
GLUT1 as a Potential Therapeutic Target in Glioblastoma

 , ,
, ,
Abstract
1. Introduction
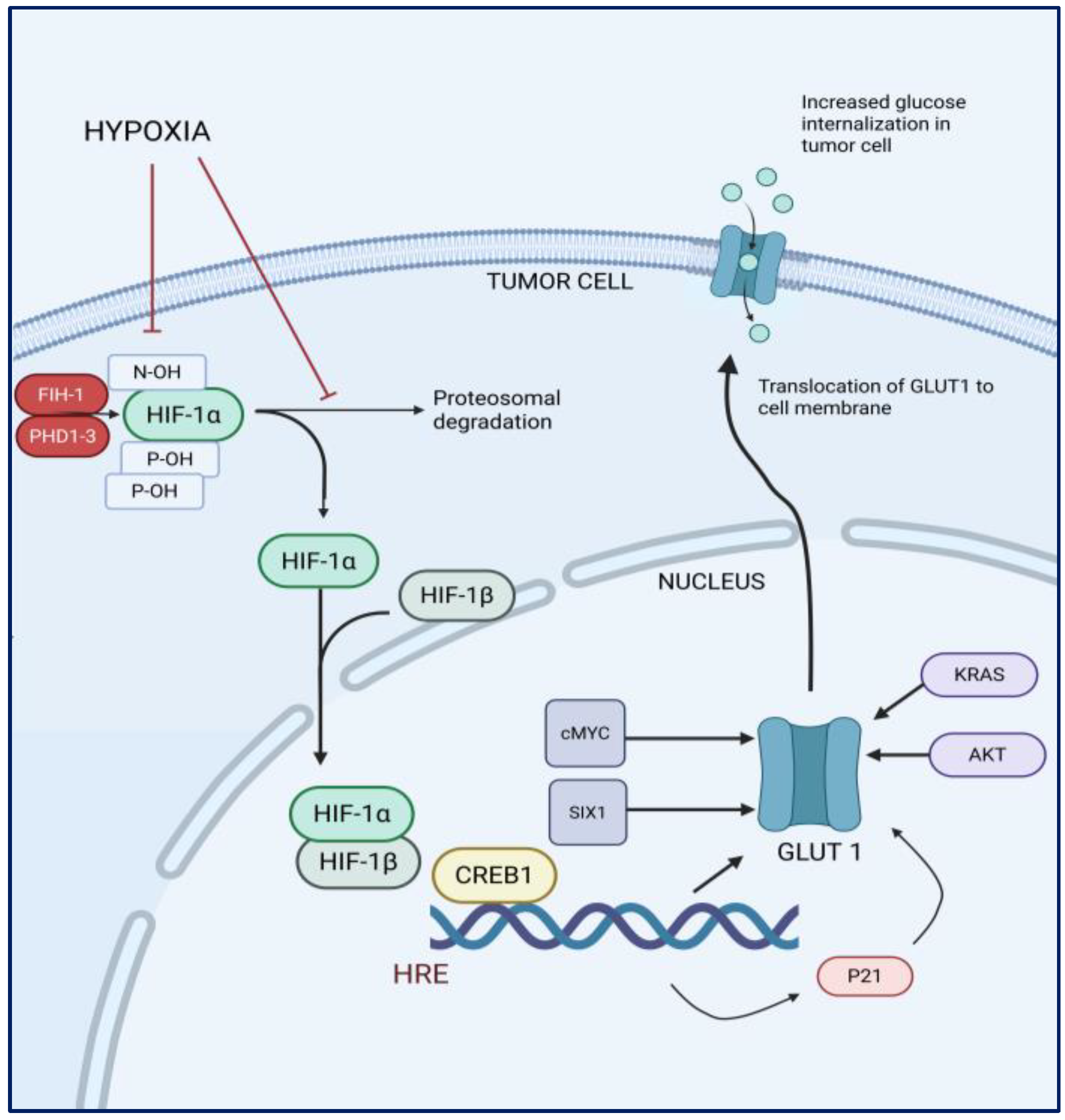
1.1. The Role of GLUT1 in Glucose Transport, Cancer Metabolism, and Tumor Progression
1.2. Hypoxia and the Expression of GLUT1 in Cancer Cells
1.3. The Role of GLUT1 in Glioblastoma
1.4. GLUT1 as a Therapeutic Target in Glioblastoma
1.5. Clinical and Prognostic Implications of GLUT1 in GBM Therapy
2. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Price, M.; Ryan, K.; Shoaf, M.L.; Neff, C.; Iorgulescu, J.B.; Landi, D.B.; Cioffi, G.; A Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S.; et al. Childhood, adolescent, and adult primary brain and central nervous system tumor statistics for practicing healthcare providers in neuro-oncology, CBTRUS 2015–2019. Neurooncol. Pract. 2023, 11, 5–25. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Xue, Q.; Liu, K.; Ge, W.; Liu, W.; Wang, J.; Zhang, M.; Li, Q.-Y.; Cai, D.; Shan, C.; et al. Dimethylaminomicheliolide (DMAMCL) suppresses the proliferation of glioblastoma cells via targeting pyruvate kinase 2 (PKM2) and rewiring aerobic glycolysis. Front. Oncol. 2019, 9, 993. [Google Scholar] [CrossRef]
- Miranda-Gonçalves, V.; Gonçalves, C.S.; Granja, S.; de Castro, J.V.; Reis, R.M.; Costa, B.M.; Baltazar, F. MCT1 is a new prognostic biomarker and its therapeutic inhibition boosts response to temozolomide in human glioblastoma. Cancers 2021, 13, 3468. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-García, A.; Samsó, P.; Fontova, P.; Simon-Molas, H.; Manzano, A.; Castaño, E.; Rosa, J.L.; Martinez-Outshoorn, U.; Ventura, F.; Navarro-Sabaté, À.; et al. TGF-β1 targets smad, p38 MAPK, and PI 3K/akt signaling pathways to induce PFKFB 3 gene expression and glycolysis in glioblastoma cells. FEBS J. 2017, 284, 3437–3454. [Google Scholar] [CrossRef]
- Wolf, A.; Agnihotri, S.; Micallef, J.; Mukherjee, J.; Sabha, N.; Cairns, R.; Hawkins, C.; Guha, A. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J. Exp. Med. 2011, 208, 313–326. [Google Scholar] [CrossRef]
- Li, J.; Zhu, S.; Tong, J.; Hao, H.; Yang, J.; Liu, Z.; Wang, Y. Suppression of lactate dehydrogenase A compromises tumor progression by downregulation of the warburg effect in glioblastoma. Neuroreport 2016, 27, 110–115. [Google Scholar] [CrossRef]
- Sanzey, M.; Abdul Rahim, S.A.; Oudin, A.; Dirkse, A.; Kaoma, T.; Vallar, L.; Herold-Mende, C.; Bjerkvig, R.; Golebiewska, A.; Niclou, S.P. Comprehensive analysis of glycolytic enzymes as therapeutic targets in the treatment of glioblastoma. PLoS ONE 2015, 10, e0123544. [Google Scholar] [CrossRef]
- Yao, Z.; Zhang, Q.; Guo, F.; Guo, S.; Yang, B.; Liu, B.; Li, P.; Li, J.; Guan, S.; Liu, X. Long noncoding RNA PCED1B-AS1 promotes the warburg effect and tumorigenesis by upregulating HIF-1α in glioblastoma. Cell Transpl. 2020, 29, 0963689720906777. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Liao, Y.; Zhao, H.; Zhang, J.; Mu, K. ANXA2P2/miR-9/LDHA axis regulates warburg effect and affects glioblastoma proliferation and apoptosis. Cell Signal 2020, 74, 109718. [Google Scholar] [CrossRef]
- Bache, M.; Rot, S.; Keßler, J.; Güttler, A.; Wichmann, H.; Greither, T.; Wach, S.; Taubert, H.; Söling, A.; Bilkenroth, U.; et al. mRNA expression levels of hypoxia-induced and stem cell-associated genes in human glioblastoma. Oncol. Rep. 2015, 33, 3155–3161. [Google Scholar] [CrossRef] [PubMed]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, X. Greasy GLUT1 maintains glioblastoma malignancy. Mol. Cell Oncol. 2021, 8, 2009423. [Google Scholar] [CrossRef]
- Guda, M.R.; Labak, C.M.; Omar, S.I.; Asuthkar, S.; Airala, S.; Tuszynski, J.; Tsung, A.J.; Velpula, K.K. GLUT1 and TUBB4 in glioblastoma could be efficacious targets. Cancers 2019, 11, 1308. [Google Scholar] [CrossRef]
- Deng, D.; Xu, C.; Sun, P.; Wu, J.; Yan, C.; Hu, M.; Yan, N. Crystal structure of the human glucose transporter GLUT1. Nature 2014, 510, 121–125. [Google Scholar] [CrossRef]
- Egert, S.; Nguyen, N.; Schwaiger, M. Myocardial glucose transporter GLUT1: Translocation induced by insulin and ischemia. J. Mol. Cell Cardiol. 1999, 31, 1337–1344. Available online: https://www.sciencedirect.com/science/article/pii/S0022282899909653 (accessed on 28 September 2024). [CrossRef]
- Rudlowski, C.; Moser, M.; Becker, A.J.; Rath, W.; Buttner, R.; Schroder, W.; Schurmann, A. GLUT1 mRNA and protein expression in ovarian borderline tumors and cancer. Oncology 2004, 66, 404–410. [Google Scholar] [CrossRef]
- Meziou, S.; Ringuette Goulet, C.; Hovington, H.; Lefebvre, V.; Lavallée, E.; Bergeron, M.; Brisson, H.; Champagne, A.; Neveu, B.; Lacombe, D.; et al. GLUT1 expression in high-risk prostate cancer: Correlation with 18F-FDG-PET/CT and clinical outcome. Prostate Cancer Prostatic Dis. 2020, 23, 441–448. [Google Scholar] [CrossRef]
- Krzeslak, A.; Wojcik-Krowiranda, K.; Forma, E.; Jozwiak, P.; Romanowicz, H.; Bienkiewicz, A.; Brys, M. Expression of GLUT1 and GLUT3 glucose transporters in endometrial and breast cancers. Pathol. Oncol. Res. 2012, 18, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Yongzhi, H.; Chen, S.; Luo, X.; Lin, Y.; Zhou, Y.; Jin, H.; Hou, B.; Deng, Y.; Tu, L.; et al. The prognostic value of GLUT1 in cancers: A systematic review and meta-analysis. Oncotarget 2017, 8, 43356–43367. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Gao, P.; Liu, Y.; Semenza, G.L.; Dang, C.V. Hypoxia-inducible factor 1 and dysregulated c-myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol. Cell Biol. 2007, 27, 7381–7393. [Google Scholar] [CrossRef]
- Gordan, J.D.; Bertout, J.A.; Hu, C.; Diehl, J.A.; Simon, M.C. HIF-2α promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell 2007, 11, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Koshiji, M.; Kageyama, Y.; Pete, E.A.; Horikawa, I.; Barrett, J.C.; Huang, L.E. HIF-1α induces cell cycle arrest by functionally counteracting myc. EMBO J. 2004, 23, 1949–1956. [Google Scholar] [CrossRef]
- Zeng, M.; Kikuchi, H.; Pino, M.S.; Chung, D.C. Hypoxia activates the K-ras proto-oncogene to stimulate angiogenesis and inhibit apoptosis in colon cancer cells. PLoS ONE 2010, 5, e10966. [Google Scholar] [CrossRef]
- Lee, S.; Lee, C.; Kim, Y.W.; Han, S.K.; Shim, Y.; Yoo, C. Hypoxia confers protection against apoptosis via PI3K/akt and ERK pathways in lung cancer cells. Cancer Lett. 2006, 242, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Jiao, M.; Nan, K. Activation of PI3 kinase/akt/HIF-1α pathway contributes to hypoxia-induced epithelial-mesenchymal transition and chemoresistance in hepatocellular carcinoma. Int. J. Oncol. 2012, 40, 461–468. [Google Scholar]
- Wofford, J.A.; Wieman, H.L.; Jacobs, S.R.; Zhao, Y.; Rathmell, J.C. IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of akt to support T-cell survival. Blood J. Am. Soc. Hematol. 2008, 111, 2101–2111. [Google Scholar] [CrossRef]
- Melstrom, L.G.; Salabat, M.R.; Ding, X.-Z.; Milam, B.M.; Strouch, M.; Pelling, J.C.; Bentrem, D.J. Apigenin inhibits the GLUT-1 glucose transporter and the phosphoinositide 3-kinase/akt pathway in human pancreatic cancer cells. Pancreas 2008, 37, 426–431. [Google Scholar] [CrossRef]
- Grimm, M.; Munz, A.; Teriete, P.; Nadtotschi, T.; Reinert, S. GLUT-1/TKTL1 coexpression predicts poor outcome in oral squamous cell carcinoma. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2014, 117, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.E.; Jung, W.; Koo, J.S. The expression of metabolism-related proteins in phyllodes tumors. Tumor Biol. 2013, 34, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Komaki, S.; Sugita, Y.; Furuta, T.; Yamada, K.; Moritsubo, M.; Abe, H.; Akiba, J.; Miyagi, N.; Nakamura, H.; Miyoshi, H.; et al. Expression of GLUT1 in pseudopalisaded and perivascular tumor cells is an independent prognostic factor for patients with glioblastomas. J. Neuropathol. Exp. Neurol. 2019, 78, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Khan, F.; Lin, Y.; Ali, H.; Pang, L.; Dunterman, M.; Hsu, W.-H.; Frenis, K.; Rowe, R.G.; Wainwright, D.A.; McCortney, K.; et al. Lactate dehydrogenase A regulates tumor-macrophage symbiosis to promote glioblastoma progression. Nat. Commun. 2024, 15, 1987. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Chaumeil, M.M.; Ward, C.S.; Haas-Kogan, D.A.; James, C.D.; Ronen, S.M. Reduced phosphocholine and hyperpolarized lactate provide magnetic resonance biomarkers of PI3K/akt/mTOR inhibition in glioblastoma. Neurooncology 2012, 14, 315–325. [Google Scholar] [CrossRef]
- Lim, K.S.; Lim, K.J.; Price, A.C.; Orr, B.A.; Eberhart, C.G.; Bar, E.E. Inhibition of monocarboxylate transporter-4 depletes stem-like glioblastoma cells and inhibits HIF transcriptional response in a lactate-independent manner. Oncogene 2014, 33, 4433–4441. [Google Scholar] [CrossRef]
- Semenza, G.L.; Jiang, B.-H.; Leung, S.W.; Passantino, R.; Concordet, J.-P.; Maire, P.; Giallongo, A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J. Biol. Chem. 1996, 271, 32529–32537. [Google Scholar] [CrossRef]
- Fong, G.; Takeda, K. Role and regulation of prolyl hydroxylase domain proteins. Cell Death Differ. 2008, 15, 635–641. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci. STKE 2007, 2007, cm8. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1 and tumor progression: Pathophysiology and therapeutics. Trends Mol. Med. 2002, 8, S62–S67. [Google Scholar] [CrossRef] [PubMed]
- Lal, A.; Peters, H.; St Croix, B.; Haroon, Z.A.; Dewhirst, M.W.; Strausberg, R.L.; Kaanders, J.H.; Van Der Kogel, A.J.; Riggins, G.J. Transcriptional response to hypoxia in human tumors. J. Natl. Cancer Inst. 2001, 93, 1337–1343. [Google Scholar]
- Schwartzenberg-Bar-Yoseph, F.; Armoni, M.; Karnieli, E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004, 64, 2627–2633. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liang, Y.; Kang, L.; Liu, Y.; Gao, S.; Chen, S.; Li, Y.; You, W.; Dong, Q.; Hong, T.; et al. Transcriptional regulation of the warburg effect in cancer by SIX1. Cancer Cell 2018, 33, 368–385.e7. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, C.; Mi, Y.; Chen, F.; Du, D. CREB1 regulates glucose transport of glioma cell line U87 by targeting GLUT1. Mol Cell Biochem. 2017, 436, 79–86. [Google Scholar] [CrossRef]
- Jin, X.; Kuang, Y.; Li, L.; Li, H.; Zhao, T.; He, Y.; Di, C.; Kang, J.; Yuan, L.; Yu, B.; et al. A positive feedback circuit comprising p21 and HIF-1α aggravates hypoxia-induced radioresistance of glioblastoma by promoting Glut1/LDHA-mediated glycolysis. FASEB J. 2022, 36, e22229. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, X.; Yang, F.; Chen, C.; Liu, P.; Ren, Y.; Sun, P.; Wang, Z.; You, Y.; Zeng, Y.-X.; et al. DHHC9-mediated GLUT1 S-palmitoylation promotes glioblastoma glycolysis and tumorigenesis. Nat. Commun. 2021, 12, 5872–5874. [Google Scholar] [CrossRef]
- Guo, H.; Nan, Y.; Zhen, Y.; Zhang, Y.; Guo, L.; Yu, K.; Huang, Q.; Zhong, Y. miRNA-451 inhibits glioma cell proliferation and invasion by downregulating glucose transporter 1. Tumor Biol. 2016, 37, 13751–13761. [Google Scholar] [CrossRef]
- Nie, S.; Li, K.; Huang, Y.; Hu, Q.; Gao, X.; Jie, S. miR-495 mediates metabolic shift in glioma cells via targeting Glut1. J. Craniofacial Surg. 2015, 26, e155–e158. [Google Scholar] [CrossRef]
- Zhang, R.; Luo, H.; Wang, S.; Chen, W.; Chen, Z.; Wang, H.-W.; Chen, Y.; Yang, J.; Zhang, X.; Wu, W.; et al. MicroRNA-377 inhibited proliferation and invasion of human glioblastoma cells by directly targeting specificity protein 1. Neurooncology 2014, 16, 1510–1522. [Google Scholar] [CrossRef]
- Yin, J.; Shi, Z.; Wei, W.; Lu, C.; Wei, Y.; Yan, W.; Li, R.; Zhang, J.; You, Y.; Wang, X. MiR-181b suppress glioblastoma multiforme growth through inhibition of SP1-mediated glucose metabolism. Cancer Cell Int. 2020, 20, 69. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Y.; Zhu, H.; Chen, X.; Tian, M.; Wei, Y.; Gong, Y.; Jiang, J. N-acetylglucosaminyltransferase I promotes glioma cell proliferation and migration through increasing the stability of the glucose transporter GLUT1. FEBS Lett. 2020, 594, 358–366. [Google Scholar] [CrossRef]
- Cheng, C.; Tu, J.; Hu, Z.; Chen, Y.; Wang, Y.; Zhang, T.; Zhang, C.; Li, C.; Wang, Y.; Niu, C. SREBP2/Rab11s/GLUT1/6 network regulates proliferation and migration of glioblastoma. Pathol. Res. Pract. 2022, 240, 154176. Available online: https://www.sciencedirect.com/science/article/pii/S0344033822004204 (accessed on 27 September 2024). [CrossRef] [PubMed]
- Azzalin, A.; Nato, G.; Parmigiani, E.; Garello, F.; Buffo, A.; Magrassi, L. Inhibitors of GLUT/SLC2A enhance the action of BCNU and temozolomide against high-grade gliomas. Neoplasia 2017, 19, 364–373. [Google Scholar] [CrossRef]
- Atif, F.; Yousuf, S.; Espinosa-Garcia, C.; Sergeeva, E.; Stein, D.G. Progesterone treatment attenuates glycolytic metabolism and induces senescence in glioblastoma. Sci. Rep. 2019, 9, 988. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Xu, D.; Ruan, Z.; Zhou, J.; Sun, W.; Rao, B.; Xu, H. Metabolism/immunity dual-regulation thermogels potentiating immunotherapy of glioblastoma through lactate-excretion inhibition and PD-1/PD-L1 blockade. Adv. Sci. 2024, 11, e2310163. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, K.; Okada, M.; Suzuki, S.; Seino, M.; Seino, S.; Takeda, H.; Kitanaka, C. Targeting the facilitative glucose transporter GLUT1 inhibits the self-renewal and tumor-initiating capacity of cancer stem cells. Oncotarget 2015, 6, 651–661. [Google Scholar] [CrossRef]
- Landis, C.J.; Zhang, S.; Benavides, G.A.; Scott, S.E.; Li, Y.; Redmann, M.; Tran, A.N.; Otamias, A.; Darley-Usmar, V.; Napierala, M.; et al. Identification of compounds that decrease glioblastoma growth and glucose uptake in vitro. ACS Chem. Biol. 2018, 13, 2048–2057. [Google Scholar] [CrossRef]
- Leone, S.; Fiore, M.; Lauro, M.G.; Pino, S.; Cornetta, T.; Cozzi, R. Resveratrol and X rays affect gap junction intercellular communications in human glioblastoma cells. Mol. Carcinog. 2008, 47, 587–598. [Google Scholar] [CrossRef]
- Yang, Y.; Chang, Y.; Huang, P.; Chiou, G.; Tseng, L.; Chiou, S.; Chen, M.; Shih, Y.; Chang, C.; Hsu, C.; et al. Resveratrol suppresses tumorigenicity and enhances radiosensitivity in primary glioblastoma tumor initiating cells by inhibiting the STAT3 axis. J. Cell Physiol. 2012, 227, 976–993. [Google Scholar] [CrossRef]
- Wang, L.; Long, L.; Wang, W.; Liang, Z. Resveratrol, a potential radiation sensitizer for glioma stem cells both in vitro and in vivo. J. Pharmacol. Sci. 2015, 129, 216–225. [Google Scholar] [CrossRef]
- Khoei, S.; Shoja, M.; Mostaar, A.; Faeghi, F. Effects of resveratrol and methoxyamine on the radiosensitivity of iododeoxyuridine in U87MG glioblastoma cell line. Exp. Biol. Med. 2016, 241, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Öztürk, Y.; Günaydın, C.; Yalçın, F.; Nazıroğlu, M.; Braidy, N. Resveratrol enhances apoptotic and oxidant effects of paclitaxel through TRPM2 channel activation in DBTRG glioblastoma cells. Oxidative Med. Cell. Longev. 2019, 2019, 4619865. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Lin, H.; Zhang, X.; Li, J. Resveratrol reverses temozolomide resistance by downregulation of MGMT in T98G glioblastoma cells by the NF-κB-dependent pathway. Oncol. Rep. 2012, 27, 2050–2056. [Google Scholar] [PubMed]
- Li, H.; Liu, Y.; Jiao, Y.; Guo, A.; Xu, X.; Qu, X.; Wang, S.; Zhao, J.; Li, Y.; Cao, Y. Resveratrol sensitizes glioblastoma-initiating cells to temozolomide by inducing cell apoptosis and promoting differentiation. Oncol. Rep. 2016, 35, 343–351. [Google Scholar] [CrossRef]
- Yang, H.; Wang, J.; Bu, X.; Yang, B.; Wang, B.; Hu, S.; Yan, Z.; Gao, Y.; Han, S.; Qu, M. Resveratrol restores sensitivity of glioma cells to temozolamide through inhibiting the activation of wnt signaling pathway. J. Cell Physiol. 2019, 234, 6783–6800. [Google Scholar] [CrossRef]
- Liu, Y.; Song, X.; Wu, M.; Wu, J.; Liu, J. Synergistic effects of resveratrol and temozolomide against glioblastoma cells: Underlying mechanism and therapeutic implications. Cancer Manag. Res. 2020, 12, 8341–8354. [Google Scholar] [CrossRef]
- Yuan, Y.; Xue, X.; Guo, R.; Sun, X.; Hu, G. Resveratrol enhances the antitumor effects of temozolomide in glioblastoma via ROS-dependent AMPK-TSC-mTOR signaling pathway. CNS Neurosci. Ther. 2012, 18, 536–546. [Google Scholar] [CrossRef]
- Lin, C.-J.; Lee, C.-C.; Shih, Y.-L.; Lin, T.-Y.; Wang, S.-H.; Lin, Y.-F.; Shih, C.-M. Resveratrol enhances the therapeutic effect of temozolomide against malignant glioma in vitro and in vivo by inhibiting autophagy. Free Radic. Biol. Med. 2012, 52, 377–391. [Google Scholar] [CrossRef]
- Arabzadeh, A.; Mortezazadeh, T.; Aryafar, T.; Gharepapagh, E.; Majdaeen, M.; Farhood, B. Therapeutic potentials of resveratrol in combination with radiotherapy and chemotherapy during glioblastoma treatment: A mechanistic review. Cancer Cell Int. 2021, 21, 391. [Google Scholar] [CrossRef]
- Liu, Y.; Tong, L.; Luo, Y.; Li, X.; Chen, G.; Wang, Y. Resveratrol inhibits the proliferation and induces the apoptosis in ovarian cancer cells via inhibiting glycolysis and targeting AMPK/mTOR signaling pathway. J. Cell Biochem. 2018, 119, 6162–6172. [Google Scholar] [CrossRef] [PubMed]
- Kueck, A.; Opipari, A.W., Jr.; Griffith, K.A.; Tan, L.; Choi, M.; Huang, J.; Wahl, H.; Liu, J.R. Resveratrol inhibits glucose metabolism in human ovarian cancer cells. Gynecol. Oncol. 2007, 107, 450–457. [Google Scholar] [CrossRef]
- Salas, M.; Obando, P.; Ojeda, L.; Ojeda, P.; Pérez, A.; Vargas-Uribe, M.; Rivas, C.I.; Vera, J.C.; Reyes, A.M. Resolution of the direct interaction with and inhibition of the human GLUT1 hexose transporter by resveratrol from its effect on glucose accumulation. Am. J. Physiol. Cell Physiol. 2013, 305, 90. [Google Scholar] [CrossRef] [PubMed]
- Kleszcz, R.; Paluszczak, J.; Krajka-Kuźniak, V.; Baer-Dubowska, W. The inhibition of c-MYC transcription factor modulates the expression of glycolytic and glutaminolytic enzymes in FaDu hypopharyngeal carcinoma cells. Adv. Clin. Exp. Med. 2018, 27, 735–742. [Google Scholar] [CrossRef]
- Howells, L.M.; Berry, D.P.; Elliott, P.J.; Jacobson, E.W.; Hoffmann, E.; Hegarty, B.; Brown, K.; Steward, W.P.; Gescher, A.J. Phase I randomized, double-blind pilot study of micronized resveratrol (SRT501) in patients with hepatic metastases--safety, pharmacokinetics, and pharmacodynamics. Cancer Prev. Res. 2011, 4, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Holcombe, R.F.; Nguyen, A.V.; Martinez, M.; Stamos, M.J.; Moyer, M.P.; Planutis, K.; Hope, C. Results of a phase I pilot clinical trial examining the effect of plant-derived resveratrol and grape powder on wnt pathway target gene expression in colonic mucosa and colon cancer. Cancer Manag. Res. 2009, 1, 25–37. [Google Scholar] [CrossRef]
- Popat, R.; Plesner, T.; Davies, F.; Cook, G.; Cook, M.; Elliott, P.; Jacobson, E.; Gumbleton, T.; Oakervee, H.; Cavenagh, J. A phase 2 study of SRT501 (resveratrol) with bortezomib for patients with relapsed and or refractory multiple myeloma. Br. J. Haematol. 2013, 160, 714–717. [Google Scholar] [CrossRef]
- Resveratrol in Treating Patients with Colorectal Cancer That Can be Removed by Surgery. Available online: https://clinicaltrials.gov/study/NCT00433576 (accessed on 28 September 2024).
- Prosniak, M.; Kenyon, L.C.; Hooper, D.C. Glioblastoma contains topologically distinct proliferative and metabolically defined subpopulations of nestin- and Glut1-expressing cells. J. Neuropathol. Exp. Neurol. 2021, 80, 674–684. [Google Scholar] [CrossRef]
- Shen, H.; Hau, E.; Joshi, S.; Dilda, P.J.; McDonald, K.L. Sensitization of glioblastoma cells to irradiation by modulating the glucose metabolism. Mol. Cancer Ther. 2015, 14, 1794–1804. [Google Scholar] [CrossRef]
- Lan, F.; Qin, Q.; Yu, H.; Yue, X. Effect of glycolysis inhibition by miR-448 on glioma radiosensitivity. J. Neurosurg. JNS 2020, 132, 1456–1464. Available online: https://thejns.org/view/journals/j-neurosurg/132/5/article-p1456.xml (accessed on 28 September 2024). [CrossRef]
- Li, Y.; Ge, Y.; Zhao, M.; Ding, F.; Wang, X.; Shi, Z.; Ge, X.; Wang, X.; Qian, X. HSP90B1-mediated plasma membrane localization of GLUT1 promotes radioresistance of glioblastomas. J. Biomed. Res. 2023, 37, 326. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, C.; Kamitori, K.; Hossain, A.; Hoshikawa, H.; Katagi, A.; Dong, Y.; Sui, L.; Tokuda, M.; Yamaguchi, F. D-allose inhibits cancer cell growth by reducing GLUT1 expression. Tohoku J. Exp. Med. 2016, 238, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Ning, K.; Lu, T.; Hua, D. Elevated expression of TrpC5 and GLUT1 is associated with chemoresistance in colorectal cancer. Oncol. Rep. 2017, 37, 1059–1065. [Google Scholar] [CrossRef]
- Song, K.; Li, M.; Xu, X.J.; Xuan, L.; Huang, G.N.; Song, X.L.; Liu, Q.F. HIF-1α and GLUT1 gene expression is associated with chemoresistance of acute myeloid leukemia. Asian Pac. J. Cancer Prev. 2014, 15, 1823–1829. [Google Scholar] [CrossRef] [PubMed]
- Van Wijk, R.; Van Solinge, W.W. The energy-less red blood cell is lost: Erythrocyte enzyme abnormalities of glycolysis. Blood 2005, 106, 4034–4042. [Google Scholar] [CrossRef]
- Evans, A.; Bates, V.; Troy, H.; Hewitt, S.; Holbeck, S.; Chung, Y.-L.; Phillips, R.; Stubbs, M.; Griffiths, J.; Airley, R. Glut-1 as a therapeutic target: Increased chemoresistance and HIF-1-independent link with cell turnover is revealed through COMPARE analysis and metabolomic studies. Cancer Chemother. Pharmacol. 2008, 61, 377–393. [Google Scholar] [CrossRef]
- Jackson, C.; Cherry, C.; Bom, S.; Dykema, A.G.; Wang, R.; Thompson, E.; Zhang, M.; Li, R.; Ji, Z.; Hou, W.; et al. Distinct myeloid-derived suppressor cell populations in human glioblastoma. Science 2025, 387, eabm5214. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Study, Year | Type of Study | Cell Line | Animal Model | Drug | Mechanism of Action | Dose | Results |
|---|---|---|---|---|---|---|---|
| Guda et al., 2019 [16] | In vitro | GSC33 and GSC28 | N/A | Fasentin | TUBB4 inhibitor | Fasentin at 25 µM and 50 µM dose | Decreased sphere formation over 15 days of treatment |
| Azzalin et al., 2017 [54] | In vitro and In vivo |
U87MG, Hu197 (stable human GBM cell line) and GBM-P1 (primary human GBM cell culture) And GL261 (mouse GBM cell line) | C57BlC inoculated with GL261 GBM cells orthotopic GBM mouse model |
Indinavir (IDV) and Ritonavir (RTV) in combination with TMZ and BCNU | Inhibition of GLUT1/SLC2A transporter | 45 µM and 55 µM of RTV in vitro and BCNU alone (2.5 mg/kg), a combination of RTV (100 mg/kg) and BCNU (1.5 mg/kg), or a combination of RTV (100 mg/kg) and BCNU (2.5 mg/kg) in vivo | Median survival was approximately 18 days for the BCNU-only group and about 37 days for the BCNU 1 + RTV group. RTV but not IDV decreased glycolytic activity and cell growth in vitro. Mice treated with RTV + BCNU have improved survival than BCNU alone, also enhanced |
| Atif et al., 2019 [55] | In vivo | U118MG (Human GBM cell line), U87dEGFR (modified human cell line) and U87MG-luc | Adult male athymic nude mice inoculated with U87MG-luc orthotopic GBM mouse model | Progesterone | Inhibition of GLUT1, GAPDH and cytoplasmic FoxO1 activity | 10–80 µM in vitro and low dose (8 mg/kg) and high dose (100 mg/kg) in vivo | Progesterone treatment significantly enhanced the survival of tumor-bearing mice by 43% compared to vehicle-treated mice without toxicity |
| Tianliang et al., 2024 [56] | In vitro and In vivo | Mouse GBM cells of GL261, Human GBM cell lines of U87 and U251, Mouse macrophage cell lines of RAW264.7 | C57BL/6J mice subcutaneously injected with GL261 cells And C57BL/6J mice inoculated with GL261 or GL261-luc orthotopic GBM mouse model | BAY-876 and PD-1/PD-l1 blocker BMS-1 | Inhibitor of GLUT1 | 1, 2, 4 ng mL−1 of BAY-876 in vitro and 2 nm of BAY-876 in vivo | The median survival of tumor-bearing mice treated with BAY-876 increased to 36.5 days compared to 28.5 days in the control group. Treatment with BMS-1 further prolonged survival to 45 days (endpoint) |
| Shibuya et al., 2015 [57] | In vitro and In vivo | GS-Y03 GBM stem cell and other cancer stem cells (PANC-1 CSLC, and A2780) | male BALB/cAJcl-nu/nu mice subcutaneously implanted with GS-Y03 cell line and other cancer stem cells | WZB117 | GLUT1 inhibitor | WZB117 4 mg/mL in DMSO was diluted in PBS to prepare 200 μL for each injection in vivo | In vivo, the tumor size between control and WZB117-treated tumor-bearing mice was statistically significant, with the treated group having a smaller tumor volume. |
| Landis et al., 2018 [58] | In vitro | GBM (PDX) D456, 1016, JX12, and JX14 | N/A | SRI-37683 and SRI-37684 | GLUT 1 inhibitor | 50 µM SRI-37683 and SRI-37684 | There was a 40–50% inhibition of glucose uptake in all tested cell lines |
| Leone S. et al., 2008 [59] | In vitro | U-87MG | N/A | Resveratrol | GLUT1, STAT3 inhibitor | 20 µM of resveratrol and 5 Gy; 180 KV X-rays | 20 µM of resveratrol and 5 Gy; 180 KV X-rays |
| Yand YP et al., 2012 [60] | In vitro and In vivo | CD133 and CD133+ cells from a patient | SCID mice (BALB/c strain) orthotopic GBM mouse model | Resveratrol | GLUT1, STAT3 inhibitor | RV 100 µM 2, 4, 6, 8 and 10 Gy | Suppression of STAT3 and induction of apoptosis of tumor cells. Treatment group median survival was 10 weeks vs. 4 weeks in the control group |
| Wang L et al., 2015 [61] | In vitro and in vivo | Human Glioma SU-2 patient derived | male nude (BALB/c) mice | Resveratrol | GLUT1, STAT3 inhibitor | RV 75 µmol/L (for in vitro) and 150 mg/kg/day (for in vivo) ip, and 2, 4 and 6 Gy; 6 MV X-rays | Increased radiosensitivity of cells, prevention of renewal and stemness, increased apoptosis, inhibition of DNA repair. Decreased relative tumor volume in nude mice in treated group |
| Khoei S et al., 2016 [62] | In vitro | U87MG GBM cell line | N/A | Resveratrol | GLUT1, STAT3 inhibitor | RV 20 µM and 2 Gy; 1.25 MeV | Decreased colony number, increased DNA damage and increased radiosensitivity |
| Ozturk Y et al., 2019 [63] | In vitro | DBTRG GBM cell line | N/A | Resveratrol | GLUT1, STAT3 inhibitor | RV 50 µM and 50 µM of paclitaxal | Increased mitochondrial ROS levels and activation of TRMP2 channel, increased caspase activity and Ca+ influx through TRMP2 channel |
| Huang H. et al., 2012 [64] | In vitro | T98G GBM cell line | N/A | Resveratrol | GLUT1, STAT3 inhibitor | RV 100 µM and TMZ 100 µM | Increased chemosensitivity, increased apoptosis, decreased intracellular translocation of NF-kB, and repression of MGMT expression |
| Li H et al., 2016 [65] | In vitro and in vivo | GBM initiating cells (GICs) from 2 patients | female NOD/SCID mice inoculated by GICs | Resveratrol | GLUT1, STAT3 inhibitor | RV 20 and 40 µM and TMZ; 200 and 400 µM (for in vitro); RV 12.5 mg/kg/day ip and TMZ 68 mg/kg/day (in vivo); oral | Induction of apoptosis and activation of p53/pATR/pATM/DSBs pathways, inhibition of self renewal and decreased cell stemness, inactivation of STAT3 and decreased tumor volume |
| Yang HC et al., 2019 [66] | In vitro and in vivo | U251, T98G, U138, A172, LN229, and normal human astrocytes | N/A | Resveratrol | GLUT1, STAT3 inhibitor | RV 2, 4, 8, 10, 16 and 32 µM and TMZ; 400 µM (in vitro); RV 10 mg/kg/day; ip and TMZ 25 mg/kg//day (in vivo); ip | Decreased cell viability and proliferation, increased apoptosis, supression of Wnt pathway and repression of MGMT expression |
| Liu Y et al., 2020 [67] | In vitro | RG-2, LN-18, LN-428 | NA | Resveratrol | GLUT1, STAT3 inhibitor | RV 25, 50, 75 and 100 µM and TMZ 250, 500, 750 and 1000 µM | Inhibition of cells growth repression of MGMT expression, decreased STAT3 decreased Bcl2 |
| Yuan Y et al., 2012 [68] | In vitro and in vivo | SHG44 GBM cell line | Female BALB/cA nude mice inoculated with SHG44 orthotropic glioma model | Resveratrol | GLUT1, STAT3 inhibitor | RV 10 µM and TMZ 100 µM (in vitro); RV 40 mg/kg/day ip and TMZ 68 mg/kg/day (in vivo) oral | Cell cycle arrest, downregulation of MMP9 expression, inhibition of cell migration, increased mitochondrial ROS, downregulation og Bcl2, inhibition of mTOR and increased expression of GFAP. Decreased tumor volume |
| Lin CJ et al., 2012 [69] | In vitro and in vivo | U87 MG and GBM8401 GBM cell lines | female nude mice BALB/c nu/nu subcutaneously injected with U87MG cells | Resveratrol | GLUT1, STAT3 inhibitor | RV 10 µM and TMZ; 100–400 µM (in vitro); RV 12.5 mg/kg/day ip and TMZ 10 mg/kg/day (in vivo) ip | Increased cell death, increased apoptosis, chemosensitivity. Decreased tumor volume. Decreased ERK activity and LC3-II protein levels and increased cleavage of PARP |
| Trial ID | Year | Cancer Type | Resveretrol Dose | Phase | Country | Results |
|---|---|---|---|---|---|---|
| NCT01476592 | 2011 | Low grade gastrointestinal tumors | 2.5 g/2 times a day for three cycles, oral | Not applicable | USA | Study completed in 2017 but results are unavailable |
| NCT00920803 [75] | 2008 | Colorectal cancer and hepatic metastasis | SRT501 5.0 g daily for 14 days, oral | Phase 1 | UK | Hepatic tissue was found to contain resveratrol |
| NCT00256334 [76] | 2005 | Colon cancer | Four dose cohorts—plant derived resveratrol tablets 80 mg/day or 20 mg/day or Freeze dried grape powder (GP) (containing reveretrol) at 120 mg/day or 80 mg/day, oral | Phase 1 | USA | GP was successful in tarteting and inhibiting Wnt target gene, |
| NCT00920556 [77] | 2009 | Multiple Myeloma | SRT501 5 g/day for 20 days in a 21 day cycle for upto 12 cycles, oral | Phase 2 | UK | Unacceptable safety profile with minimal efficacy in recurrent multiple myeloma. |
| NCT00433576 [78] | 2006 | Colorectal cancer | Oral resveretrol for 9 days | Phase 1 | USA | Study completed in 2009 but results are unavailable. The objective of the study was to determine oral dose and colon mucosal levels and correlate levels of COX-2 and M1G in colorectal cancer tissue and WBCs |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruchika, F.; Suvarnapathaki, S.; Serrano-Farias, A.; Bettegowda, C.; Rincon-Torroella, J. GLUT1 as a Potential Therapeutic Target in Glioblastoma. Brain Sci. 2025, 15, 585. https://doi.org/10.3390/brainsci15060585
Ruchika F, Suvarnapathaki S, Serrano-Farias A, Bettegowda C, Rincon-Torroella J. GLUT1 as a Potential Therapeutic Target in Glioblastoma. Brain Sciences. 2025; 15(6):585. https://doi.org/10.3390/brainsci15060585
Chicago/Turabian StyleRuchika, FNU, Sanika Suvarnapathaki, Antolin Serrano-Farias, Chetan Bettegowda, and Jordina Rincon-Torroella. 2025. "GLUT1 as a Potential Therapeutic Target in Glioblastoma" Brain Sciences 15, no. 6: 585. https://doi.org/10.3390/brainsci15060585
APA StyleRuchika, F., Suvarnapathaki, S., Serrano-Farias, A., Bettegowda, C., & Rincon-Torroella, J. (2025). GLUT1 as a Potential Therapeutic Target in Glioblastoma. Brain Sciences, 15(6), 585. https://doi.org/10.3390/brainsci15060585