


Sleep Deprivation and Alzheimer’s Disease: A Review of the Bidirectional Interactions and Therapeutic Potential of Omega-3


Abstract
1. Introduction
2. Sleep-Related Cognitive Changes in Healthy Aging
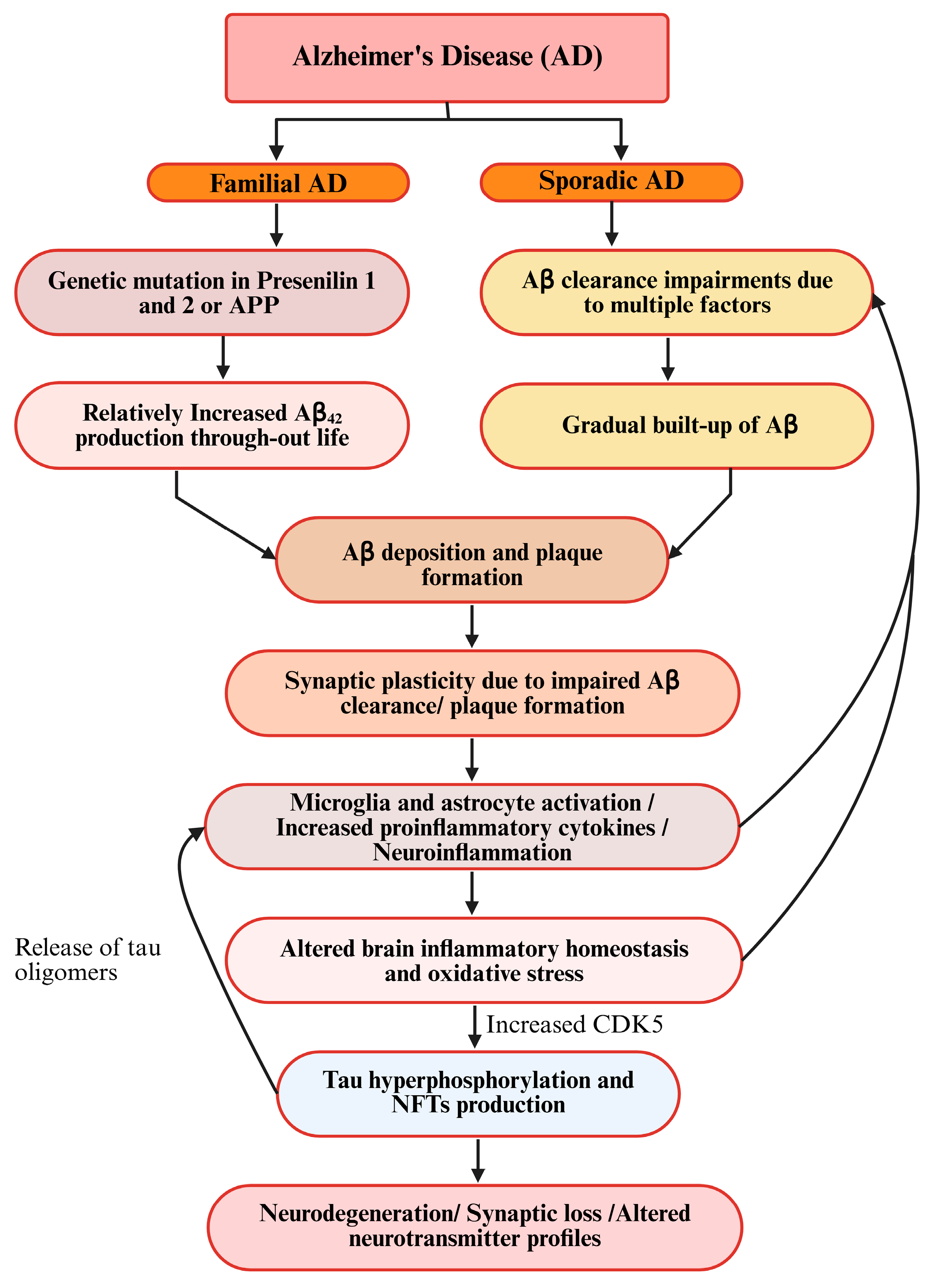
3. Pathogenesis and Hypotheses of AD
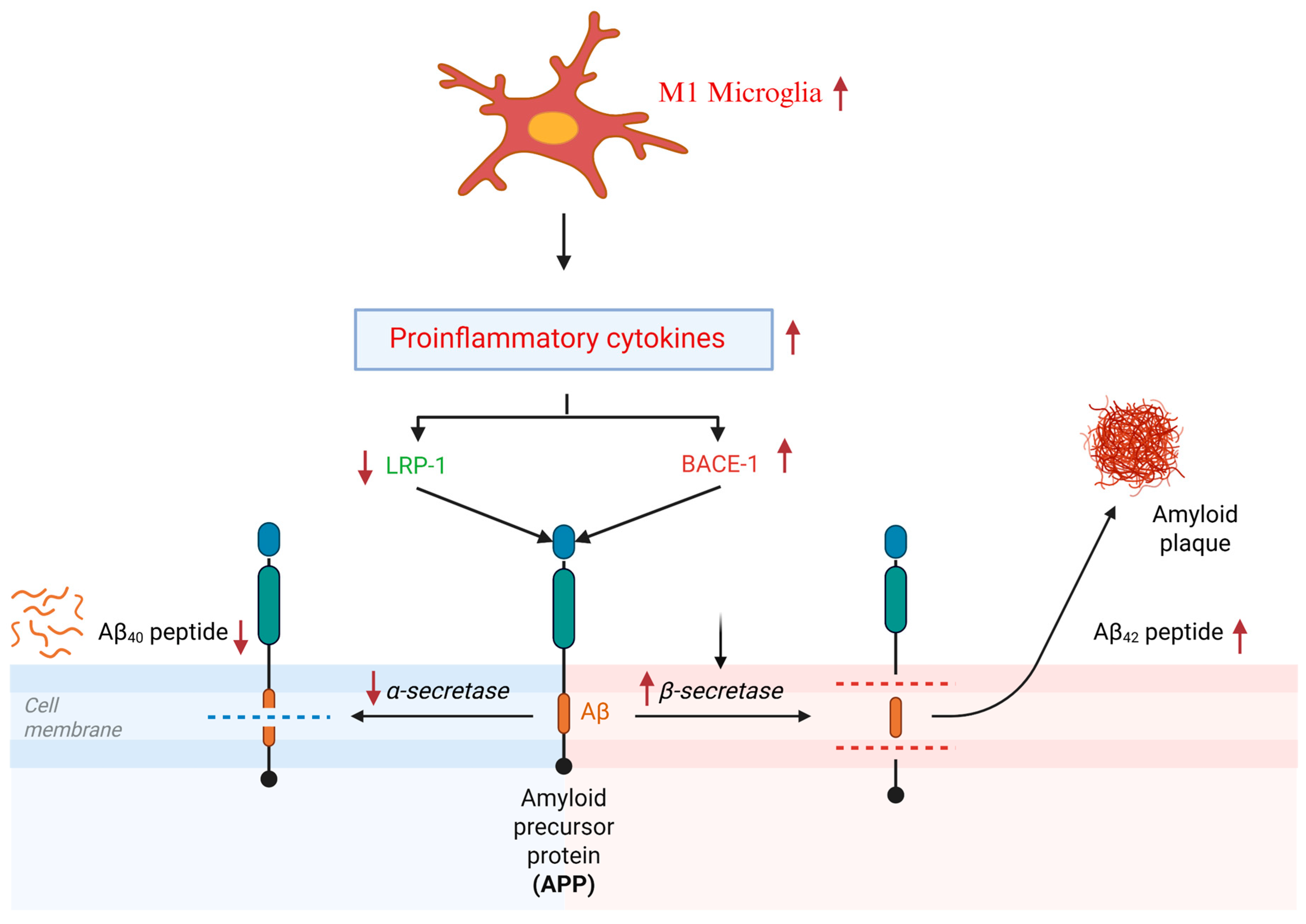
3.1. Neuroinflammatory Hypothesis of AD
3.2. Aβ Hypothesis of AD
3.3. Tau Hyperphosphorylation in AD
3.4. Other Hypotheses for AD
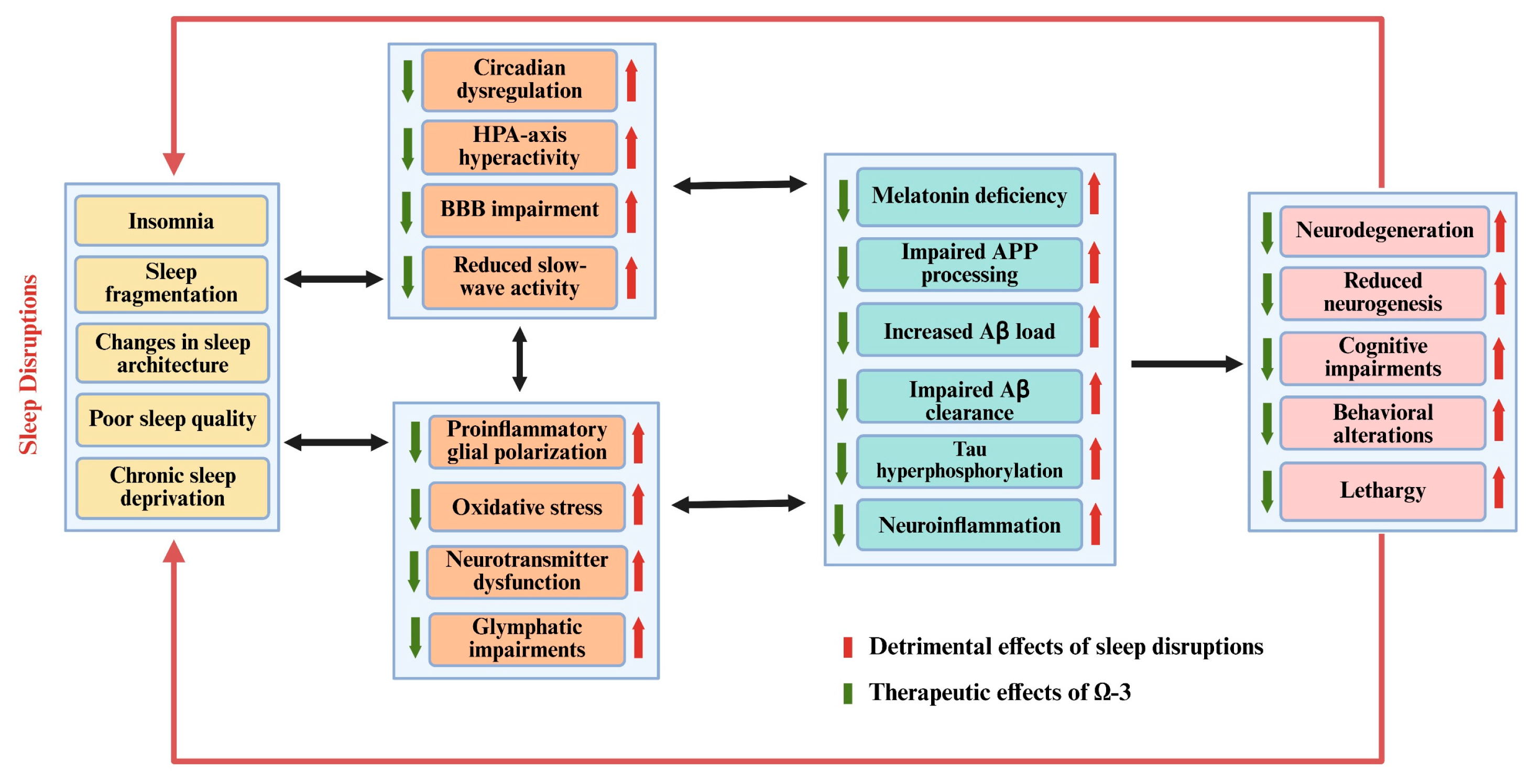
4. The Neuropathological Comorbidity Between SD and AD
5. Current Therapeutic Strategies for Sleep Disorders and AD
5.1. Therapeutic Status and Medications for Sleep Disorders
5.2. Therapeutic Status and Medications for AD
6. Therapeutic Potential of n-3 PUFAs in Sleep Disorders and AD
6.1. Dietary Sources of n-3 PUFAs
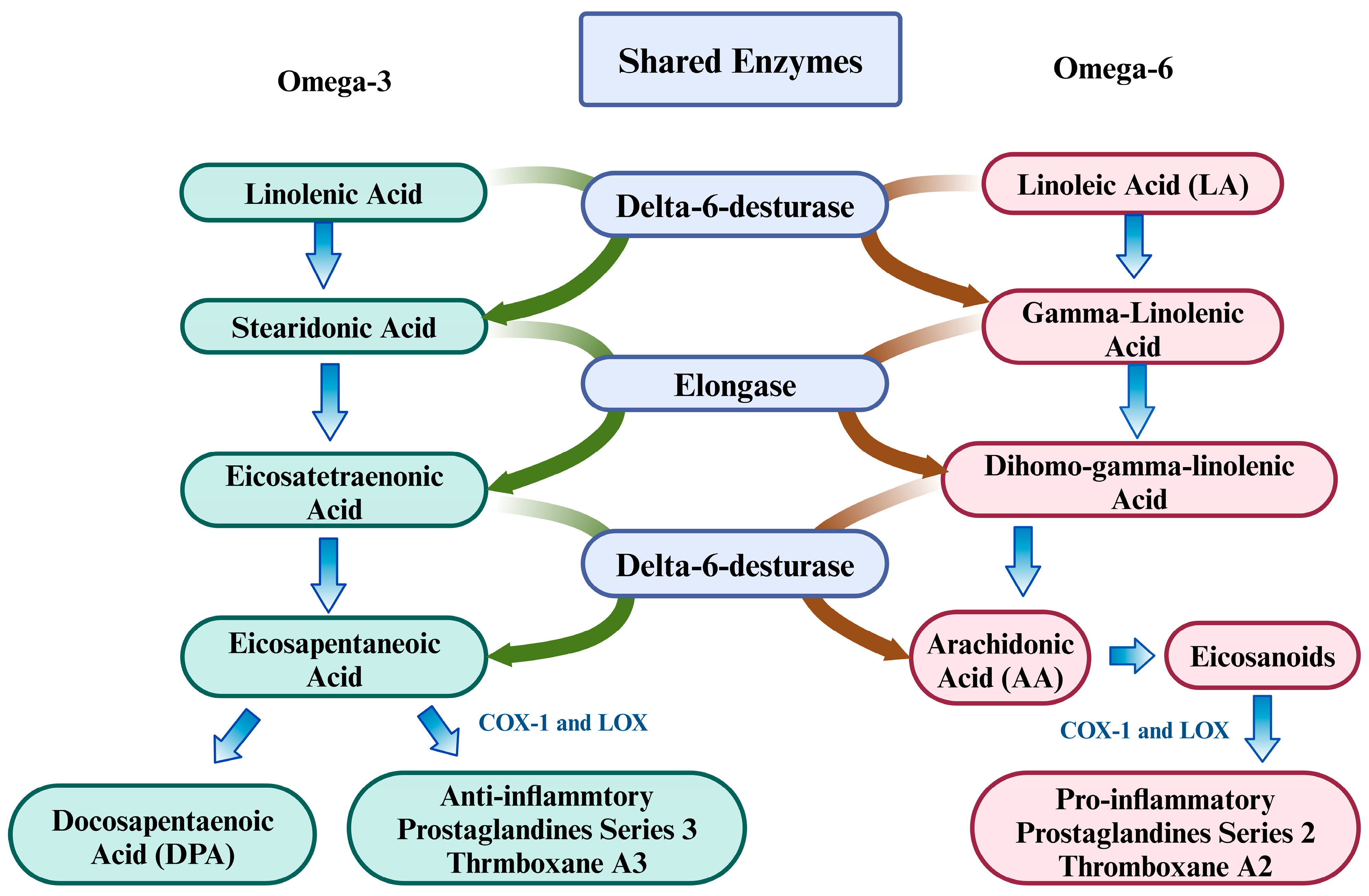
6.2. Importance of Homeostatic Balance Between Omega-3 and Omega-6 PUFAs
6.3. Therapeutic Potential of Omega-3 Fatty Acids
6.4. Omega-3 as a Possible Therapeutic Agent for Treating the Pathogenesis of AD
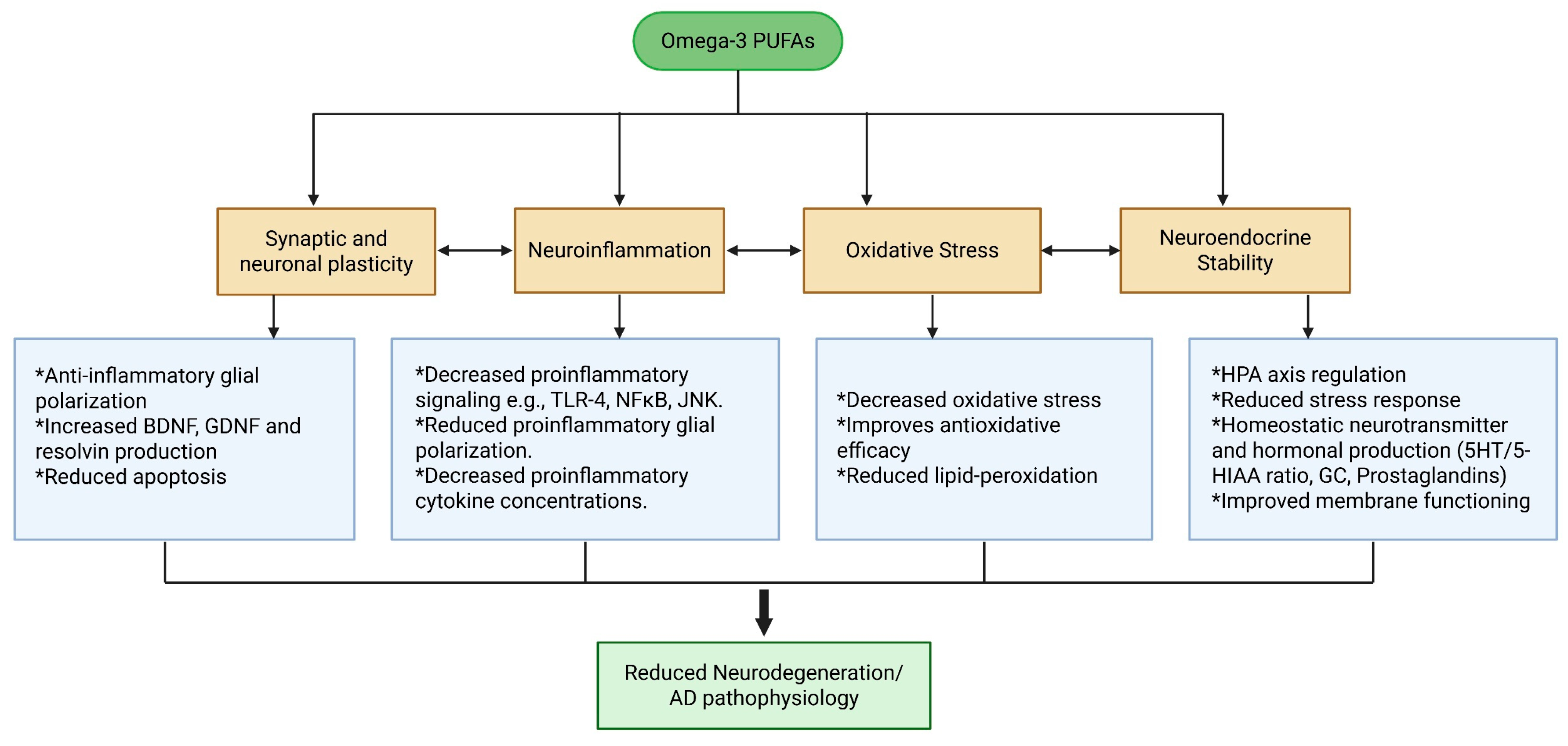
6.4.1. The Effects of Omega-3 on Oxidative Stress
6.4.2. The Effects of Omega-3 on Neuroinflammation and Aβ Clearance
6.4.3. The Effects of Omega-3 on Tau Hyperphosphorylation
6.4.4. Other Therapeutic Effects of Omega-3 on Pathophysiology of AD
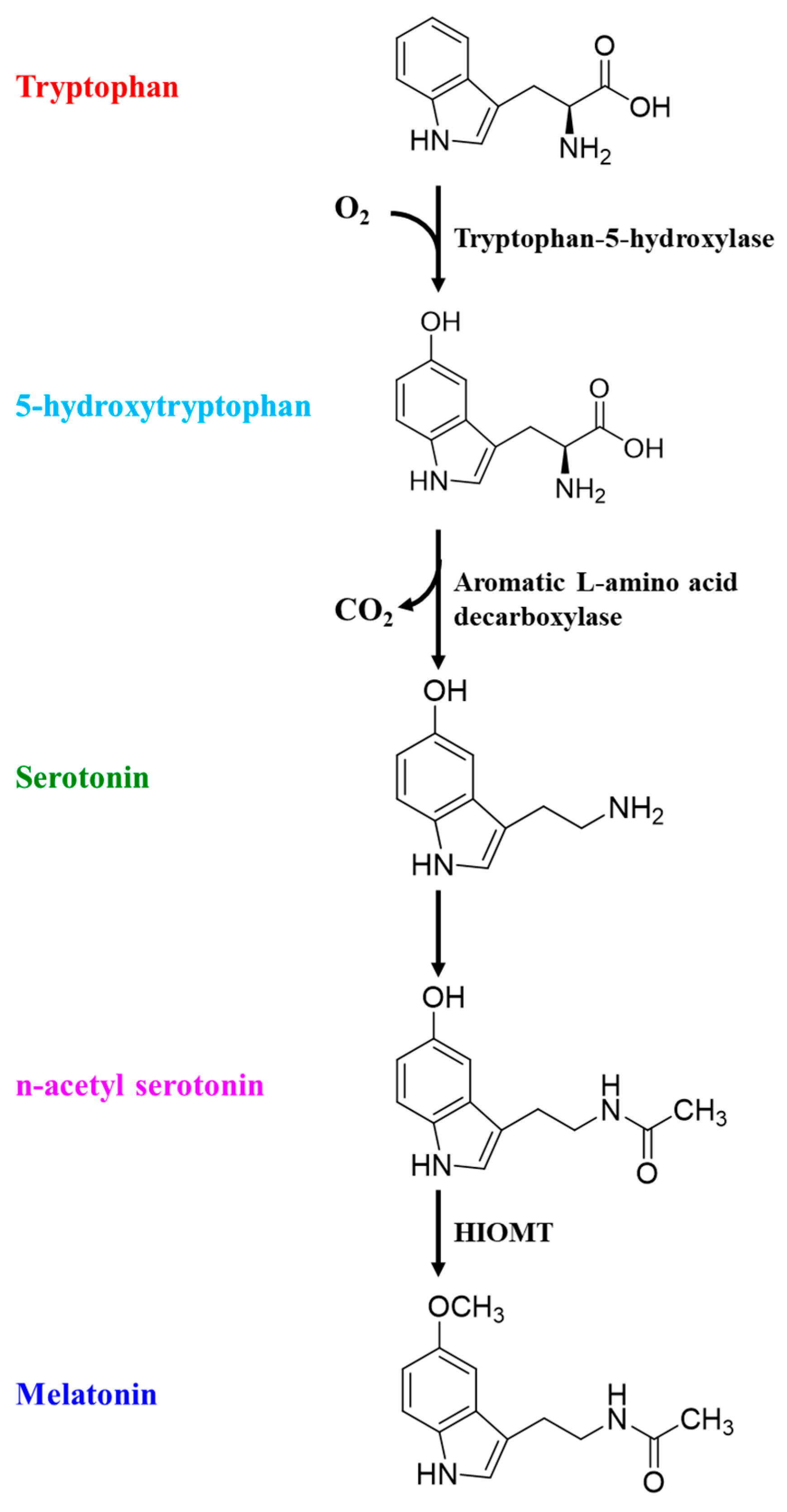
6.5. The Possible Therapeutic Roles of Omega-3 for Treating Sleep Disorders
7. Conclusions, Limitations, and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| SD | Sleep deprivation |
| Aβ | Amyloid-beta |
| LC n-3 PUFAs | Long-chain omega-3 polyunsaturated fatty acids |
| IL | Interleukin |
| PLs | Phospholipids |
| SWA | Slow-wave activity |
| APP | Amyloid precursor protein |
| ROS | Reactive oxygen species |
| LRP-1 | Low-density lipoprotein receptor-related protein 1 |
| BACE-1 | Beta-secretase 1 |
| RAGE | Receptor for advanced glycation end products |
| TNFR | Tumor necrosis factor receptor |
| NMDA | N-methyl-D-aspartate |
| ApoE | Apolipoprotein E |
| NFTs | Neurofibrillary tangles |
| TREM | Triggering receptor expressed on myeloid cells |
References
- Mander, B.A.; Winer, J.R.; Walker, M.P. Sleep and Human Aging. Neuron 2017, 94, 19–36. [Google Scholar] [CrossRef] [PubMed]
- Lacerda, R.A.V.; Desio, J.A.F.; Kammers, C.M.; Henkes, S.; Freitas de Sá, M.; de Souza, E.F.; da Silva, D.M.; Teixeira Pinheiro Gusmão, C.; dos Santos, J.C.C. Sleep Disorders and Risk of Alzheimer’s Disease: A Two-Way Road. Ageing Res. Rev. 2024, 101, 102514. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Ferreira, D.; Smedby, Ö.; Moreno, R. Decomposing the Effect of Normal Aging and Alzheimer’s Disease in Brain Morphological Changes via Learned Aging Templates. Sci. Rep. 2025, 15, 11813. [Google Scholar] [CrossRef]
- Bang, J.; Spina, S.; Miller, B.L. Frontotemporal Dementia. Lancet 2015, 386, 1672–1682. [Google Scholar] [CrossRef]
- Graff-Radford, N.R.; Woodruff, B.K. Frontotemporal Dementia. Semin. Neurol. 2007, 27, 48–57. [Google Scholar] [CrossRef]
- Ferreira, D.; Perestelo-Pérez, L.; Westman, E.; Wahlund, L.O.; Sarrisa, A.; Serrano-Aguilar, P. Meta-Review of CSF Core Biomarkers in Alzheimer’s Disease: The State-of-the-Art after the New Revised Diagnostic Criteria. Front. Aging Neurosci. 2014, 6, 47. [Google Scholar] [CrossRef]
- Dubois, B.; Villain, N.; Schneider, L.; Fox, N.; Campbell, N.; Galasko, D.; Kivipelto, M.; Jessen, F.; Hanseeuw, B.; Boada, M.; et al. Alzheimer Disease as a Clinical-Biological Construct-An International Working Group Recommendation. JAMA Neurol. 2024, 81, 1304–1311. [Google Scholar] [CrossRef]
- Wong, R.; Lovier, M.A. Sleep Disturbances and Dementia Risk in Older Adults: Findings From 10 Years of National U.S. Prospective Data. Am. J. Prev. Med. 2023, 64, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ren, R.; Yang, L.; Zhang, H.; Shi, Y.; Okhravi, H.R.; Vitiello, M.V.; Sanford, L.D.; Tang, X. Sleep in Alzheimer’s Disease: A Systematic Review and Meta-Analysis of Polysomnographic Findings. Transl. Psychiatry 2022, 12, 136. [Google Scholar] [CrossRef]
- Kabeshita, Y.; Adachi, H.; Matsushita, M.; Kanemoto, H.; Sato, S.; Suzuki, Y.; Yoshiyama, K.; Shimomura, T.; Yoshida, T.; Shimizu, H.; et al. Sleep Disturbances Are Key Symptoms of Very Early Stage Alzheimer Disease with Behavioral and Psychological Symptoms: A Japan Multi-Center Cross-Sectional Study (J-BIRD). Int. J. Geriatr. Psychiatry 2017, 32, 222–230. [Google Scholar] [CrossRef]
- Musiek, E.S.; Xiong, D.D.; Holtzman, D.M. Sleep, Circadian Rhythms, and the Pathogenesis of Alzheimer Disease. Exp. Mol. Med. 2015, 47, e148. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, R.F.; Lutsey, P.L.; Benveniste, H.; Brown, D.L.; Full, K.M.; Lee, J.M.; Osorio, R.S.; Pase, M.P.; Redeker, N.S.; Redline, S.; et al. Impact of Sleep Disorders and Disturbed Sleep on Brain Health: A Scientific Statement from the American Heart Association. Stroke 2024, 55, E61–E76. [Google Scholar] [CrossRef]
- Anghel, L.; Ciubară, A.; Nechita, A.; Nechita, L.; Manole, C.; Baroiu, L.; Ciubară, A.B.; Mușat, C.L. Sleep Disorders Associated with Neurodegenerative Diseases. Diagnostics 2023, 13, 2898. [Google Scholar] [CrossRef] [PubMed]
- Casagrande, M.; Forte, G.; Favieri, F.; Corbo, I. Sleep Quality and Aging: A Systematic Review on Healthy Older People, Mild Cognitive Impairment and Alzheimer’s Disease. Int. J. Environ. Res. Public Health 2022, 19, 8457. [Google Scholar] [CrossRef] [PubMed]
- Joo, H.J.; Joo, J.H.; Kwon, J.; Jang, B.N.; Park, E.C. Association between Quality and Duration of Sleep and Subjective Cognitive Decline: A Cross-Sectional Study in South Korea. Sci. Rep. 2021, 11, 16989. [Google Scholar] [CrossRef]
- Romanella, S.M.; Roe, D.; Tatti, E.; Cappon, D.; Paciorek, R.; Testani, E.; Rossi, A.; Rossi, S.; Santarnecchi, E. The Sleep Side of Aging and Alzheimer’s Disease. Sleep Med. 2021, 77, 209–225. [Google Scholar] [CrossRef]
- Djonlagic, I.; Mariani, S.; Fitzpatrick, A.L.; Van Der Klei, V.M.G.T.H.; Johnson, D.A.; Wood, A.C.; Seeman, T.; Nguyen, H.T.; Prerau, M.J.; Luchsinger, J.A.; et al. Macro and Micro Sleep Architecture and Cognitive Performance in Older Adults. Nat. Hum. Behav. 2020, 5, 123–145. [Google Scholar] [CrossRef]
- Cerasuolo, M.; Di Meo, I.; Auriemma, M.C.; Paolisso, G.; Papa, M.; Rizzo, M.R. Exploring the Dynamic Changes of Brain Lipids, Lipid Rafts, and Lipid Droplets in Aging and Alzheimer’s Disease. Biomolecules 2024, 14, 1362. [Google Scholar] [CrossRef]
- Tomczyk, M.; Bidzan-Wiącek, M.; Kortas, J.A.; Kochanowicz, M.; Jost, Z.; Fisk, H.L.; Calder, P.C.; Antosiewicz, J. Omega-3 Fatty Acid Supplementation Affects Tryptophan Metabolism during a 12-Week Endurance Training in Amateur Runners: A Randomized Controlled Trial. Sci. Rep. 2024, 14, 4102. [Google Scholar] [CrossRef]
- Wichers, M.C.; Maes, M. The Role of Indoleamine 2,3-Dioxygenase (IDO) in the Pathophysiology of Interferon-α-Induced Depression. J. Psychiatry Neurosci. 2004, 29, 11–17. [Google Scholar]
- Hardeland, R. Melatonin in Aging and Disease-Multiple Consequences of Reduced Secretion, Options and Limits of Treatment. Aging Dis. 2012, 3, 194–225. [Google Scholar]
- Ouyang, M.; Chen, T.; Chen, J.; Liu, C.; Luo, H.; Yang, S.; Liao, W. The Association between Sleep Duration, Bedtime, and Cognitive Ability in Chinese Adults: Evidence from the China Family Panel Studies. Heliyon 2024, 10, e30009. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, M.; Tschiderer, L.; Stefani, A.; Heidbreder, A.; Willeit, P.; Högl, B. Sleep Quality and Daytime Sleepiness in Epilepsy: Systematic Review and Meta-Analysis of 25 Studies Including 8,196 Individuals. Sleep Med. Rev. 2021, 57, 101466. [Google Scholar] [CrossRef]
- Katsuki, F.; Gerashchenko, D.; Brown, R.E. Alterations of Sleep Oscillations in Alzheimer’s Disease: A Potential Role for GABAergic Neurons in the Cortex, Hippocampus, and Thalamus. Brain Res. Bull. 2022, 187, 181–198. [Google Scholar] [CrossRef]
- van den Berg, M.; Toen, D.; Verhoye, M.; Keliris, G.A. Alterations in Theta-Gamma Coupling and Sharp Wave-Ripple, Signs of Prodromal Hippocampal Network Impairment in the TgF344-AD Rat Model. Front. Aging Neurosci. 2023, 15, 1081058. [Google Scholar] [CrossRef] [PubMed]
- Brendstrup-Brix, K.; Ulv Larsen, S.M.; Lee, H.; Knudsen, G.M. Perivascular Space Diffusivity and Brain Microstructural Measures Are Associated with Circadian Time and Sleep Quality. J. Sleep Res. 2024, 33, e14226. [Google Scholar] [CrossRef] [PubMed]
- Niazi, N.U.K.; Huang, C.; Yang, Z.; Zhang, Y.; Song, C. Comparison between Sub-Chronic and Chronic Sleep Deprivation-Induced Behavioral and Neuroimmunological Abnormalities in Mice: Focusing on Glial Cell Phenotype Polarization. Behav. Brain Res. 2024, 470, 115067. [Google Scholar] [CrossRef]
- Wisor, J.P.; Clegern, W.C. Quantification of Short-Term Slow Wave Sleep Homeostasis and Its Disruption by Minocycline in the Laboratory Mouse. Neurosci. Lett. 2011, 490, 165–169. [Google Scholar] [CrossRef]
- Chen, Y.; He, X.; Cai, J.; Li, Q. Functional Aspects of the Brain Lymphatic Drainage System in Aging and Neurodegenerative Diseases. J. Biomed. Res. 2024, 38, 206. [Google Scholar] [CrossRef]
- Kaushal, N.; Ramesh, V.; Gozal, D. TNF-α and Temporal Changes in Sleep Architecture in Mice Exposed to Sleep Fragmentation. PLoS ONE 2012, 7, e45610. [Google Scholar] [CrossRef]
- Rockstrom, M.D.; Chen, L.; Taishi, P.; Nguyen, J.T.; Gibbons, C.M.; Veasey, S.C.; Krueger, J.M. Tumor Necrosis Factor Alpha in Sleep Regulation. Sleep Med. Rev. 2018, 40, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, M.È.; Verkhratsky, A. General Pathophysiology of Microglia. In Advances in Neurobiology; Tremblay, M.-È., Verkhratsky, A., Eds.; Springer International Publishing: Cham, Switzerland, 2024; Volume 37, pp. 3–14. ISBN 978-3-031-55529-9. [Google Scholar]
- Bellier, F.; Walter, A.; Lecoin, L.; Chauveau, F.; Rouach, N.; Rancillac, A. Astrocytes at the Heart of Sleep: From Genes to Network Dynamics. Cell. Mol. Life Sci. 2025, 82, 207. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.-S.; Kim, J.-H.; Jeong, J.-Y.; Lee, M.-G.; Suk, K.; Jang, I.-S. Astrocyte-Derived Adenosine Excites Sleep-Promoting Neurons in the Ventrolateral Preoptic Nucleus: Astrocyte-Neuron Interactions in the Regulation of Sleep. Glia 2022, 70, 1864–1885. [Google Scholar] [CrossRef]
- Gorgulu, Y.; Caliyurt, O.; Kose Cinar, R.; Sonmez, M.B. Acute Sleep Deprivation Immediately Increases Serum GDNF, BDNF and VEGF Levels in Healthy Subjects. Sleep Biol. Rhythms 2022, 20, 73–79. [Google Scholar] [CrossRef]
- Coluk, Y.; Yildirim, G.; Yildirmak, S.; Peker, E.G.G. Altered Brain-Derived Neurotrophic Factor Levels and Oxidative Stress in REM Sleep Deprivation: A Rat Model Study. BMC Neurol. 2025, 25, 122. [Google Scholar] [CrossRef] [PubMed]
- In’t Veld, B.A.; Ruitenberg, A.; Hofman, A.; Launer, L.J.; van Duijn, C.M.; Stijnen, T.; Breteler, M.M.B.; Stricker, B.H.C. Nonsteroidal Antiinflammatory Drugs and the Risk of Alzheimer’s Disease. N. Engl. J. Med. 2001, 345, 1515–1521. [Google Scholar] [CrossRef]
- Szekely, C.A.; Green, R.C.; Breitner, J.C.S.; Østbye, T.; Beiser, A.S.; Corrada, M.M.; Dodge, H.H.; Ganguli, M.; Kawas, C.H.; Kuller, L.H.; et al. No Advantage of Aβ42-Lowering NSAIDs for Prevention of Alzheimer Dementia in Six Pooled Cohort Studies Symbol. Neurology 2008, 70, 2291–2298. [Google Scholar] [CrossRef]
- de Sousa, A.A.; Rigby Dames, B.A.; Graff, E.C.; Mohamedelhassan, R.; Vassilopoulos, T.; Charvet, C.J. Going beyond Established Model Systems of Alzheimer’s Disease: Companion Animals Provide Novel Insights into the Neurobiology of Aging. Commun. Biol. 2023, 6, 655. [Google Scholar] [CrossRef]
- Lyketsos, C.G.; Breitner, J.C.S.; Green, R.C.; Martin, B.K.; Meinert, C.; Piantadosi, S.; Sabbagh, M. Naproxen and Celecoxib Do Not Prevent AD in Early Results from a Randomized Controlled Trial. Neurology 2007, 68, 1800–1808. [Google Scholar] [CrossRef]
- Song, C.; Li, X.; Leonard, B.E.; Horrobin, D.F. Effects of Dietary N-3 or n-6 Fatty Acids on Interleukin-1β-Induced Anxiety, Stress, and Inflammatory Responses in Rats. J. Lipid Res. 2003, 44, 1984–1991. [Google Scholar] [CrossRef]
- Wang, C.; Holtzman, D.M. Bidirectional Relationship between Sleep and Alzheimer’s Disease: Role of Amyloid, Tau, and Other Factors. Neuropsychopharmacology 2020, 45, 104–120. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; van der Flier, W.M.; Jessen, F.; Hoozemanns, J.; Thal, D.R.; Boche, D.; Brosseron, F.; Teunissen, C.; Zetterberg, H.; Jacobs, A.H.; et al. Neuroinflammation in Alzheimer Disease. Nat. Rev. Immunol. 2025, 25, 321–352. [Google Scholar] [CrossRef]
- Li, Z.H.; Cheng, L.; Wen, C.; Ding, L.; You, Q.Y.; Zhang, S.B. Activation of CNR1/PI3K/AKT Pathway by Tanshinone IIA Protects Hippocampal Neurons and Ameliorates Sleep Deprivation-Induced Cognitive Dysfunction in Rats. Front. Pharmacol. 2022, 13, 823732. [Google Scholar] [CrossRef]
- Awogbindin, I.; Wanklin, M.; Verkhratsky, A.; Tremblay, M.È. Microglia in Neurodegenerative Diseases. In Advances in Neurobiology; Tremblay, M.-È., Verkhratsky, A., Eds.; Springer International Publishing: Cham, Switzerland, 2024; Volume 37, pp. 497–512. ISBN 978-3-031-55529-9. [Google Scholar]
- Huang, W.; Huang, J.; Huang, N.; Luo, Y. The Role of TREM2 in Alzheimer’s Disease: From the Perspective of Tau. Front. Cell Dev. Biol. 2023, 11, 1280257. [Google Scholar] [CrossRef] [PubMed]
- Tran, K.M.; Kawauchi, S.; Kramár, E.A.; Rezaie, N.; Liang, H.Y.; Sakr, J.S.; Gomez-Arboledas, A.; Arreola, M.A.; da Cunha, C.; Phan, J.; et al. A Trem2 R47H Mouse Model without Cryptic Splicing Drives Age- and Disease-Dependent Tissue Damage and Synaptic Loss in Response to Plaques. Mol. Neurodegener. 2023, 18, 12. [Google Scholar] [CrossRef]
- Cai, W.; Wu, T.; Chen, N. The Amyloid-Beta Clearance: From Molecular Targets to Glial and Neural Cells. Biomolecules 2023, 13, 313. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Zhang, X.; So, K.-F.; Jiang, W.; Chiu, K. Targeting Microglia in Alzheimer’s Disease: Pathogenesis and Potential Therapeutic Strategies. Biomolecules 2024, 14, 833. [Google Scholar] [CrossRef]
- Storck, S.E.; Meister, S.; Nahrath, J.; Meißner, J.N.; Schubert, N.; Di Spiezio, A.; Baches, S.; Vandenbroucke, R.E.; Bouter, Y.; Prikulis, I.; et al. Endothelial LRP1 Transports Amyloid-Β1-42 across the Blood-Brain Barrier. J. Clin. Investig. 2016, 126, 123–136. [Google Scholar] [CrossRef]
- von Einem, B.; Schwanzar, D.; Rehn, F.; Beyer, A.S.; Weber, P.; Wagner, M.; Schneckenburger, H.; von Arnim, C.A.F. The Role of Low-Density Receptor-Related Protein 1 (LRP1) as a Competitive Substrate of the Amyloid Precursor Protein (APP) for BACE1. Exp. Neurol. 2010, 225, 85–93. [Google Scholar] [CrossRef]
- Doecke, J.D.; Pérez-Grijalba, V.; Fandos, N.; Fowler, C.; Villemagne, V.L.; Masters, C.L.; Pesini, P.; Sarasa, M. Total Aβ42/Aβ40 Ratio in Plasma Predicts Amyloid-PET Status, Independent of Clinical AD Diagnosis. Neurology 2020, 94, E1580–E1591. [Google Scholar] [CrossRef]
- Cho, H.J.; Son, S.M.; Jin, S.M.; Hong, H.S.; Shin, D.H.; Kim, S.J.; Huh, K.; Mook-Jung, I. RAGE Regulates BACE1 and Aβ Generation via NFAT1 Activation in Alzheimer’s Disease Animal Model. FASEB J. 2009, 23, 2639–2649. [Google Scholar] [CrossRef] [PubMed]
- Tobon-Velasco, J.; Cuevas, E.; Torres-Ramos, M. Receptor for AGEs (RAGE) as Mediator of NF-KB Pathway Activation in Neuroinflammation and Oxidative Stress. CNS Neurol. Disord. Drug Targets 2014, 13, 1615–1626. [Google Scholar] [CrossRef]
- Li, L.; Guo, J.; Liang, X.; Huang, Y.; Wang, Q.; Luo, Y.; King, L.; Chen, L.; Peng, X.; Yan, H.; et al. Associations of Advanced Glycation End Products with Sleep Disorders in Chinese Adults. Nutrients 2024, 16, 3282. [Google Scholar] [CrossRef]
- Zhu, L.; Liu, X.; Nemeth, D.P.; DiSabato, D.J.; Witcher, K.G.; Mckim, D.B.; Oliver, B.; Le, X.; Gorantla, G.; Berdysz, O.; et al. Interleukin-1 Causes CNS Inflammatory Cytokine Expression via Endothelia-Microglia Bi-Cellular Signaling. Brain. Behav. Immun. 2019, 81, 292–304. [Google Scholar] [CrossRef]
- Mandache, E.; Negoescu, A. The Ultrastructure of the Abdominal Lymph Nodes Cortex in Rats during Primary and Secondary Immune Response (a Special Reference to Dendritic Reticulum Cells and Interdigitating Cells). Morphol. Embryol. 1990, 36, 49–58. [Google Scholar]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a Central Mechanism in Alzheimer’s Disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Zhao, R.; Zhou, H.; Su, S.B. A Critical Role for Interleukin-1β in the Progression of Autoimmune Diseases. Int. Immunopharmacol. 2013, 17, 658–669. [Google Scholar] [CrossRef]
- Wong, M.Y.; Lewis, M.; Doherty, J.J.; Shi, Y.; Cashikar, A.G.; Amelianchik, A.; Tymchuk, S.; Sullivan, P.M.; Qian, M.; Covey, D.F.; et al. 25-Hydroxycholesterol Amplifies Microglial IL-1β Production in an ApoE Isoform-Dependent Manner. J. Neuroinflammation 2020, 17, 192. [Google Scholar] [CrossRef] [PubMed]
- Kong, Q.; Peterson, T.S.; Baker, O.; Stanley, E.; Camden, J.; Seye, C.I.; Erb, L.; Simonyi, A.; Wood, W.G.; Sun, G.Y.; et al. Interleukin-1beta Enhances Nucleotide-Induced and Alpha-Secretase-Dependent Amyloid Precursor Protein Processing in Rat Primary Cortical Neurons via up-Regulation of the P2Y(2) Receptor. J. Neurochem. 2009, 109, 1300–1310. [Google Scholar] [CrossRef]
- Tanabe, K.; Kozawa, O.; Iida, H. CAMP/PKA Enhances Interleukin-1β-Induced Interleukin-6 Synthesis through STAT3 in Glial Cells. Cell. Signal. 2016, 28, 19–24. [Google Scholar] [CrossRef]
- Yadav, V.; Mythri, C.; Kumarasamy, M. Natural Products as Potential Modulators of Pro-Inflammatory Cytokines Signalling in Alzheimer’s Disease. Brain Behav. Immun. Integr. 2024, 5, 100048. [Google Scholar] [CrossRef]
- Steeland, S.; Gorlé, N.; Vandendriessche, C.; Balusu, S.; Brkic, M.; Van Cauwenberghe, C.; Van Imschoot, G.; Van Wonterghem, E.; De Rycke, R.; Kremer, A.; et al. Counteracting the Effects of TNF Receptor-1 Has Therapeutic Potential in Alzheimer’s Disease. EMBO Mol. Med. 2018, 10, e8300. [Google Scholar] [CrossRef]
- Raffaele, S.; Lombardi, M.; Verderio, C.; Fumagalli, M. Tnf Production and Release from Microglia via Extracellular Vesicles: Impact on Brain Functions. Cells 2020, 9, 2145. [Google Scholar] [CrossRef] [PubMed]
- Pillai, J.A.; Bebek, G.; Khrestian, M.; Bena, J.; Bergmann, C.C.; Bush, W.S.; Leverenz, J.B.; Bekris, L.M. TNFRSF1B Gene Variants and Related Soluble TNFR2 Levels Impact Resilience in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 638922. [Google Scholar] [CrossRef]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef]
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and Cellular Mechanisms Underlying the Pathogenesis of Alzheimer’s Disease. Mol. Neurodegener. 2020, 15, 40. [Google Scholar] [CrossRef] [PubMed]
- Armato, U.; Chiarini, A.; Chakravarthy, B.; Chioffi, F.; Pacchiana, R.; Colarusso, E.; Whitfield, J.F.; Dal Prà, I. Calcium-Sensing Receptor Antagonist (Calcilytic) NPS 2143 Specifically Blocks the Increased Secretion of Endogenous Aβ42 Prompted by Exogenous Fibrillary or Soluble Aβ25-35 in Human Cortical Astrocytes and Neurons-Therapeutic Relevance to Alzheimer’s Dise. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 1634–1652. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Rao, Y.L.; Ganaraja, B.; Murlimanju, B.V.; Joy, T.; Krishnamurthy, A.; Agrawal, A. Hippocampus and Its Involvement in Alzheimer’s Disease: A Review. 3 Biotech 2022, 12, 55. [Google Scholar] [CrossRef]
- Wirths, O.; Bayer, T.A. Intraneuronal Aβ Accumulation and Neurodegeneration: Lessons from Transgenic Models. Life Sci. 2012, 91, 1148–1152. [Google Scholar] [CrossRef]
- Li, S.; Wang, J.; Guo, Q.; Bai, Y.; Liu, W.; Hodgetts, K.J.; Rosenberg, P.A.; Selkoe, D.J. Impairment of Hippocampal Long-Term Potentiation by Soluble Amyloid-β Oligomers Is Mediated by Glutamate Transporter 1 Expressed in Neurons. Neural Regen. Res. 2025. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jiao, G.; Song, C.; Gu, S.; Brown, R.E.; Zhang, J.; Zhang, P.; Gagnon, J.; Locke, S.; Stefanova, R.; et al. An Extract from Shrimp Processing By-Products Protects SH-SY5Y Cells from Neurotoxicity Induced by Aβ25-35. Mar. Drugs 2017, 15, 83. [Google Scholar] [CrossRef] [PubMed]
- Sedighi, M.; Baluchnejadmojarad, T.; Roghani, M. Linagliptin Protects Human Sh-Sy5y Neuroblastoma Cells against Amyloid-β Cytotoxicity via the Activation of Wnt1 and Suppression of Il-6 Release. Iran. Biomed. J. 2021, 25, 343–348. [Google Scholar] [CrossRef]
- Qin, Z.X.; Zhu, H.Y.; Hu, Y.H. Effects of Lysophosphatidylcholine on Β-Amyloid-Induced Neuronal Apoptosis. Acta Pharmacol. Sin. 2009, 30, 388–395. [Google Scholar] [CrossRef]
- Dou, R.X.; Zhang, Y.M.; Hu, X.J.; Gao, F.L.; Zhang, L.L.; Liang, Y.H.; Zhang, Y.Y.; Yao, Y.P.; Yin, L.; Zhang, Y.; et al. Aβ1-42 Promotes Microglial Activation and Apoptosis in the Progression of AD by Binding to TLR4. Redox Biol. 2024, 78, 103428. [Google Scholar] [CrossRef] [PubMed]
- Sallaberry, C.A.; Voss, B.J.; Majewski, J.; Biernat, J.; Mandelkow, E.; Chi, E.Y.; Vander Zanden, C.M. Tau and Membranes: Interactions That Promote Folding and Condensation. Front. Cell Dev. Biol. 2021, 9, 725241. [Google Scholar] [CrossRef]
- Rawat, P.; Sehar, U.; Bisht, J.; Selman, A.; Culberson, J.; Reddy, P.H. Phosphorylated Tau in Alzheimer’s Disease and Other Tauopathies. Int. J. Mol. Sci. 2022, 23, 12841. [Google Scholar] [CrossRef]
- Protto, V.; Miteva, M.T.; Iannuzzi, F.; Marcocci, M.E.; Li Puma, D.D.; Piacentini, R.; Belli, M.; Sansone, L.; Pietrantoni, A.; Grassi, C.; et al. HSV-1 Infection Induces Phosphorylated Tau Propagation among Neurons via Extracellular Vesicles. MBio 2024, 15, e01522-24. [Google Scholar] [CrossRef]
- Rajmohan, R.; Reddy, P.H. Amyloid-Beta and Phosphorylated Tau Accumulations Cause Abnormalities at Synapses of Alzheimer’s Disease Neurons. J. Alzheimer’s Dis. 2017, 57, 975–999. [Google Scholar] [CrossRef]
- Bloom, G.S. Amyloid-β and Tau: The Trigger and Bullet in Alzheimer Disease Pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef]
- Busche, M.A.; Hyman, B.T. Synergy between Amyloid-β and Tau in Alzheimer’s Disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Ashton, N.J.; Brum, W.S.; Molfetta, G.D.; Benedet, A.L.; Arslan, B.; Jonaitis, E.; Langhough, R.E.; Cody, K.; Wilson, R.; Carlsson, C.M.; et al. Diagnostic Accuracy of a Plasma Phosphorylated Tau 217 Immunoassay for Alzheimer Disease Pathology. JAMA Neurol. 2024, 81, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Janelidze, S.; Mattsson, N.; Palmqvist, S.; Smith, R.; Beach, T.G.; Serrano, G.E.; Chai, X.; Proctor, N.K.; Eichenlaub, U.; Zetterberg, H.; et al. Plasma P-Tau181 in Alzheimer’s Disease: Relationship to Other Biomarkers, Differential Diagnosis, Neuropathology and Longitudinal Progression to Alzheimer’s Dementia. Nat. Med. 2020, 26, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Kepp, K.P.; Robakis, N.K.; Høilund-Carlsen, P.F.; Sensi, S.L.; Vissel, B. The Amyloid Cascade Hypothesis: An Updated Critical Review. Brain 2023, 146, 3969–3990. [Google Scholar] [CrossRef]
- Antonioni, A.; Raho, E.M.; Manzoli, L.; Koch, G.; Flacco, M.E.; Di Lorenzo, F. Blood Phosphorylated Tau181 Reliably Differentiates Amyloid-positive from Amyloid-negative Subjects in the Alzheimer’s Disease Continuum: A Systematic Review and Meta-analysis. Alzheimer’s Dement. Diagnosis, Assess. Dis. Monit. 2025, 17, e70068. [Google Scholar] [CrossRef]
- Herholz, K. Acetylcholine Esterase Activity in Mild Cognitive Impairment and Alzheimer’s Disease. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 7–11. [Google Scholar] [CrossRef]
- Terry, A.V.; Buccafusco, J.J. The Cholinergic Hypothesis of Age and Alzheimer’s Disease-Related Cognitive Deficits: Recent Challenges and Their Implications for Novel Drug Development. J. Pharmacol. Exp. Ther. 2003, 306, 821–827. [Google Scholar] [CrossRef]
- Alkhalifa, A.E.; Alkhalifa, O.; Durdanovic, I.; Ibrahim, D.R.; Maragkou, S. Oxidative Stress and Mitochondrial Dysfunction in Alzheimer’s Disease: Insights into Pathophysiology and Treatment. J. Dement. Alzheimer’s Dis. 2025, 2, 17. [Google Scholar] [CrossRef]
- Da, D.; Zhao, Q.; Zhang, H.; Wu, W.; Zeng, X.; Liang, X.; Jiang, Y.; Xiao, Z.; Yu, J.; Ding, S.; et al. Oral Microbiome in Older Adults with Mild Cognitive Impairment. J. Oral Microbiol. 2023, 15, 2173544. [Google Scholar] [CrossRef]
- L’Heureux, J.E.; Corbett, A.; Ballard, C.; Vauzour, D.; Creese, B.; Winyard, P.G.; Jones, A.M.; Vanhatalo, A. Oral Microbiome and Nitric Oxide Biomarkers in Older People with Mild Cognitive Impairment and APOE4 Genotype. PNAS Nexus 2025, 4, 543. [Google Scholar] [CrossRef]
- Rigat, L.; Ouk, K.; Kramer, A.; Priller, J. Dysfunction of Circadian and Sleep Rhythms in the Early Stages of Alzheimer’s Disease. Acta Physiol. 2023, 238, e13970. [Google Scholar] [CrossRef] [PubMed]
- Cardinali, D.P.; Vigo, D.E.; Olivar, N.; Vidal, M.F.; Furio, A.M.; Brusco, L.I. Therapeutic Application of Melatonin in Mild Cognitive Impairment. Am. J. Neurodegener. Dis. 2012, 1, 280–291. [Google Scholar]
- Zhang, Z.; Xue, P.; Bendlin, B.B.; Zetterberg, H.; De Felice, F.; Tan, X.; Benedict, C. Melatonin: A Potential Nighttime Guardian against Alzheimer’s. Mol. Psychiatry 2025, 30, 237–250. [Google Scholar] [CrossRef]
- Melhuish Beaupre, L.M.; Brown, G.M.; Gonçalves, V.F.; Kennedy, J.L. Melatonin’s Neuroprotective Role in Mitochondria and Its Potential as a Biomarker in Aging, Cognition and Psychiatric Disorders. Transl. Psychiatry 2021, 11, 339. [Google Scholar] [CrossRef] [PubMed]
- Branger, P.; Arenaza-Urquijo, E.M.; Tomadesso, C.; Mézenge, F.; André, C.; de Flores, R.; Mutlu, J.; de La Sayette, V.; Eustache, F.; Chételat, G.; et al. Relationships between Sleep Quality and Brain Volume, Metabolism, and Amyloid Deposition in Late Adulthood. Neurobiol. Aging 2016, 41, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Cankar, N.; Beschorner, N.; Tsopanidou, A.; Qvist, F.L.; Colaço, A.R.; Andersen, M.; Kjaerby, C.; Delle, C.; Lambert, M.; Mundt, F.; et al. Sleep Deprivation Leads to Non-Adaptive Alterations in Sleep Microarchitecture and Amyloid-β Accumulation in a Murine Alzheimer Model. Cell Rep. 2024, 43, 114977. [Google Scholar] [CrossRef]
- Duncan, M.J.; Smith, J.T.; Franklin, K.M.; Beckett, T.L.; Murphy, M.P.; St. Clair, D.K.; Donohue, K.D.; Striz, M.; O’Hara, B.F. Effects of Aging and Genotype on Circadian Rhythms, Sleep, and Clock Gene Expression in APPxPS1 Knock-in Mice, a Model for Alzheimer’s Disease. Exp. Neurol. 2012, 236, 249–258. [Google Scholar] [CrossRef]
- Toor, B.; van den Berg, N.; Ray, L.B.; Fogel, S.M. Sleep Spindles and Slow Waves Are Physiological Markers for Age-Related Changes in Gray Matter in Brain Regions Supporting Problem-Solving Skills. Learn. Mem. 2023, 30, 12–24. [Google Scholar] [CrossRef]
- Zavecz, Z.; Nagy, T.; Galkó, A.; Nemeth, D.; Janacsek, K. The Relationship between Subjective Sleep Quality and Cognitive Performance in Healthy Young Adults: Evidence from Three Empirical Studies. Sci. Rep. 2020, 10, 4855. [Google Scholar] [CrossRef]
- Drew, V.J.; Wang, C.; Kim, T. Progressive Sleep Disturbance in Various Transgenic Mouse Models of Alzheimer’s Disease. Front. Aging Neurosci. 2023, 15, 1119810. [Google Scholar] [CrossRef]
- Sato, T.; Ochiishi, T.; Higo-Yamamoto, S.; Oishi, K. Circadian and Sleep Phenotypes in a Mouse Model of Alzheimer’s Disease Characterized by Intracellular Accumulation of Amyloid β Oligomers. Exp. Anim. 2024, 73, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Li, H.; Gowravaram, N.; Quan, M.; Kausar, N.; Gomperts, S.N. Disruption of Hippocampal Neuronal Circuit Function Depends upon Behavioral State in the APP/PS1 Mouse Model of Alzheimer’s Disease. Sci. Rep. 2022, 12, 21022. [Google Scholar] [CrossRef]
- Sperling, R.A.; Donohue, M.C.; Rissman, R.A.; Johnson, K.A.; Rentz, D.M.; Grill, J.D.; Heidebrink, J.L.; Jenkins, C.; Jimenez-Maggiora, G.; Langford, O.; et al. Amyloid and Tau Prediction of Cognitive and Functional Decline in Unimpaired Older Individuals: Longitudinal Data from the A4 and LEARN Studies. J. Prev. Alzheimer’s Dis. 2024, 11, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.J.K.; Robinson, P.A.; Kedziora, D.J.; Abeysuriya, R.G. Mammalian Sleep Dynamics: How Diverse Features Arise from a Common Physiological Framework. PLoS Comput. Biol. 2010, 6, e1000826. [Google Scholar] [CrossRef]
- Eagleman, D.M.; Vaughn, D.A. The Defensive Activation Theory: REM Sleep as a Mechanism to Prevent Takeover of the Visual Cortex. Front. Neurosci. 2021, 15, 632853. [Google Scholar] [CrossRef]
- Le Bon, O. Relationships between REM and NREM in the NREM-REM Sleep Cycle: A Review on Competing Concepts. Sleep Med. 2020, 70, 6–16. [Google Scholar] [CrossRef]
- Kocagoncu, E.; Klimovich-Gray, A.; Hughes, L.E.; Rowe, J.B. Evidence and Implications of Abnormal Predictive Coding in Dementia. Brain 2021, 144, 3311–3321. [Google Scholar] [CrossRef]
- Hertenstein, E.; Trinca, E.; Wunderlin, M.; Schneider, C.L.; Züst, M.A.; Fehér, K.D.; Su, T.; Straten, A.V.; Berger, T.; Baglioni, C.; et al. Cognitive Behavioral Therapy for Insomnia in Patients with Mental Disorders and Comorbid Insomnia: A Systematic Review and Meta-Analysis. Sleep Med. Rev. 2022, 62, 101597. [Google Scholar] [CrossRef] [PubMed]
- Dubovsky, S.L.; Marshall, D. Benzodiazepines Remain Important Therapeutic Options in Psychiatric Practice. Psychother. Psychosom. 2022, 91, 307–334. [Google Scholar] [CrossRef]
- Lappas, A.S.; Polyzopoulou, Z.A.; Christodoulou, N.; Bozikas, V.-P.; Samara, M.T. Effects of Antidepressants on Sleep in Post-Traumatic Stress Disorder: An Overview of Reviews. Curr. Neuropharmacol. 2024, 22, 749–805. [Google Scholar] [CrossRef]
- Woodbury, A.; Yu, S.P.; Wei, L.; García, P. Neuro-Modulating Effects of Honokiol: A Review. Front. Neurol. 2013, 4, 130. [Google Scholar] [CrossRef] [PubMed]
- Usach, I.; Alaimo, A.; Fernández, J.; Ambrosini, A.; Mocini, S.; Ochiuz, L.; Peris, J.E. Magnolol and Honokiol: Two Natural Compounds with Similar Chemical Structure but Different Physicochemical and Stability Properties. Pharmaceutics 2021, 13, 224. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, S.; Misrani, A.; Tang, B.L.; Chen, J.; Yang, L.; Long, C. Jujuboside A Prevents Sleep Loss-Induced Disturbance of Hippocampal Neuronal Excitability and Memory Impairment in Young APP/PS1 Mice. Sci. Rep. 2019, 9, 4512. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.H.; Ha, T.W.; Hong, J.T.; Oh, K.W. Sinomenine, an Alkaloid Derived from Sinomenium Acutum Potentiates Pentobarbital-Induced Sleep Behaviors and Non-Rapid Eye Movement (NREM) Sleep in Rodents. Biomol. Ther. 2017, 25, 586–592. [Google Scholar] [CrossRef]
- Woo, J.H.; Ha, T.W.; Kang, J.S.; Hong, J.T.; Oh, K.W. Potentiation of Decursinol Angelate on Pentobarbital-Induced Sleeping Behaviors via the Activation of GABAA-Ergic Systems in Rodents. Korean J. Physiol. Pharmacol. 2017, 21, 27–36. [Google Scholar] [CrossRef]
- Ma, Y.; Eun, J.S.; Lee, K.S.; Lee, E.S.; Kim, C.S.; Hwang, B.Y.; Oh, K.W. Methanol Extract of Longanae Arillus Regulates Sleep Architecture and EEG Power Spectra in Restraint-Stressed Rats. Nat. Prod. Sci. 2009, 15, 213–221. [Google Scholar]
- Chen, X.; Yan, X.; Jing, C.; Fu, B.; Jin, W.; Zhang, S.; Wang, M.; Liu, F.; Sun, L. Ginsenoside Rc Maintains Sleep Rhythm Homeostasis by Alleviating Oxidative Stress. Phytomedicine 2025, 141, 156634. [Google Scholar] [CrossRef]
- Kim, T.H.; Custodio, R.J.; Cheong, J.H.; Kim, H.J.; Jung, Y.S. Sleep Promoting Effect of Luteolin in Mice via Adenosine A1 and A2A Receptors. Biomol. Ther. 2019, 27, 584–590. [Google Scholar] [CrossRef]
- Avallone, R.; Zanoli, P.; Puia, G.; Kleinschnitz, M.; Schreier, P.; Baraldi, M. Pharmacological Profile of Apigenin, a Flavonoid Isolated from Matricaria Chamomilla. Biochem. Pharmacol. 2000, 59, 1387–1394. [Google Scholar] [CrossRef]
- Viola, H.; Wasowski, C.; Levi De Stein, M.; Wolfman, C.; Silveira, R.; Dajas, F.; Medina, J.H.; Paladini, A.C. Apigenin, a Component of Matricaria Recutita Flowers, Is a Central Benzodiazepine Receptors-Ligand with Anxiolytic Effects. Planta Med. 1995, 61, 213–216. [Google Scholar] [CrossRef]
- Lee, Y.-J.; Lee, Y.M.; Lee, C.-K.; Jung, J.K.; Han, S.B.; Hong, J.T. Therapeutic Applications of Compounds in the Magnolia Family. Pharmacol. Ther. 2011, 130, 157–176. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.X.; Zhang, Q.Y.; Cui, S.Y.; Cui, X.Y.; Zhang, J.; Zhang, Y.H.; Bai, Y.J.; Zhao, Y.Y. Hypnotic Effect of Jujubosides from Semen Ziziphi Spinosae. J. Ethnopharmacol. 2010, 130, 163–166. [Google Scholar] [CrossRef]
- Chen, S.W.; Xiao, J.M.; Wang, R.; Wen, J.W.; Wei, X.K.; Yi, J.Z.; Yu, L.L. Behavioral Effects of Sinomenine in Murine Models of Anxiety. Life Sci. 2005, 78, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Grosso, C.; Santos, M.; Barroso, M.F. From Plants to Psycho-Neurology: Unravelling the Therapeutic Benefits of Bioactive Compounds in Brain Disorders. Antioxidants 2023, 12, 1603. [Google Scholar] [CrossRef]
- Sørensen, A.T.; Ledri, M.; Melis, M.; Ledri, L.N.; Andersson, M.; Kokaia, M. Altered Chloride Homeostasis Decreases the Action Potential Threshold and Increases Hyperexcitability in Hippocampal Neurons. eNeuro 2017, 4, e0172. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.M. Modulators of GABAA Receptor-Mediated Inhibition in the Treatment of Neuropsychiatric Disorders: Past, Present, and Future. Neuropsychopharmacology 2024, 49, 83–95. [Google Scholar] [CrossRef]
- Oishi, Y.; Saito, Y.C.; Sakurai, T. GABAergic Modulation of Sleep-Wake States. Pharmacol. Ther. 2023, 249, 108505. [Google Scholar] [CrossRef]
- Hepsomali, P.; Groeger, J.A.; Nishihira, J.; Scholey, A. Effects of Oral Gamma-Aminobutyric Acid (GABA) Administration on Stress and Sleep in Humans: A Systematic Review. Front. Neurosci. 2020, 14, 923. [Google Scholar] [CrossRef]
- Zhu, W.; Huang, L.; Cheng, H.; Li, N.; Zhang, B.; Dai, W.; Wu, X.; Zhang, D.; Feng, W.; Li, S.; et al. GABA and Its Receptors’ Mechanisms in the Treatment of Insomnia. Heliyon 2024, 10, e40665. [Google Scholar] [CrossRef]
- Eldufani, J.; Blaise, G. The Role of Acetylcholinesterase Inhibitors Such as Neostigmine and Rivastigmine on Chronic Pain and Cognitive Function in Aging: A Review of Recent Clinical Applications. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2019, 5, 175–183. [Google Scholar] [CrossRef]
- Marucci, G.; Buccioni, M.; Ben, D.D.; Lambertucci, C.; Volpini, R.; Amenta, F. Efficacy of Acetylcholinesterase Inhibitors in Alzheimer’s Disease. Neuropharmacology 2021, 190, 108352. [Google Scholar] [CrossRef] [PubMed]
- Lalo, U.; Pankratov, Y.; Kirchhoff, F.; North, R.A.; Verkhratsky, A. NMDA Receptors Mediate Neuron-to-Glia Signaling in Mouse Cortical Astrocytes. J. Neurosci. 2006, 26, 2673–2683. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chang, L.; Song, Y.; Li, H.; Wu, Y. The Role of NMDA Receptors in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 43. [Google Scholar] [CrossRef]
- Moore, A.H.; Wu, M.; Shaftel, S.S.; Graham, K.A.; O’Banion, M.K. Sustained Expression of Interleukin-1β in Mouse Hippocampus Impairs Spatial Memory. Neuroscience 2009, 164, 1484–1495. [Google Scholar] [CrossRef] [PubMed]
- Taepavarapruk, P.; Song, C. Reductions of Acetylcholine Release and Nerve Growth Factor Expression Are Correlated with Memory Impairment Induced by Interleukin-1β Administrations: Effects of Omega-3 Fatty Acid EPA Treatment. J. Neurochem. 2010, 112, 1054–1064. [Google Scholar] [CrossRef]
- Mojzych, I.; Zawadzka, A.; Andrzejewski, K.; Jampolska, M.; Bednarikova, Z.; Gancar, M.; Gazova, Z.; Mazur, M.; Kaczyńska, K. A Novel Tetrahydroacridine Derivative with Potent Acetylcholinesterase Inhibitory Properties and Dissociative Capability against Aβ42 Fibrils Confirmed by In Vitro Studies. Int. J. Mol. Sci. 2024, 25, 10072. [Google Scholar] [CrossRef]
- Tari, A.R.; Walker, T.L.; Huuha, A.M.; Sando, S.B.; Wisloff, U. Neuroprotective Mechanisms of Exercise and the Importance of Fitness for Healthy Brain Ageing. Lancet 2025, 405, 1093–1118. [Google Scholar] [CrossRef]
- Vecchio, L.M.; Meng, Y.; Xhima, K.; Lipsman, N.; Hamani, C.; Aubert, I. The Neuroprotective Effects of Exercise: Maintaining a Healthy Brain Throughout Aging. Brain Plast. 2018, 4, 17–52. [Google Scholar] [CrossRef]
- Castro, C.B.; Costa, L.M.; Dias, C.B.; Chen, J.; Hillebrandt, H.; Gardener, S.L.; Brown, B.M.; Loo, R.L.; Garg, M.L.; Rainey-Smith, S.R.; et al. Multi-Domain Interventions for Dementia Prevention–A Systematic Review. J. Nutr. Heal. Aging 2023, 27, 1271–1280. [Google Scholar] [CrossRef]
- Xu, H.; Turchini, G.M.; Francis, D.S.; Liang, M.; Mock, T.S.; Rombenso, A.; Ai, Q. Are Fish What They Eat? A Fatty Acid’s Perspective. Prog. Lipid Res. 2020, 80, 101064. [Google Scholar] [CrossRef]
- Zhu, P.; Fan, L.; Yan, X.; Li, J. Advances of α-Linolenic Acid: Sources, Extraction, Biological Activity and Its Carrier. Trends Food Sci. Technol. 2024, 152, 104676. [Google Scholar] [CrossRef]
- Bishehkolaei, M.; Pathak, Y. Influence of Omega N-6/n-3 Ratio on Cardiovascular Disease and Nutritional Interventions. Hum. Nutr. Metab. 2024, 37, 200275. [Google Scholar] [CrossRef]
- Peredo-Lovillo, A.; Romero-Luna, H.E.; Jiménez-Fernández, M. Health Promoting Microbial Metabolites Produced by Gut Microbiota after Prebiotics Metabolism. Food Res. Int. 2020, 136, 109473. [Google Scholar] [CrossRef] [PubMed]
- Niazi, N.U.K.; Cai, S. Omega-3 Fatty Acids and Neurodegenerative Diseases: Focus on Alzheimer’s Disease. In Phospholipases in Physiology and Pathology: Volumes 1–7; Chakraborti, S., Ed.; Elsevier: Amsterdam, The Netherlands, 2023; Volume 7, pp. V7-167–V7-188. ISBN 9780323956871. [Google Scholar]
- Isseroff, R.R.; Ziboh, V.A.; Chapkin, R.S.; Martinez, D.T. Conversion of Linoleic Acid into Arachidonic Acid by Cultured Murine and Human Keratinocytes. J. Lipid Res. 1987, 28, 1342–1349. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Chen, C.; Dong, Y.; You, C.; Wang, S.; Monroig, Ó.; Tocher, D.R.; Li, Y. Regulation of Long-Chain Polyunsaturated Fatty Acid Biosynthesis in Teleost Fish. Prog. Lipid Res. 2021, 82, 101095. [Google Scholar] [CrossRef]
- Harizi, H.; Corcuff, J.B.; Gualde, N. Arachidonic-Acid-Derived Eicosanoids: Roles in Biology and Immunopathology. Trends Mol. Med. 2008, 14, 461–469. [Google Scholar] [CrossRef]
- Patterson, E.; Wall, R.; Fitzgerald, G.F.; Ross, R.P.; Stanton, C. Health Implications of High Dietary Omega-6 Polyunsaturated Fatty Acids. J. Nutr. Metab. 2012, 2012, 539426. [Google Scholar] [CrossRef]
- Mazidi, M.; Gao, H.-K.; Vatanparast, H.; Kengne, A.P. Impact of the Dietary Fatty Acid Intake on C-Reactive Protein Levels in US Adults. Medicine 2017, 96, e5736. [Google Scholar] [CrossRef]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism Pathways of Arachidonic Acids: Mechanisms and Potential Therapeutic Targets. Signal Transduct. Target. Ther. 2021, 6, 94. [Google Scholar] [CrossRef]
- Rådmark, O.; Werz, O.; Steinhilber, D.; Samuelsson, B. 5-Lipoxygenase, a Key Enzyme for Leukotriene Biosynthesis in Health and Disease. Biochim. Biophys. Acta 2015, 1851, 331–339. [Google Scholar] [CrossRef]
- Pandey, M.K.; Von Suskil, M.; Chitren, R.; Al-Odat, O.; Jonnalagadda, S.C.; Aggarwal, B.B. Cancer on Fire: Role of Inflammation in Prevention and Treatment. In Current Advances for Development of Functional Foods Modulating Inflammation and Oxidative Stress; Elsevier: Amsterdam, The Netherlands, 2022; pp. 605–626. ISBN 9780128234822. [Google Scholar]
- Majumdar, R.; Tameh, A.T.; Arya, S.B.; Parent, C.A. Exosomes Mediate LTB4 Release during Neutrophil Chemotaxis. PLoS Biol. 2021, 19, e3001271. [Google Scholar] [CrossRef] [PubMed]
- Afonso, P.V.; Janka-Junttila, M.; Lee, Y.J.; McCann, C.P.; Oliver, C.M.; Aamer, K.A.; Losert, W.; Cicerone, M.T.; Parent, C.A. LTB4 Is a Signal-Relay Molecule during Neutrophil Chemotaxis. Dev. Cell 2012, 22, 1079–1091. [Google Scholar] [CrossRef] [PubMed]
- Mirowska, W.; Bartusik-Aebisher, D.; Aebisher, D. Leukotrienes. In The Biochemical Guide to Hormones; Nova Science Publishers: Hauppauge, NY, USA, 2023; Volume 357, pp. 179–183. [Google Scholar]
- Efstathia, K.; Panayiotis, V. Dietary Neurolipidomics in Alzheimer’s Disease. J. Nutr. Med. Diet Care 2022, 8, 10–23937. [Google Scholar] [CrossRef]
- Luo, S.; Hou, H.; Wang, Y.; Li, Y.; Zhang, L.; Zhang, H.; Jin, Q.; Wu, G.; Wang, X. Effects of Omega-3, Omega-6, and Total Dietary Polyunsaturated Fatty Acid Supplementation in Patients with Atherosclerotic Cardiovascular Disease: A Systematic Review and Meta-Analysis. Food Funct. 2024, 15, 1208–1222. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Zhang, Y.; Li, H.; Wang, F.; Yao, S. Toll-like Receptor 4: A Potential Therapeutic Target for Multiple Human Diseases. Biomed. Pharmacother. 2023, 166, 115338. [Google Scholar] [CrossRef]
- Fahrmann, J.F.; Ballester, O.F.; Ballester, G.; Witte, T.R.; Salazar, A.J.; Kordusky, B.; Cowen, K.G.; Ion, G.; Primerano, D.A.; Boskovic, G.; et al. Inhibition of Nuclear Factor Kappa B Activation in Early-Stage Chronic Lymphocytic Leukemia by Omega-3 Fatty Acids. Cancer Investig. 2013, 31, 24–38. [Google Scholar] [CrossRef]
- Hwang, D.H.; Kim, J.A.; Lee, J.Y. Mechanisms for the Activation of Toll-like Receptor 2/4 by Saturated Fatty Acids and Inhibition by Docosahexaenoic Acid. Eur. J. Pharmacol. 2016, 785, 24–35. [Google Scholar] [CrossRef]
- Stulnig, T.M. Immunomodulation by Polyunsaturated Fatty Acids: Mechanisms and Effects. Int. Arch. Allergy Immunol. 2003, 132, 310–321. [Google Scholar] [CrossRef]
- Kuo, H.C.; Lee, K.F.; Chen, S.L.; Chiu, S.C.; Lee, L.Y.; Chen, W.P.; Chen, C.C.; Chu, C.H. Neuron–Microglia Contacts Govern the PGE2 Tolerance through TLR4-Mediated de Novo Protein Synthesis. Biomedicines 2022, 10, 419. [Google Scholar] [CrossRef]
- Soon, A.L.; Hye, J.K.; Ki, C.C.; Jong, C.B.; Ji, K.P.; Jeong, K.S.; Won, J.C.; Jong, H.L.; Won, Y.P. DHA and EPA Down-Regulate COX-2 Expression through Suppression of NF-ΚB Activity in LPS-Treated Human Umbilical Vein Endothelial Cells. Korean J. Physiol. Pharmacol. 2009, 13, 301–307. [Google Scholar] [CrossRef]
- Cui, J.; Shan, K.; Yang, Q.; Qi, Y.; Qu, H.; Li, J.; Wang, R.; Jia, L.; Chen, W.; Feng, N.; et al. Prostaglandin E3 Attenuates Macrophage-Associated Inflammation and Prostate Tumour Growth by Modulating Polarization. J. Cell. Mol. Med. 2021, 25, 5586–5601. [Google Scholar] [CrossRef] [PubMed]
- Bagga, D.; Wang, L.; Farias-Eisner, R.; Glaspy, J.A.; Reddy, S.T. Differential Effects of Prostaglandin Derived from ω-6 and ω-3 Polyunsaturated Fatty Acids on COX-2 Expression and IL-6 Secretion. Proc. Natl. Acad. Sci. USA 2003, 100, 1751–1756. [Google Scholar] [CrossRef] [PubMed]
- Norris, P.C.; Dennis, E.A. Omega-3 Fatty Acids Cause Dramatic Changes in TLR4 and Purinergic Eicosanoid Signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 8517–8522. [Google Scholar] [CrossRef]
- Torres, M.; Parets, S.; Fernández-Díaz, J.; Beteta-Göbel, R.; Rodríguez-Lorca, R.; Román, R.; Lladó, V.; Rosselló, C.A.; Fernández-García, P.; Escribá, P.V. Lipids in Pathophysiology and Development of the Membrane Lipid Therapy: New Bioactive Lipids. Membranes 2021, 11, 919. [Google Scholar] [CrossRef]
- Fan, Y.Y.; McMurray, D.N.; Ly, L.H.; Chapkin, R.S. Dietary (n-3) Polyunsaturated Fatty Acids Remodel Mouse T-Cell Lipid Rafts. J. Nutr. 2003, 133, 1913–1920. [Google Scholar] [CrossRef]
- Wei, B.Z.; Li, L.; Dong, C.W.; Tan, C.C.; Xu, W. The Relationship of Omega-3 Fatty Acids with Dementia and Cognitive Decline: Evidence from Prospective Cohort Studies of Supplementation, Dietary Intake, and Blood Markers. Am. J. Clin. Nutr. 2023, 117, 1096–1109. [Google Scholar] [CrossRef]
- Rapoport, S.I.; Ramadan, E.; Basselin, M. Docosahexaenoic Acid (DHA) Incorporation into the Brain from Plasma, as an in Vivo Biomarker of Brain DHA Metabolism and Neurotransmission. Prostaglandins Other Lipid Mediat. 2011, 96, 109–113. [Google Scholar] [CrossRef]
- Kidd, P.M. Omega-3 DHA and EPA for Cognition, Behavior, and Mood: Clinical Findings and Structural-Functional Synergies with Cell Membrane Phospholipids. Altern. Med. Rev. 2007, 12, 207–227. [Google Scholar]
- Schilke, R.M.; Blackburn, C.M.R.; Bamgbose, T.T.; Woolard, M.D. Interface of Phospholipase Activity, Immune Cell Function, and Atherosclerosis. Biomolecules 2020, 10, 1449. [Google Scholar] [CrossRef]
- Calder, P.C. Mechanisms of Action of (n-3) Fatty Acids. J. Nutr. 2012, 142, 592S–599S. [Google Scholar] [CrossRef]
- Risé, P.; Eligini, S.; Ghezzi, S.; Colli, S.; Galli, C. Fatty Acid Composition of Plasma, Blood Cells and Whole Blood: Relevance for the Assessment of the Fatty Acid Status in Humans. Prostaglandins Leukot. Essent. Fat. Acids 2007, 76, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi Fard, S.; Wang, F.; Sinclair, A.J.; Elliott, G.; Turchini, G.M. How Does High DHA Fish Oil Affect Health? A Systematic Review of Evidence. Crit. Rev. Food Sci. Nutr. 2019, 59, 1684–1727. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, M.; Rockwell, M.S.; Wentz, L.M. The Influence of Dietary and Supplemental Omega-3 Fatty Acids on the Omega-3 Index: A Scoping Review. Front. Nutr. 2023, 10, 1072653. [Google Scholar] [CrossRef]
- Joffre, C.; Dinel, A.L.; Chataigner, M.; Pallet, V.; Layé, S. N-3 Polyunsaturated Fatty Acids and Their Derivates Reduce Neuroinflammation during Aging. Nutrients 2020, 12, 647. [Google Scholar] [CrossRef]
- Sidhu, V.K.; Huang, B.X.; Desai, A.; Kevala, K.; Kim, H.Y. Role of DHA in Aging-Related Changes in Mouse Brain Synaptic Plasma Membrane Proteome. Neurobiol. Aging 2016, 41, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Yurko-Mauro, K.; Alexander, D.D.; Van Elswyk, M.E. Docosahexaenoic Acid and Adult Memory: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0120391. [Google Scholar] [CrossRef]
- Wood, A.H.R.; Chappell, H.F.; Zulyniak, M.A. Dietary and Supplemental Long-Chain Omega-3 Fatty Acids as Moderators of Cognitive Impairment and Alzheimer’s Disease. Eur. J. Nutr. 2022, 61, 589–604. [Google Scholar] [CrossRef]
- Conquer, J.A.; Tierney, M.C.; Zecevic, J.; Bettger, W.J.; Fisher, R.H. Fatty Acid Analysis of Blood Plasma of Patients with Alzheimer’s Disease, Other Types of Dementia, and Cognitive Impairment. Lipids 2000, 35, 1305–1312. [Google Scholar] [CrossRef]
- Zhang, X.; Yuan, T.; Chen, X.; Liu, X.; Hu, J.; Liu, Z. Effects of DHA on Cognitive Dysfunction in Aging and Alzheimer’s Disease: The Mediating Roles of ApoE. Prog. Lipid Res. 2024, 93, 101256. [Google Scholar] [CrossRef]
- Sala-Vila, A.; Satizabal, C.L.; Tintle, N.; Melo van Lent, D.; Vasan, R.S.; Beiser, A.S.; Seshadri, S.; Harris, W.S. Red Blood Cell DHA Is Inversely Associated with Risk of Incident Alzheimer’s Disease and All-Cause Dementia: Framingham Offspring Study. Nutrients 2022, 14, 2408. [Google Scholar] [CrossRef] [PubMed]
- Stavrinou, P.S.; Andreou, E.; Aphamis, G.; Pantzaris, M.; Ioannou, M.; Patrikios, I.S.; Giannaki, C.D. The Effects of a 6-Month High Dose Omega-3 and Omega-6 Polyunsaturated Fatty Acids and Antioxidant Vitamins Supplementation on Cognitive Function and Functional Capacity in Older Adults with Mild Cognitive Impairment. Nutrients 2020, 12, 325. [Google Scholar] [CrossRef]
- Heshmati, J.; Morvaridzadeh, M.; Maroufizadeh, S.; Akbari, A.; Yavari, M.; Amirinejad, A.; Maleki-Hajiagha, A.; Sepidarkish, M. Omega-3 Fatty Acids Supplementation and Oxidative Stress Parameters: A Systematic Review and Meta-Analysis of Clinical Trials. Pharmacol. Res. 2019, 149, 104462. [Google Scholar] [CrossRef]
- Lee, J.C.M.; Simonyi, A.; Sun, A.Y.; Sun, G.Y. Phospholipases A2 and Neural Membrane Dynamics: Implications for Alzheimer’s Disease. J. Neurochem. 2011, 116, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Luchtman, D.W.; Meng, Q.; Wang, X.; Shao, D.; Song, C. Omega-3 Fatty Acid Eicospentaenoic Acid Attenuates MPP+-Induced Neurodegeneration in Fully Differentiated Human SH-SY5Y and Primary Mesencephalic Cells. J. Neurochem. 2013, 124, 855–868. [Google Scholar] [CrossRef]
- Davinelli, S.; Medoro, A.; Intrieri, M.; Saso, L.; Scapagnini, G.; Kang, J.X. Targeting NRF2–KEAP1 Axis by Omega-3 Fatty Acids and Their Derivatives: Emerging Opportunities against Aging and Diseases. Free Radic. Biol. Med. 2022, 193, 736–750. [Google Scholar] [CrossRef]
- Shao, J.; Wang, S.; Liu, L. Maternal Omega-3 Fatty Acid Supplementation against Prenatal Lead Exposure Induced Cognitive Impairment in Offspring Mice. J. Toxicol. Sci. 2022, 47, 183–192. [Google Scholar] [CrossRef]
- Gu, M.; Li, X.; Yan, L.; Zhang, Y.; Yang, L.; Li, S.; Song, C. Endogenous ω-3 Fatty Acids in Fat-1 Mice Attenuated Depression-like Behaviors, Spatial Memory Impairment and Relevant Changes Induced by Olfactory Bulbectomy. Prostaglandins Leukot. Essent. Fat. Acids 2021, 171, 102313. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Li, Y.; Tang, H.; Zhang, C.; Li, W.; Zhang, Y.; Li, Y.; Zhao, Y.; Song, C. Endogenous Omega (N)-3 Fatty Acids in Fat-1 Mice Attenuated Depression-like Behavior, Imbalance between Microglial M1 and M2 Phenotypes, and Dysfunction of Neurotrophins Induced by Lipopolysaccharide Administration. Nutrients 2018, 10, 1351. [Google Scholar] [CrossRef]
- Majou, D.; Dermenghem, A.L. Effects of DHA (Omega-3 Fatty Acid) and Estradiol on Amyloid β-Peptide Regulation in the Brain. Brain Res. 2024, 1823, 148681. [Google Scholar] [CrossRef]
- Park, Y.H.; Shin, S.J.; Kim, H.S.; Hong, S.B.; Kim, S.; Nam, Y.; Kim, J.J.; Lim, K.; Kim, J.S.; Kim, J.-i.; et al. Omega-3 Fatty Acid-Type Docosahexaenoic Acid Protects against Aβ-Mediated Mitochondrial Deficits and Pathomechanisms in Alzheimer’s Disease-Related Animal Model. Int. J. Mol. Sci. 2020, 21, 3879. [Google Scholar] [CrossRef] [PubMed]
- Sharman, M.J.; Gyengesi, E.; Liang, H.; Chatterjee, P.; Karl, T.; Li, Q.X.; Wenk, M.R.; Halliwell, B.; Martins, R.N.; Münch, G. Assessment of Diets Containing Curcumin, Epigallocatechin-3-Gallate, Docosahexaenoic Acid and α-Lipoic Acid on Amyloid Load and Inflammation in a Male Transgenic Mouse Model of Alzheimer’s Disease: Are Combinations More Effective? Neurobiol. Dis. 2019, 124, 505–519. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Li, X.; Kang, Z.; Kadotomi, Y. Omega-3 Fatty Acid Ethyl-Eicosapentaenoate Attenuates IL-1β-Induced Changes in Dopamine and Metabolites in the Shell of the Nucleus Accumbens: Involved with PLA2 Activity and Corticosterone Secretion. Neuropsychopharmacology 2007, 32, 736–744. [Google Scholar] [CrossRef]
- Nunan, J.; Small, D.H. Regulation of APP Cleavage by α-, β- and γ-Secretases. FEBS Lett. 2000, 483, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Amtul, Z.; Uhrig, M.; Rozmahel, R.F.; Beyreuther, K. Structural Insight into the Differential Effects of Omega-3 and Omega-6 Fatty Acids on the Production of Aβ Peptides and Amyloid Plaques. J. Biol. Chem. 2011, 286, 6100–6107. [Google Scholar] [CrossRef]
- Dubal, D.B. The Way of Tau: Secretion and Synaptic Dysfunction. Trends Mol. Med. 2018, 24, 595–597. [Google Scholar] [CrossRef]
- Fertan, E.; Stover, K.R.J.; Brant, M.G.; Stafford, P.M.; Kelly, B.; Diez-Cecilia, E.; Wong, A.A.; Weaver, D.F.; Brown, R.E. Effects of the Novel IDO Inhibitor DWG-1036 on the Behavior of Male and Female 3xTg-AD Mice. Front. Pharmacol. 2019, 10, 1044. [Google Scholar] [CrossRef]
- Pukoli, D.; Polyák, H.; Rajda, C.; Vécsei, L. Kynurenines and Neurofilament Light Chain in Multiple Sclerosis. Front. Neurosci. 2021, 15, 658202. [Google Scholar] [CrossRef]
- Belkouch, M.; Hachem, M.; Elgot, A.; Van, A.L.; Picq, M.; Guichardant, M.; Lagarde, M.; Bernoud-Hubac, N. The Pleiotropic Effects of Omega-3 Docosahexaenoic Acid on the Hallmarks of Alzheimer’s Disease. J. Nutr. Biochem. 2016, 38, 1–11. [Google Scholar] [CrossRef]
- De Felice, F.G.; Gonçalves, R.A.; Ferreira, S.T. Impaired Insulin Signalling and Allostatic Load in Alzheimer Disease. Nat. Rev. Neurosci. 2022, 23, 215–230. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, S.; Zhang, Q.; Shao, W.; Han, X.; Wang, Y.; Du, Y. Endoplasmic Reticulum Stress Mediates JNK-Dependent IRS-1 Serine Phosphorylation and Results in Tau Hyperphosphorylation in Amyloid β Oligomer-Treated PC12 Cells and Primary Neurons. Gene 2016, 587, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Yavari, M.; Kalupahana, N.S.; Harris, B.N.; Ramalingam, L.; Zu, Y.; Kahathuduwa, C.N.; Moustaid-Moussa, N. Mechanisms Linking Obesity, Insulin Resistance, and Alzheimer’s Disease: Effects of Polyphenols and Omega-3 Polyunsaturated Fatty Acids. Nutrients 2025, 17, 1203. [Google Scholar] [CrossRef] [PubMed]
- Ganai, S.A.; Mohan, S.; Padder, S.A. Exploring Novel and Potent Glycogen Synthase Kinase-3β Inhibitors through Systematic Drug Designing Approach. Sci. Rep. 2025, 15, 4118. [Google Scholar] [CrossRef] [PubMed]
- Hoost, S.S.; Honig, L.S.; Kang, M.S.; Bahl, A.; Lee, A.J.; Sanchez, D.; Reyes-Dumeyer, D.; Lantigua, R.A.; Dage, J.L.; Brickman, A.M.; et al. Association of Dietary Fatty Acids with Longitudinal Change in Plasma-Based Biomarkers of Alzheimer’s Disease. J. Prev. Alzheimer’s Dis. 2025, 12, 100117. [Google Scholar] [CrossRef]
- Reitz, C.; Tang, M.X.; Schupf, N.; Manly, J.J.; Mayeux, R.; Luchsinger, J.A. Association of Higher Levels of High-Density Lipoprotein Cholesterol in Elderly Individuals and Lower Risk of Late-Onset Alzheimer Disease. Arch. Neurol. 2010, 67, 1491–1497. [Google Scholar] [CrossRef]
- Kuo, Y.M.; Emmerling, M.R.; Bisgaier, C.L.; Essenburg, A.D.; Lampert, H.C.; Drumm, D.; Roher, A.E. Elevated Low-Density Lipoprotein in Alzheimer’s Disease Correlates with Brain Aβ 1-42 Levels. Biochem. Biophys. Res. Commun. 1998, 252, 711–715. [Google Scholar] [CrossRef]
- Belaidi, A.A.; Bush, A.I.; Ayton, S. Apolipoprotein E in Alzheimer’s Disease: Molecular Insights and Therapeutic Opportunities. Mol. Neurodegener. 2025, 20, 47. [Google Scholar] [CrossRef]
- Ebright, B.; Assante, I.; Poblete, R.A.; Wang, S.; Duro, M.V.; Bennett, D.A.; Arvanitakis, Z.; Louie, S.G.; Yassine, H.N. Eicosanoid Lipidome Activation in Post-Mortem Brain Tissues of Individuals with APOE4 and Alzheimer’s Dementia. Alzheimer’s Res. Ther. 2022, 14, 152. [Google Scholar] [CrossRef]
- Liu, J.J.; Green, P.; John Mann, J.; Rapoport, S.I.; Sublette, M.E. Pathways of Polyunsaturated Fatty Acid Utilization: Implications for Brain Function in Neuropsychiatric Health and Disease. Brain Res. 2015, 1597, 220–246. [Google Scholar] [CrossRef]
- Simopoulos, A.P. Evolutionary Aspects of Diet: The Omega-6/Omega-3 Ratio and the Brain. Mol. Neurobiol. 2011, 44, 203–215. [Google Scholar] [CrossRef]
- Freund-Levi, Y.; Eriksdotter-Jönhagen, M.; Cederholm, T.; Basun, H.; Faxén-Irving, G.; Garlind, A.; Vedin, I.; Vessby, B.; Wahlund, L.O.; Palmblad, J. ω-3 Fatty Acid Treatment in 174 Patients with Mild to Moderate Alzheimer Disease: OmegAD Study—A Randomized Double-Blind Trial. Arch. Neurol. 2006, 63, 1402–1408. [Google Scholar] [CrossRef]
- Liu, X.; Zhuang, P.; Li, Y.; Wu, F.; Wan, X.; Zhang, Y.; Jiao, J. Association of Fish Oil Supplementation with Risk of Incident Dementia: A Prospective Study of 215,083 Older Adults. Clin. Nutr. 2022, 41, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.Y.; Cheng, C.; Satyanarayanan, S.K.; Chiu, L.T.; Chien, Y.C.; Chuu, C.P.; Lan, T.H.; Su, K.P. Omega-3 Fatty Acids and Blood-Based Biomarkers in Alzheimer’s Disease and Mild Cognitive Impairment: A Randomized Placebo-Controlled Trial. Brain. Behav. Immun. 2022, 99, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Shinto, L.; Quinn, J.; Montine, T.; Dodge, H.H.; Woodward, W.; Baldauf-Wagner, S.; Waichunas, D.; Bumgarner, L.; Bourdette, D.; Silbert, L.; et al. A Randomized Placebo-Controlled Pilot Trial of Omega-3 Fatty Acids and Alpha Lipoic Acid in Alzheimer’s Disease. J. Alzheimer’s Dis. 2014, 38, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, D.R.; Bäckman, K.; Scarmeas, N.; Stern, Y.; Manly, J.J.; Mayeux, R.; Gu, Y. Dietary Fatty Acids and Risk of Alzheimer’s Disease and Related Dementias: Observations from the Washington Heights-Hamilton Heights-Inwood Columbia Aging Project (WHICAP). Alzheimer’s Dement. 2020, 16, 1638–1649. [Google Scholar] [CrossRef]
- Gu, Y.; Luchsinger, J.A.; Stern, Y.; Scarmeas, N. Mediterranean Diet, Inflammatory and Metabolic Biomarkers, and Risk of Alzheimer’s Disease. J. Alzheimer’s Dis. 2010, 22, 483–492. [Google Scholar] [CrossRef]
- Devore, E.E.; Grodstein, F.; Van Rooij, F.J.A.; Hofman, A.; Rosner, B.; Stampfer, M.J.; Witteman, J.C.M.; Breteler, M.M.B. Dietary Intake of Fish and Omega-3 Fatty Acids in Relation to Long-Term Dementia Risk. Am. J. Clin. Nutr. 2009, 90, 170–176. [Google Scholar] [CrossRef]
- Danthiir, V.; Hosking, D.E.; Nettelbeck, T.; Vincent, A.D.; Wilson, C.; O’Callaghan, N.; Calvaresi, E.; Clifton, P.; Wittert, G.A. An 18-Mo Randomized, Double-Blind, Placebo-Controlled Trial of DHA-Rich Fish Oil to Prevent Age-Related Cognitive Decline in Cognitively Normal Older Adults. Am. J. Clin. Nutr. 2018, 107, 754–762. [Google Scholar] [CrossRef]
- Cai, W.; Haddad, M.; Haddad, R.; Kesten, I.; Hoffman, T.; Laan, R.; Westfall, S.; Defaye, M.; Abdullah, N.S.; Wong, C.; et al. The Gut Microbiota Promotes Pain in Fibromyalgia. Neuron 2025, 13, 1079–1091. [Google Scholar] [CrossRef]
- Sublette, M.E.; Daray, F.M.; Ganança, L.; Shaikh, S.R. The Role of Polyunsaturated Fatty Acids in the Neurobiology of Major Depressive Disorder and Suicide Risk. Mol. Psychiatry 2024, 29, 269–286. [Google Scholar] [CrossRef]
- He, C.; Qu, X.; Cui, L.; Wang, J.; Kang, J.X. Improved Spatial Learning Performance of Fat-1 Mice Is Associated with Enhanced Neurogenesis and Neuritogenesis by Docosahexaenoic Acid. Proc. Natl. Acad. Sci. USA 2009, 106, 11370–11375. [Google Scholar] [CrossRef] [PubMed]
- Dyall, S.C. Long-Chain Omega-3 Fatty Acids and the Brain: A Review of the Independent and Shared Effects of EPA, DPA and DHA. Front. Aging Neurosci. 2015, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Farooqui, A.A.; Siddiqi, N.J.; Alhomida, A.S.; Ong, W.Y. Effects of Docosahexaenoic Acid on Neurotransmission. Biomol. Ther. 2012, 20, 152–157. [Google Scholar] [CrossRef]
- Checa-Ros, A.; D’Marco, L. Role of Omega-3 Fatty Acids as Non-Photic Zeitgebers and Circadian Clock Synchronizers. Int. J. Mol. Sci. 2022, 23, 12162. [Google Scholar] [CrossRef]
- Iob, E.; Kirschbaum, C.; Steptoe, A. Persistent Depressive Symptoms, HPA-Axis Hyperactivity, and Inflammation: The Role of Cognitive-Affective and Somatic Symptoms. Mol. Psychiatry 2020, 25, 1130–1140. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, M.; Walker, C. Circadian Clock, Glucocorticoids and NF-ΚB Signaling in Neuroinflammation- Implicating Glucocorticoid Induced Leucine Zipper as a Molecular Link. ASN Neuro 2022, 14, 17590914221120190. [Google Scholar] [CrossRef]
- Dickmeis, T. Glucocorticoids and the Circadian Clock. J. Endocrinol. 2009, 200, 3–22. [Google Scholar] [CrossRef]
- Gao, K.; Mu, C.L.; Farzi, A.; Zhu, W.Y. Tryptophan Metabolism: A Link between the Gut Microbiota and Brain. Adv. Nutr. 2020, 11, 709–723. [Google Scholar] [CrossRef]
- De Carvalho, C.C.C.R.; Caramujo, M.J. The Various Roles of Fatty Acids. Molecules 2018, 23, 2583. [Google Scholar] [CrossRef]
- Maulucci, G.; Cohen, O.; Daniel, B.; Sansone, A.; Petropoulou, P.I.; Filou, S.; Spyridonidis, A.; Pani, G.; De Spirito, M.; Chatgilialoglu, C.; et al. Fatty Acid-Related Modulations of Membrane Fluidity in Cells: Detection and Implications. Free Radic. Res. 2016, 50, S40–S50. [Google Scholar] [CrossRef]
- Korkutata, M.; Lazarus, M. Adenosine A2A Receptors and Sleep. Int. Rev. Neurobiol. 2023, 170, 155–178. [Google Scholar] [CrossRef] [PubMed]
- Dashti, H.S.; Follis, J.L.; Smith, C.E.; Tanaka, T.; Cade, B.E.; Gottlieb, D.J.; Hruby, A.; Jacques, P.F.; Lamon-Fava, S.; Richardson, K.; et al. Habitual Sleep Duration Is Associated with BMI and Macronutrient Intake and May Be Modified by CLOCK Genetic Variants. Am. J. Clin. Nutr. 2015, 101, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Judge, M.P.; Cong, X.; Harel, O.; Courville, A.B.; Lammi-Keefe, C.J. Maternal Consumption of a DHA-Containing Functional Food Benefits Infant Sleep Patterning: An Early Neurodevelopmental Measure. Early Hum. Dev. 2012, 88, 531–537. [Google Scholar] [CrossRef]
- Boone, K.M.; Rausch, J.; Pelak, G.; Li, R.; Turner, A.N.; Klebanoff, M.A.; Keim, S.A. Docosahexaenoic Acid and Arachidonic Acid Supplementation and Sleep in Toddlers Born Preterm: Secondary Analysis of a Randomized Clinical Trial. J. Clin. Sleep Med. 2019, 15, 1197–1208. [Google Scholar] [CrossRef]
- Hysing, M.; Kvestad, I.; Kjellevold, M.; Midtbø, L.K.; Graff, I.E.; Lie, Ø.; Hurum, H.; Stormark, K.M.; Øyen, J. Fatty Fish Intake and the Effect on Mental Health and Sleep in Preschool Children in FINS-KIDS, a Randomized Controlled Trial. Nutrients 2018, 10, 1478. [Google Scholar] [CrossRef] [PubMed]
- Patan, M.J.; Kennedy, D.O.; Husberg, C.; Hustvedt, S.O.; Calder, P.C.; Middleton, B.; Khan, J.; Forster, J.; Jackson, P.A. Differential Effects of Dha-and Epa-Rich Oils on Sleep in Healthy Young Adults: A Randomized Controlled Trial. Nutrients 2021, 13, 248. [Google Scholar] [CrossRef]
- Yokoi-Shimizu, K.; Yanagimoto, K.; Hayamizu, K. Effect of Docosahexaenoic Acid and Eicosapentaenoic Acid Supplementation on Sleep Quality in Healthy Subjects: A Randomized, Double-Blinded, Placebo-Controlled Trial. Nutrients 2022, 14, 4136. [Google Scholar] [CrossRef]
- Murphy, R.A.; Devarshi, P.P.; Mun, J.G.; Marshall, K.; Mitmesser, S.H. Association of Omega-3 Levels and Sleep in US Adults, National Health and Nutrition Examination Survey, 2011–2012. Sleep Health 2022, 8, 294–297. [Google Scholar] [CrossRef]
- Yehuda, S.; Rabinovitz, S.; Mostofsky, D.I. Mixture of Essential Fatty Acids Lowers Test Anxiety. Nutr. Neurosci. 2005, 8, 265–267. [Google Scholar] [CrossRef]
- Dretsch, M.N.; Johnston, D.; Bradley, R.S.; MacRae, H.; Deuster, P.A.; Harris, W.S. Effects of Omega-3 Fatty Acid Supplementation on Neurocognitive Functioning and Mood in Deployed U.S. Soldiers: A Pilot Study. Mil. Med. 2014, 179, 396–403. [Google Scholar] [CrossRef]
- Ford, P.A.; Jaceldo-Siegl, K.; Lee, J.W.; Tonstad, S. Trans Fatty Acid Intake Is Related to Emotional Affect in the Adventist Health Study-2. Nutr. Res. 2016, 36, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Christian, L.M.; Blair, L.M.; Porter, K.; Lower, M.; Cole, R.M.; Belury, M.A. Polyunsaturated Fatty Acid (PUFA) Status in Pregnant Women: Associations with Sleep Quality, Inflammation, and Length of Gestation. PLoS ONE 2016, 11, e0148752. [Google Scholar] [CrossRef] [PubMed]
- Lotrich, F.E.; Sears, B.; McNamara, R.K. Polyunsaturated Fatty Acids Moderate the Effect of Poor Sleep on Depression Risk. Prostaglandins Leukot. Essent. Fat. Acids 2016, 106, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cui, Y.; Li, L.; Wu, L.; Hanlon, A.; Pinto-Martin, J.; Raine, A.; Hibbeln, J.R. The Mediating Role of Sleep in the Fish Consumption-Cognitive Functioning Relationship: A Cohort Study. Sci. Rep. 2017, 7, 17961. [Google Scholar] [CrossRef]
- Hawkins, M.; Marcus, B.; Pekow, P.; Rosal, M.C.; Tucker, K.L.; Spencer, R.M.C.; Chasan-Taber, L. The Impact of a Randomized Controlled Trial of a Lifestyle Intervention on Sleep among Latina Postpartum Women. Ann. Behav. Med. 2019, 55, 892–903. [Google Scholar] [CrossRef]
- Sanguansri, L.; Augustin, M.A.; Lockett, T.J.; Abeywardena, M.Y.; Royle, P.J.; Mano, M.T.; Patten, G.S. Bioequivalence of N-3 Fatty Acids from Microencapsulated Fish Oil Formulations in Human Subjects. Br. J. Nutr. 2015, 113, 822–831. [Google Scholar] [CrossRef]
- Bowen, K.J.; Harris, W.S.; Kris-Etherton, P.M. Omega-3 Fatty Acids and Cardiovascular Disease: Are There Benefits? Curr. Treat. Options Cardiovasc. Med. 2016, 18, 69. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Study | Subjects | Study Type | Treatment/Source | Duration | Results |
|---|---|---|---|---|---|---|
| 1 | Freund-Levi et al. [215] | 174 AD patients | Randomized double blind trial | DHA: 1.7 g; EPA: 0.6 g | 6 months | n-3 intake improved AD-induced cognitive decline in very mild cases |
| 2 | Liu et al. [216] | 215,083 healthy adults (≥60 y) | Prospective cohort study | Intake of fish oil supplements | 7.92 years | A marginal interaction between ApoE gene variants, n-3, and risk of cognitive decline was found |
| 3 | Lin et al. [217] | 163 MCI or AD patients | Randomized placebo-controlled trial | DHA: 0.7 g; EPA: 1.6 g; EPA + DHA: 0.8 + 0.35 g | 2 years | EPA reduced CCL4 levels and the constructional praxis. Both EPA and DHA improved the speaking ability score |
| 4 | Shinto et al. [218] | 39 AD patients | Randomized placebo-controlled trial | DHA: 675 mg; EPA: 975 mg; LA: 600 mg | 1 year | The combination reduced the rate of decline in MMSE scores in AD patients |
| 5 | Gustafon et al. [219] | 2612 healthy elderly (≥65 y) | Prospective cohort study | n-3 PUFA dietary supplement | 7 years | n-3 intake is positively correlated with lowered risk of developing AD |
| 6 | Gu et al. [220] | 1219 healthy elderly (≥65 y) | Prospective cohort study | Mediterranean diet rich in n-3 intake | 4 years | Mediterranean diet intake is associated with reduced risks of AD |
| 7 | DeVore et al. [221] | 5395 healthy adults (≥55 y) | Population-based cohort study | n-3 and fish intake | 9.6 years | No significant changes in memory were found between the treatment and placebo groups |
| 8 | Danthiir et al. [222] | 403 healthy elderly (65–90 y) | Double-blind, randomized, placebo-controlled | EPA: 600 mg; DHA: 1720 mg | 18 months | n-3 intake showed a positive correlation in ApoE carriers |
| No. | Study | Subjects | Study Type | Treatment/Source | Duration | Results |
|---|---|---|---|---|---|---|
| 1 | Yokoi-Shimizu et al. [241] | 66 healthy Japanese males and females | Double-blinded, placebo-controlled trial | DHA: 576 mg; EPA: 284 mg | 12 weeks | Sleep quality improved compared to placebo |
| 2 | Murphy, Rachel A. et al. [242] | 6175 adult participants with insomnia | Observational cross-sectional study | Serum fatty acid analysis | 1 year | Poor sleep quality was associated with reduced blood EPA and DHA levels |
| 3 | Yehuda et al. [243] | 126 students suffering from anxiety | Randomized controlled trial | Fatty acid mixture treatment | One month | Fatty acid mixture improved sleep quality |
| 4 | Dretsch et al. [244] | 160 US male and female soldiers (18–55 y) | Randomized, double-blind, placebo-controlled trial | 2.5 g of EPA + DHA ethyl esters/day | 60 days | Decreased daytime sleepiness |
| 5 | Ford PA et al., [245] | 8771 healthy participants | Observational longitudinal study | n-3 PUFA dietary supplement | 4 years | n-3 intake is positively correlated with sleep duration |
| 6 | Christian LM et al. [246] | 135 pregnant females | Observational longitudinal study | RBC DHA contents | 2nd trimester till partum | RBC DHA contents were positively correlated with sleep quality and duration |
| 7 | Lotrich FE et al. [247] | 104 non-depressed hepatitis C patients | Prospective observational study | AA/EPA + DHA status | 6 months | Serum n-3 status is positively correlated with sleep quality and reduced MDD |
| 8 | Liu J. et al. [248] | 581 schoolchildren (9–11 y) | Prospective cohort study | Fish consumption | 2 years | Fish intake decreased the sleep disturbance score |
| 9 | Patan. et al. [240] | 84 young healthy adults (29–45 y) | Randomized controlled trial | DHA: 900 mg EPA: 270 mg | 26 weeks | EPA and DHA intake improved sleep latency and sleep efficiency |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niazi, N.U.K.; Jiang, J.; Ou, H.; Chen, R.; Yang, Z. Sleep Deprivation and Alzheimer’s Disease: A Review of the Bidirectional Interactions and Therapeutic Potential of Omega-3. Brain Sci. 2025, 15, 641. https://doi.org/10.3390/brainsci15060641
Niazi NUK, Jiang J, Ou H, Chen R, Yang Z. Sleep Deprivation and Alzheimer’s Disease: A Review of the Bidirectional Interactions and Therapeutic Potential of Omega-3. Brain Sciences. 2025; 15(6):641. https://doi.org/10.3390/brainsci15060641
Chicago/Turabian StyleNiazi, Nasar Ullah Khan, Jiahui Jiang, Haiyan Ou, Ruiye Chen, and Zhiyou Yang. 2025. "Sleep Deprivation and Alzheimer’s Disease: A Review of the Bidirectional Interactions and Therapeutic Potential of Omega-3" Brain Sciences 15, no. 6: 641. https://doi.org/10.3390/brainsci15060641
APA StyleNiazi, N. U. K., Jiang, J., Ou, H., Chen, R., & Yang, Z. (2025). Sleep Deprivation and Alzheimer’s Disease: A Review of the Bidirectional Interactions and Therapeutic Potential of Omega-3. Brain Sciences, 15(6), 641. https://doi.org/10.3390/brainsci15060641