [CrossRef]
- Buciuc, M.; Botha, H.; Murray, M.E.; Schwarz, C.G.; Senjem, M.L.; Jones, D.T.; Knopman, D.S.; Boeve, B.F.; Petersen, R.C.; Jack, C.R., Jr.; et al. Utility of FDG-PET in diagnosis of Alzheimer-related TDP-43 proteinopathy. Neurology 2020, 95, e23–e34. [Google Scholar] [CrossRef]
- Tanaka, M.; Diano, M.; Battaglia, S. Editorial: Insights into structural and functional organization of the brain: Evidence from neuroimaging and non-invasive brain stimulation techniques. Front. Psychiatry 2023, 14, 1225755. [Google Scholar] [CrossRef]
- Di Fazio, C.; Tamietto, M.; Stanziano, M.; Nigri, A.; Scaliti, E.; Palermo, S. Cortico-Cortical Paired Associative Stimulation (ccPAS) in Ageing and Alzheimer’s Disease: A Quali-Quantitative Approach to Potential Therapeutic Mechanisms and Applications. Brain Sci. 2025, 15, 237. [Google Scholar] [CrossRef]
- Palermo, S.; Di Fazio, C.; Scaliti, E.; Stanziano, M.; Nigri, A.; Tamietto, M. Cortical excitability and the aging brain: Toward a biomarker of cognitive resilience. Front. Psychol. 2025, 16, 1542880. [Google Scholar] [CrossRef]
- Alongi, P.; Laudicella, R.; Panasiti, F.; Stefano, A.; Comelli, A.; Giaccone, P.; Arnone, A.; Minutoli, F.; Quartuccio, N.; Cupidi, C.; et al. Radiomics Analysis of Brain [(18)F]FDG PET/CT to Predict Alzheimer’s Disease in Patients with Amyloid PET Positivity: A Preliminary Report on the Application of SPM Cortical Segmentation, Pyradiomics and Machine-Learning Analysis. Diagnostics 2022, 12, 933. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Youssef, H.; Weissmann, C.; Uruk, G.; Gatto, R.G. Looking into Abnormal Co-Expressions of Tau and TDP-43 in the Realm of Mixed Dementia Types: A Double-Punch Scenario. Brain Sci. 2025, 15, 716. https://doi.org/10.3390/brainsci15070716
Youssef H, Weissmann C, Uruk G, Gatto RG. Looking into Abnormal Co-Expressions of Tau and TDP-43 in the Realm of Mixed Dementia Types: A Double-Punch Scenario. Brain Sciences. 2025; 15(7):716. https://doi.org/10.3390/brainsci15070716
Chicago/Turabian StyleYoussef, Hossam, Carina Weissmann, Gokhan Uruk, and Rodolfo Gabriel Gatto. 2025. "Looking into Abnormal Co-Expressions of Tau and TDP-43 in the Realm of Mixed Dementia Types: A Double-Punch Scenario" Brain Sciences 15, no. 7: 716. https://doi.org/10.3390/brainsci15070716
APA StyleYoussef, H., Weissmann, C., Uruk, G., & Gatto, R. G. (2025). Looking into Abnormal Co-Expressions of Tau and TDP-43 in the Realm of Mixed Dementia Types: A Double-Punch Scenario. Brain Sciences, 15(7), 716. https://doi.org/10.3390/brainsci15070716