
Modeling Alzheimer’s Disease: A Review of Gene-Modified and Induced Animal Models, Complex Cell Culture Models, and Computational Modeling




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Genetic Factors Associated with the Development of Alzheimer’s Disease
3. Transgenic Animal Models
3.1. Mouse Models
3.2. Rat Models of Alzheimer’s Disease
3.3. Large Animal Models
3.4. Lower Vertebrate and Invertebrate Animal Models
3.4.1. Zebrafish Models
3.4.2. Drosophila melanogaster Models
3.4.3. Caenorhabditis elegans Models
4. Induced Models
4.1. Induction of Alzheimer’s Disease Symptoms by Chemicals
4.2. Stereotactic Administration of Various Forms of Tau and Aβ
4.3. Stereotactic Delivery of Vectors for Expression of Genes Associated with Alzheimer’s Disease
5. Cell Culture Models
5.1. Three-Dimensional Modeling of Cell Cultures
5.1.1. Three-Dimensional Framework Models
5.1.2. Spheroids
5.1.3. Organoids
5.1.4. Microfluidic Systems: Organs-on-Chips
6. Computational Approaches to Modeling Alzheimer’s Disease: In Silico Methods
6.1. Computational Modeling of Protein Interactions and Molecular Dynamics
6.2. Systematic Methodologies for the Analysis of Gene and Gene Expression Data
6.3. Omics Technologies
6.4. Artificial Intelligence Methods in Alzheimer’s Disease Research
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAVs | adeno-associated viruses |
| Aβ | amyloid β |
| ADNI | Alzheimer’s Disease Neuroimaging Initiative |
| AIBL | Australian Imaging Biomarkers and Lifestyle Study of Ageing |
| AI | artificial intelligence |
| AICD | APP intracellular domain |
| ANN | artificial neural network |
| APP | amyloid precursor protein |
| BBB | blood–brain barrier |
| DL | deep learning |
| FAD | familial Alzheimer’s disease |
| LOAD | late-onset Alzheimer’s disease |
| ML | machine learning |
| NACC | National Alzheimer’s Coordinating Center |
| NFT | neurofibrillary tangles |
| OASIS | Open Access Series of Imaging Studies |
| OKA | okadaic acid |
| PSEN | presenilin |
| iPSCs | induced pluripotent stem cells |
| XAI | Explainable Artificial Intelligence |
References
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; Tredici, K.D.; et al. Correlation of Alzheimer Disease Neuropathologic Changes with Cognitive Status: A Review of the Literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef]
- Duyckaerts, C.; Delatour, B.; Potier, M.C. Classification and Basic Pathology of Alzheimer Disease. Acta Neuropathol. 2009, 118, 5–36. [Google Scholar] [CrossRef] [PubMed]
- Nisbet, R.M.; Polanco, J.C.; Ittner, L.M.; Götz, J. Tau Aggregation and Its Interplay with Amyloid-β. Acta Neuropathol. 2015, 129, 207–220. [Google Scholar] [CrossRef]
- Busche, M.A.; Hyman, B.T. Synergy between Amyloid-β and Tau in Alzheimer’s Disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Mayeux, R.; Stern, Y. Epidemiology of Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006239. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Ren, R.J.; Zhong, Z.L.; Dammer, E.; Zhao, Q.H.; Shan, S.; Zhou, Z.; Li, X.; Zhang, Y.Q.; Cui, H.L.; et al. Mutation Profile of APP, PSEN1, and PSEN2 in Chinese Familial Alzheimer’s Disease. Neurobiol. Aging 2019, 77, 154–157. [Google Scholar] [CrossRef]
- Jia, L.; Fu, Y.; Shen, L.; Zhang, H.; Zhu, M.; Qiu, Q.; Wang, Q.; Yan, X.; Kong, C.; Hao, J.; et al. PSEN1, PSEN2, and APP Mutations in 404 Chinese Pedigrees with Familial Alzheimer’s Disease. Alzheimer’s Dement. 2020, 16, 178–191. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Wilke, C.; Jansen, I.E.; Schulte, C.; Simón-Sánchez, J.; Metzger, F.G.; Bender, B.; Gasser, T.; Maetzler, W.; Rizzu, P.; et al. Pilot Whole-Exome Sequencing of a German Early-Onset Alzheimer’s Disease Cohort Reveals a Substantial Frequency of PSEN2 Variants. Neurobiol. Aging 2016, 37, e11–e208.e17. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased Clearance of CNS β-Amyloid in Alzheimer’s Disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Zhao, N.; Caulfield, T.R.; Liu, C.C.; Bu, G. Apolipoprotein E and Alzheimer Disease: Pathobiology and Targeting Strategies. Nat. Rev. Neurol. 2019, 15, 501–518. [Google Scholar] [CrossRef]
- Neu, S.C.; Pa, J.; Kukull, W.; Beekly, D.; Kuzma, A.; Gangadharan, P.; Wang, L.S.; Romero, K.; Arneric, S.P.; Redolfi, A.; et al. Apolipoprotein E Genotype and Sex Risk Factors for Alzheimer Disease: A Meta-Analysis. JAMA Neurol. 2017, 74, 1178–1189. [Google Scholar] [CrossRef] [PubMed]
- Belloy, M.E.; Napolioni, V.; Greicius, M.D. A Quarter Century of APOE and Alzheimer’s Disease: Progress to Date and the Path Forward. Neuron 2019, 101, 820–838. [Google Scholar] [CrossRef]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hägg, S.; Athanasiu, L.; et al. Genome-Wide Meta-Analysis Identifies New Loci and Functional Pathways Influencing Alzheimer’s Disease Risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K. The Pathological Process Underlying Alzheimer’s Disease in Individuals under Thirty. Acta Neuropathol. 2011, 121, 171–181. [Google Scholar] [CrossRef]
- Liu, P.P.; Xie, Y.; Meng, X.Y.; Kang, J.S. History and Progress of Hypotheses and Clinical Trials for Alzheimer’s Disease. Signal Transduct. Target. Ther. 2019, 4, 29. [Google Scholar] [CrossRef]
- Mehta, D.; Jackson, R.; Paul, G.; Shi, J.; Sabbagh, M. Why Do Trials for Alzheimer’s Disease Drugs Keep Failing? A Discontinued Drug Perspective for 2010–2015. Expert Opin. Investig. Drugs 2017, 26, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Bennett, D.A.; Schneider, J.A.; Arvanitakis, Z.; Kelly, J.F.; Aggarwal, N.T.; Shah, R.C.; Wilson, R.S. Neuropathology of Older Persons without Cognitive Impairment from Two Community-Based Studies. Neurology 2006, 66, 1837–1844. [Google Scholar] [CrossRef]
- Jack, C.R.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical Model of Dynamic Biomarkers of the Alzheimer’s Pathological Cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef]
- Jack, C.R.; Lowe, V.J.; Weigand, S.D.; Wiste, H.J.; Senjem, M.L.; Knopman, D.S.; Shiung, M.M.; Gunter, J.L.; Boeve, B.F.; Kemp, B.J.; et al. Serial PIB and MRI in Normal, Mild Cognitive Impairment and Alzheimers Disease: Implications for Sequence of Pathological Events in Alzheimers Disease. Brain 2009, 132, 1355–1365. [Google Scholar] [CrossRef]
- Leow, A.D.; Yanovsky, I.; Parikshak, N.; Hua, X.; Lee, S.; Toga, A.W.; Jack, C.R.; Bernstein, M.A.; Britson, P.J.; Gunter, J.L.; et al. Alzheimer’s Disease Neuroimaging Initiative: A One-Year Follow up Study Using Tensor-Based Morphometry Correlating Degenerative Rates, Biomarkers and Cognition. Neuroimage 2009, 45, 645–655. [Google Scholar] [CrossRef]
- Schuff, N.; Woerner, N.; Boreta, L.; Kornfield, T.; Shaw, L.M.; Trojanowski, J.Q.; Thompson, P.M.; Jack, C.R.; Weiner, M.W. MRI of Hippocampal Volume Loss in Early Alzheimers Disease in Relation to ApoE Genotype and Biomarkers. Brain 2009, 132, 1067–1077. [Google Scholar] [CrossRef]
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The Genetic Landscape of Alzheimer Disease: Clinical Implications and Perspectives. Genet. Med. 2016, 18, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S.; De Los Angeles, J.; Miller, D.D.; Xia, W.; Selkoe, D.J. Are Presenilins Intramembrane-Cleaving Proteases? Implications for the Molecular Mechanism of Alzheimer’s Disease. Biochemistry 1999, 38, 11223–11230. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.W.; Quaranta, V.; Glenner, G.G. Neuritic Plaques and Cerebrovascular Amyloid in Alzheimer Disease Are Antigenically Related. Proc. Natl. Acad. Sci. USA 1985, 82, 8729–8732. [Google Scholar] [CrossRef]
- Hardy, J.; Chartier-Harlin, M.C.; Mullan, M. Alzheimer Disease: The New Agenda. Am. J. Hum. Genet. 1992, 50, 648–651. [Google Scholar]
- Pousinha, P.A.; Mouska, X.; Bianchi, D.; Temido-Ferreira, M.; Rajão-Saraiva, J.; Gomes, R.; Fernandez, S.P.; Salgueiro-Pereira, A.R.; Gandin, C.; Raymond, E.F.; et al. The Amyloid Precursor Protein C-Terminal Domain Alters CA1 Neuron Firing, Modifying Hippocampus Oscillations and Impairing Spatial Memory Encoding. Cell Rep. 2019, 29, 317–331.e5. [Google Scholar] [CrossRef]
- Pousinha, P.A.; Mouska, X.; Raymond, E.F.; Gwizdek, C.; Dhib, G.; Poupon, G.; Zaragosi, L.-E.; Giudici, C.; Bethus, I.; Pacary, E.; et al. Physiological and Pathophysiological Control of Synaptic GluN2B-NMDA Receptors by the C-Terminal Domain of Amyloid Precursor Protein. Elife 2017, 6, e25659. [Google Scholar] [CrossRef] [PubMed]
- Ghosal, K.; Vogt, D.L.; Liang, M.; Shen, Y.; Lamb, B.T.; Pimplikar, S.W. Alzheimer’s Disease-like Pathological Features in Transgenic Mice Expressing the APP Intracellular Domain. Proc. Natl. Acad. Sci. USA 2009, 106, 18367–18372. [Google Scholar] [CrossRef]
- Lauritzen, I.; Pardossi-Piquard, R.; Bauer, C.; Brigham, E.; Abraham, J.-D.; Ranaldi, S.; Fraser, P.; St-George-Hyslop, P.; Le Thuc, O.; Espin, V.; et al. The β-Secretase-Derived C-Terminal Fragment of ΒAPP, C99, But Not Aβ, Is a Key Contributor to Early Intraneuronal Lesions in Triple-Transgenic Mouse Hippocampus. J. Neurosci. 2012, 32, 16243–16255. [Google Scholar] [CrossRef]
- Karran, E.; Mercken, M.; Strooper, B. De The Amyloid Cascade Hypothesis for Alzheimer’s Disease: An Appraisal for the Development of Therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef]
- Mullan, M.; Crawford, F.; Axelman, K.; Houlden, H.; Lilius, L.; Winblad, B.; Lannfelt, L. A Pathogenic Mutation for Probable Alzheimer’s Disease in the APP Gene at the N–Terminus of β–Amyloid. Nat. Genet. 1992, 1, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Citron, M.; Oltersdorf, T.; Haass, C.; McConlogue, L.; Hung, A.Y.; Seubert, P.; Vigo-Pelfrey, C.; Lieberburg, I.; Selkoe, D.J. Mutation of the β-Amyloid Precursor Protein in Familial Alzheimer’s Disease Increases β-Protein Production. Nature 1992, 360, 672–674. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, L.; van Duijn, C.M.; Cras, P.; Cruts, M.; Van Hul, W.; van Harskamp, F.; Warren, A.; McInnis, M.G.; Antonarakis, S.E.; Martin, J.J.; et al. Presenile Dementia and Cerebral Haemorrhage Linked to a Mutation at Codon 692 of the β–Amyloid Precursor Protein Gene. Nat. Genet. 1992, 1, 218–221. [Google Scholar] [CrossRef]
- Walsh, D.M.; Hartley, D.M.; Condron, M.M.; Selkoe, D.J.; Teplow, D.B. In Vitro Studies of Amyloid β-Protein Fibril Assembly and Toxicity Provide Clues to the Aetiology of Flemish Variant (Ala692 → Gly) Alzheimer’s Disease. Biochem. J. 2001, 355, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Cras, P.; Van Harskamp, F.; Hendriks, L.; Ceuterick, C.; Van Duijn, C.M.; Stefanko, S.Z.; Hofman, A.; Kros, J.M.; Van Broeckhoven, C.; Martin, J.J. Presenile Alzheimer Dementia Characterized by Amyloid Angiopathy and Large Amyloid Core Type Senile Plaques in the APP 692Ala→Gly Mutation. Acta Neuropathol. 1998, 96, 253–260. [Google Scholar] [CrossRef]
- Zou, Z.; Liu, C.; Che, C.; Huang, H. Clinical Genetics of Alzheimer’s Disease. Biomed. Res. Int. 2014, 2014, 291862. [Google Scholar] [CrossRef]
- Levy, E.; Carman, M.D.; Fernandez-Madrid, I.J.; Power, M.D.; Lieberburg, I.; van Duinen, S.G.; Bots, G.T.A.M.; Luyendijk, W.; Frangione, B. Mutation of the Alzheimer’s Disease Amyloid Gene in Hereditary Cerebral Hemorrhage, Dutch Type. Science 1990, 248, 1124–1126. [Google Scholar] [CrossRef]
- Park, D.H.; Park, H.S.; Joh, T.H.; Anwar, M.; Ruggiero, D.A. Strain Difference in Phenylethanolamine N-Methyltransferase Activity and Immunoreactivity of Medulla Oblongata of Sprague-Dawley and Long-Evans Hooded Rats. Neurosci. Lett. 1991, 128, 240–242. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, T.J.; Cho, H.S.; Vonsattel, J.P.G.; William Rebeck, G.; Greenberg, S.M. Novel Amyloid Precursor Protein Mutation in an Iowa Family with Dementia and Severe Cerebral Amyloid Angiopathy. Ann. Neurol. 2001, 49, 697–705. [Google Scholar] [CrossRef]
- Pasalar, P.; Najmabadi, H.; Noorian, A.R.; Moghimi, B.; Jannati, A.; Soltanzadeh, A.; Krefft, T.; Crook, R.; Hardy, J. An Iranian Family with Alzheimer’s Disease Caused by a Novel APP Mutation (THr714ALa). Neurology 2002, 58, 1574–1575. [Google Scholar] [CrossRef]
- Kumar-Singh, S.; De Jonghe, C.; Cruts, M.; Kleinert, R.; Wang, R.; Mercken, M.; De Strooper, B.; Vanderstichele, H.; Löfgren, A.; Vanderhoeven, I.; et al. Nonfibrillar Diffuse Amyloid Deposition Due to a γ 42-Secretase Site Mutation Points to an Essential Role for N-Truncated Aβ 42 in Alzheimer’s Disease. Hum. Mol. Genet. 2000, 9, 2589–2598. [Google Scholar] [CrossRef]
- Bornebroek, M.; De Jonghe, C.; Haan, J.; Kumar-Singh, S.; Younkin, S.; Roos, R.; Van Broeckhoven, C. Hereditary Cerebral Hemorrhage with Amyloidosis Dutch Type (AβPP 693): Decreased Plasma Amyloid-β 42 Concentration. Neurobiol. Dis. 2003, 14, 619–623. [Google Scholar] [CrossRef] [PubMed]
- Ancolio, K.; Dumanchin, C.; Barelli, H.; Warter, J.M.; Brice, A.; Campion, D.; Frebourg, T.; Checler, F. Unusual Phenotypic Alteration of β Amyloid Precursor Protein (ΒAPP) Maturation by a New Val-715 → Met ΒAPP-770 Mutation Responsible for Probable Early-Onset Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 1999, 96, 4119–4124. [Google Scholar] [CrossRef] [PubMed]
- Cruts, M.; Dermaut, B.; Rademakers, R.; Van Den Broeck, M.; Stögbauer, F.; Van Broeckhoven, C. Novel APP Mutation V715A Associated with Presenile Alzheimer’s Disease in a German Family. J. Neurol. 2003, 250, 1374–1375. [Google Scholar] [CrossRef]
- Eckman, C.B.; Mehta, N.D.; Crook, R.; Perez-tur, J.; Prihar, G.; Pfeiffer, E.; Graff-Radford, N.; Hinder, P.; Yager, D.; Zenk, B.; et al. A New Pathogenic Mutation in the APP Gene (1716V) Increases the Relative Proportion of Aβ42(43). Hum. Mol. Genet. 1997, 6, 2087–2089. [Google Scholar] [CrossRef] [PubMed]
- Goate, A.; Chartier-Harlin, M.C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a Missense Mutation in the Amyloid Precursor Protein Gene with Familial Alzheimer’s Disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef]
- Murrell, J.R.; Hake, A.M.; Quaid, K.A.; Farlow, M.R.; Ghetti, B. Early-Onset Alzheimer Disease Caused by a New Mutation (V717L) in the Amyloid Precursor Protein Gene. Arch. Neurol. 2000, 57, 885–887. [Google Scholar] [CrossRef]
- Sun, Q.; Xie, N.; Tang, B.; Li, R.; Shen, Y. Alzheimer’s Disease: From Genetic Variants to the Distinct Pathological Mechanisms. Front. Mol. Neurosci. 2017, 10, 319. [Google Scholar] [CrossRef]
- Bagyinszky, E.; Youn, Y.C.; An, S.S.A.; Kim, S. The Genetics of Alzheimer’s Disease. Clin. Interv. Aging 2014, 9, 535–551. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Zheng, H. Practical Considerations for Choosing a Mouse Model of Alzheimer’s Disease. Mol. Neurodegener. 2017, 12, 89. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-Avidity Binding to β-Amyloid and Increased Frequency of Type 4 Allele in Late-Onset Familial Alzheimer Disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.J.; Lohmann, E.; Kinsella, E.; Brás, J.M.; Luu, N.; Gurunlian, N.; Dursun, B.; Bilgic, B.; Santana, I.; Hanagasi, H.; et al. Exome Sequencing Reveals an Unexpected Genetic Cause of Disease: NOTCH3 Mutation in a Turkish Family with Alzheimer’s Disease. Neurobiol. Aging 2012, 33, 1008.e17–1008.e23. [Google Scholar] [CrossRef] [PubMed]
- Pottier, C.; Hannequin, D.; Coutant, S.; Rovelet-Lecrux, A.; Wallon, D.; Rousseau, S.; Legallic, S.; Paquet, C.; Bombois, S.; Pariente, J.; et al. High Frequency of Potentially Pathogenic SORL1 Mutations in Autosomal Dominant Early-Onset Alzheimer Disease. Mol. Psychiatry 2012, 17, 875–879. [Google Scholar] [CrossRef]
- Lupton, M.K.; Proitsi, P.; Danillidou, M.; Tsolaki, M.; Hamilton, G.; Wroe, R.; Pritchard, M.; Lord, K.; Martin, B.M.; Kloszewska, I.; et al. Deep Sequencing of the Nicastrin Gene in Pooled DNA, the Identification of Genetic Variants That Affect Risk of Alzheimer’s Disease. PLoS ONE 2011, 6, e17298. [Google Scholar] [CrossRef]
- Pottier, C.; Wallon, D.; Rousseau, S.; Rovelet-Lecrux, A.; Richard, A.C.; Rollin-Sillaire, A.; Frebourg, T.; Campion, D.; Hannequin, D. TREM2 R47H Variant as a Risk Factor for Early-Onset Alzheimer’s Disease. J. Alzheimer’s Dis. 2013, 35, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 Associated with the Risk of Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.K.; Younkin, S.; et al. TREM2 Variants in Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common Variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP Are Associated with Alzheimer’s Disease. Nat. Genet. 2011, 43, 429–436. [Google Scholar] [CrossRef]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common Variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 Are Associated with Late-Onset Alzheimer’s Disease. Nat. Genet. 2011, 43, 436–443. [Google Scholar] [CrossRef]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic Meta-Analysis of Diagnosed Alzheimer’s Disease Identifies New Risk Loci and Implicates Aβ, Tau, Immunity and Lipid Processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef]
- Liu, J.Z.; Erlich, Y.; Pickrell, J.K. Case-Control Association Mapping by Proxy Using Family History of Disease. Nat. Genet. 2017, 49, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Marioni, R.E.; Harris, S.E.; Zhang, Q.; McRae, A.F.; Hagenaars, S.P.; Hill, W.D.; Davies, G.; Ritchie, C.W.; Gale, C.R.; Starr, J.M.; et al. GWAS on Family History of Alzheimer’s Disease. Transl. Psychiatry 2018, 8, 99. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Cooper, S.; Liu, J.Z.; Barrio-Hernandez, I.; Bello, E.; Kumasaka, N.; Young, A.M.H.; Franklin, R.J.M.; Johnson, T.; Estrada, K.; et al. Genome-Wide Meta-Analysis, Fine-Mapping and Integrative Prioritization Implicate New Alzheimer’s Disease Risk Genes. Nat. Genet. 2021, 53, 392–402. [Google Scholar] [CrossRef] [PubMed]
- de Rojas, I.; Moreno-Grau, S.; Tesi, N.; Grenier-Boley, B.; Andrade, V.; Jansen, I.E.; Pedersen, N.L.; Stringa, N.; Zettergren, A.; Hernández, I.; et al. Common Variants in Alzheimer’s Disease and Risk Stratification by Polygenic Risk Scores. Nat. Commun. 2021, 12, 3417. [Google Scholar] [CrossRef]
- de la Fuente, J.; Grotzinger, A.D.; Marioni, R.E.; Nivard, M.G.; Tucker-Drob, E.M. Integrated Analysis of Direct and Proxy Genome Wide Association Studies Highlights Polygenicity of Alzheimer’s Disease Outside of the APOE Region. PLoS Genet. 2022, 18, e1010208. [Google Scholar] [CrossRef] [PubMed]
- Escott-Price, V.; Hardy, J. Genome-Wide Association Studies for Alzheimer’s Disease: Bigger Is Not Always Better. Brain Commun. 2022, 4, fcac125. [Google Scholar] [CrossRef]
- Andrews, S.J.; Fulton-Howard, B.; Goate, A. Interpretation of Risk Loci from Genome-Wide Association Studies of Alzheimer’s Disease. Lancet Neurol. 2020, 19, 326–335. [Google Scholar] [CrossRef]
- Braidy, N.; Muñoz, P.; Palacios, A.G.; Castellano-Gonzalez, G.; Inestrosa, N.C.; Chung, R.S.; Sachdev, P.; Guillemin, G.J. Recent Rodent Models for Alzheimer’s Disease: Clinical Implications and Basic Research. J. Neural Transm. 2012, 119, 173–195. [Google Scholar] [CrossRef]
- Chen, R.; Tilley, M.R.; Wei, H.; Zhou, F.; Zhou, F.M.; Ching, S.; Quan, N.; Stephens, R.L.; Hill, E.R.; Nottoli, T.; et al. Abolished Cocaine Reward in Mice with a Cocaine-Insensitive Dopamine Transporter. Proc. Natl. Acad. Sci. USA 2006, 103, 9333–9338. [Google Scholar] [CrossRef]
- Hickman-Davis, J.M.; Davis, I.C. Transgenic Mice. Paediatr. Respir. Rev. 2006, 7, 49–53. [Google Scholar] [CrossRef]
- Nolan, M.F.; Malleret, G.; Dudman, J.T.; Buhl, D.L.; Santoro, B.; Gibbs, E.; Vronskaya, S.; Buzsáki, G.; Siegelbaum, S.A.; Kandel, E.R.; et al. A Behavioral Role for Dendritic Integration: HCN1 Channels Constrain Spatial Memory and Plasticity at Inputs to Distal Dendrites of CA1 Pyramidal Neurons. Cell 2004, 119, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Picciotto, M.R.; Wickman, K. Using Knockout and Transgenic Mice to Study Neurophysiology and Behavior. Physiol. Rev. 1998, 78, 1131–1163. [Google Scholar] [CrossRef]
- Available online: https://www.Alzforum.Org/Research-Models/Alzheimers-Disease (accessed on 23 November 2024).
- Jankowsky, J.L.; Xu, G.; Fromholt, D.; Gonzales, V.; Borchelt, D.R. Environmental Enrichment Exacerbates Amyloid Plaque Formation in a Transgenic Mouse Model of Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2003, 62, 1220–1227. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-Transgenic Model of Alzheimer’s Disease with Plaques and Tangles: Intracellular Aβ and Synaptic Dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal β-Amyloid Aggregates, Neurodegeneration, and Neuron Loss in Transgenic Mice with Five Familial Alzheimer’s Disease Mutations: Potential Factors in Amyloid Plaque Formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef]
- Sasaguri, H.; Hashimoto, S.; Watamura, N.; Sato, K.; Takamura, R.; Nagata, K.; Tsubuki, S.; Ohshima, T.; Yoshiki, A.; Sato, K.; et al. Recent Advances in the Modeling of Alzheimer’s Disease. Front. Neurosci. 2022, 16, 807473. [Google Scholar] [CrossRef]
- Ameen-Ali, K.E.; Wharton, S.B.; Simpson, J.E.; Heath, P.R.; Sharp, P.; Berwick, J. Review: Neuropathology and Behavioural Features of Transgenic Murine Models of Alzheimer’s Disease. Neuropathol. Appl. Neurobiol. 2017, 43, 553–570. [Google Scholar] [CrossRef]
- Sethi, P.; Bhaskar, R.; Singh, K.K.; Gupta, S.; Han, S.S.; Avinash, D.; Abomughaid, M.M.; Koul, A.; Rani, B.; Ghosh, S.; et al. Exploring Advancements in Early Detection of Alzheimer’s Disease with Molecular Assays and Animal Models. Ageing Res. Rev. 2024, 100, 102411. [Google Scholar] [CrossRef]
- Drummond, E.; Wisniewski, T. Alzheimer’s Disease: Experimental Models and Reality. Acta Neuropathol. 2017, 133, 155–175. [Google Scholar] [CrossRef]
- Games, D.; Adams, D.; Alessandrini, R.; Barbour, R.; Borthelette, P.; Blackwell, C.; Carr, T.; Clemens, J.; Donaldson, T.; Gillespie, F.; et al. Alzheimer-Type Neuropathology in Transgenic Mice Overexpressing V717F β-Amyloid Precursor Protein. Nature 1995, 373, 523–527. [Google Scholar] [CrossRef]
- Chen, G.; Chen, K.S.; Knox, J.; Inglis, J.; Bernard, A.; Martin, S.J.; Justice, A.; McConlogue, L.; Games, D.; Freedman, S.B.; et al. A Learning Deficit Related to Age and β-Amyloid Plaques in a Mouse Model of Alzheimer’s Disease. Nature 2000, 408, 975–979. [Google Scholar] [CrossRef]
- Holcomb, L.; Gordon, M.N.; Mcgowan, E.; Yu, X.; Benkovic, S.; Jantzen, P.; Wright, K.; Saad, I.; Mueller, R.; Morgan, D.; et al. Accelerated Alzheimer-Type Phenotype in Transgenic Mice Carrying Both Mutant Amyloid Precursor Protein and Presenilin 1 Transgenes. Nat. Med. 1998, 4, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative Memory Deficits, Aβ Elevation, and Amyloid Plaques in Transgenic Mice. Science 1996, 274, 99–102. [Google Scholar] [CrossRef]
- Rockenstein, E.; Mallory, M.; Mante, M.; Sisk, A.; Masliaha, E. Early Formation of Mature Amyloid-β Protein Deposits in a Mutant APP Transgenic Model Depends on Levels of Aβ1-42. J. Neurosci. Res. 2001, 66, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Mucke, L.; Masliah, E.; Yu, G.Q.; Mallory, M.; Rockenstein, E.M.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Johnson-Wood, K.; McConlogue, L. High-Level Neuronal Expression of Aβ(1-42) in Wild-Type Human Amyloid Protein Precursor Transgenic Mice: Synaptotoxicity without Plaque Formation. J. Neurosci. 2000, 20, 4050–4058. [Google Scholar] [CrossRef]
- Chishti, M.A.; Yang, D.S.; Janus, C.; Phinney, A.L.; Horne, P.; Pearson, J.; Strome, R.; Zuker, N.; Loukides, J.; French, J.; et al. Early-Onset Amyloid Deposition and Cognitive Deficits in Transgenic Mice Expressing a Double Mutant Form of Amyloid Precursor Protein 695. J. Biol. Chem. 2001, 276, 21562–21570. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.M.; Roberson, E.D. Mouse Models of Alzheimer’s Disease. Brain Res. Bull. 2012, 88, 3–12. [Google Scholar] [CrossRef]
- Yang, C.; Sun, Z.P.; Jiang, J.; Cai, X.L.; Wang, Y.; Wang, H.; Che, C.; Tu, E.; Pan, A.H.; Zhang, Y.; et al. Increased Expression of the Proapoptotic Presenilin Associated Protein Is Involved in Neuronal Tangle Formation in Human Brain. Sci. Rep. 2024, 14, 25274. [Google Scholar] [CrossRef]
- Zhao, H.; Mu, Y.; Liang, A.; Wei, J.; Lai, S.; Li, X.; Chen, P.; Li, H.; He, H.; Liu, X.; et al. Suppressing DUSP16 Overexpression Induced by ELK1 Promotes Neural Progenitor Cell Differentiation in Mouse Models of Alzheimer’s Disease. Aging Cell 2024, 24, e14372. [Google Scholar] [CrossRef]
- McNerney, M.W.; Kraybill, E.P.; Narayanan, S.; Mojabi, F.S.; Venkataramanan, V.; Heath, A. Memory-Related Hippocampal Brain-Derived Neurotrophic Factor Activation Pathways from Repetitive Transcranial Magnetic Stimulation in the 3xTg-AD Mouse Line. Exp. Gerontol. 2023, 183, 112323. [Google Scholar] [CrossRef]
- Westi, E.W.; Molhemi, S.; Hansen, C.T.; Skoven, C.S.; Knopper, R.W.; Ahmad, D.A.; Rindshøj, M.B.; Ameen, A.O.; Hansen, B.; Kohlmeier, K.A.; et al. Comprehensive Analysis of the 5xFAD Mouse Model of Alzheimer’s Disease Using DMRI, Immunohistochemistry, and Neuronal and Glial Functional Metabolic Mapping. Biomolecules 2024, 14, 1294. [Google Scholar] [CrossRef]
- Wong, S.Q.; Ouellette, A.; McNamara, A.; Tam, R.A.; Alexandrov, A.; Nawrocik-Madrid, A.; Sanchez, J.J.; Ginsburg, B.C.; Andrade, A.A.; Lapierre, L.R. Spatial Memory in Alzheimer’s Disease 5XFAD Mice Is Enhanced by XPO1 Inhibitor KPT-330. bioRxiv 2024. [Google Scholar] [CrossRef]
- Zhu, Y.; Nwabuisi-Heath, E.; Dumanis, S.B.; Tai, L.M.; Yu, C.; Rebeck, G.W.; Ladu, M.J. APOE Genotype Alters Glial Activation and Loss of Synaptic Markers in Mice. Glia 2012, 60, 559–569. [Google Scholar] [CrossRef]
- Wang, C.; Wilson, W.A.; Moore, S.D.; Mace, B.E.; Maeda, N.; Schmechel, D.E.; Sullivan, P.M. Human ApoE4-Targeted Replacement Mice Display Synaptic Deficits in the Absence of Neuropathology. Neurobiol. Dis. 2005, 18, 390–398. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Bales, K.R.; Wu, S.; Bhat, P.; Parsadanian, M.; Fagan, A.M.; Chang, L.K.; Sun, Y.; Paul, S.M. Expression of Human Apolipoprotein E Reduces Amyloid-β Deposition in a Mouse Model of Alzheimer’s Disease. J. Clin. Investig. 1999, 103, R15–R21. [Google Scholar] [CrossRef]
- Wang, Z.H.; Xia, Y.; Wu, Z.; Kang, S.S.; Zhang, J.C.; Liu, P.; Liu, X.; Song, W.; Huin, V.; Dhaenens, C.M.; et al. Neuronal ApoE4 Stimulates C/EBPβ Activation, Promoting Alzheimer’s Disease Pathology in a Mouse Model. Prog. Neurobiol. 2022, 209, 102212. [Google Scholar] [CrossRef]
- Fryer, J.D.; Taylor, J.W.; DeMattos, R.B.; Bales, K.R.; Paul, S.M.; Parsadanian, M.; Holtzman, D.M. Apolipoprotein E Markedly Facilitates Age-Dependent Cerebral Amyloid Angiopathy and Spontaneous Hemorrhage in Amyloid Precursor Protein Transgenic Mice. J. Neurosci. 2003, 23, 7889–7896. [Google Scholar] [CrossRef]
- Lewis, J.; Dickson, D.W.; Lin, W.L.; Chisholm, L.; Corral, A.; Jones, G.; Yen, S.H.; Sahara, N.; Skipper, L.; Yager, D.; et al. Enhanced Neurofibrillary Degeneration in Transgenic Mice Expressing Mutant Tau and APP. Science 2001, 293, 1487–1491. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Kitazawa, M.; Tseng, B.P.; LaFerla, F.M. Amyloid Deposition Precedes Tangle Formation in a Triple Transgenic Model of Alzheimer’s Disease. Neurobiol. Aging 2003, 24, 1063–1070. [Google Scholar] [CrossRef]
- Billings, L.M.; Oddo, S.; Green, K.N.; McGaugh, J.L.; LaFerla, F.M. Intraneuronal Aβ Causes the Onset of Early Alzheimer’s Disease-Related Cognitive Deficits in Transgenic Mice. Neuron 2005, 45, 675–688. [Google Scholar] [CrossRef]
- Andorfer, C.; Kress, Y.; Espinoza, M.; De Silva, R.; Tucker, K.L.; Barde, Y.A.; Duff, K.; Davies, P. Hyperphosphorylation and Aggregation of Tau in Mice Expressing Normal Human Tau Isoforms. J. Neurochem. 2003, 86, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Duff, K.; Knight, H.; Refolo, L.M.; Sanders, S.; Yu, X.; Picciano, M.; Malester, B.; Hutton, M.; Adamson, J.; Goedert, M.; et al. Characterization of Pathology in Transgenic Mice Over-Expressing Human Genomic and CDNA Tau Transgenes. Neurobiol. Dis. 2000, 7, 87–98. [Google Scholar] [CrossRef]
- Zhong, M.Z.; Peng, T.; Duarte, M.L.; Wang, M.; Cai, D. Updates on Mouse Models of Alzheimer’s Disease. Mol. Neurodegener. 2024, 19, 23. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, Y.; Sekiya, M.; Saito, T.; Saido, T.C.; Iijima, K.M. Cognitive and Emotional Alterations in App Knock-in Mouse Models of Aβ Amyloidosis. BMC Neurosci. 2018, 19, 46. [Google Scholar] [CrossRef]
- Saito, T.; Mihira, N.; Matsuba, Y.; Sasaguri, H.; Hashimoto, S.; Narasimhan, S.; Zhang, B.; Murayama, S.; Higuchi, M.; Lee, V.M.Y.; et al. Humanization of the Entire Murine Mapt Gene Provides a Murine Model of Pathological Human Tau Propagation. J. Biol. Chem. 2019, 294, 12754–12765. [Google Scholar] [CrossRef]
- Liraz, O.; Boehm-Cagan, A.; Michaelson, D.M. ApoE4 Induces Aβ42, Tau, and Neuronal Pathology in the Hippocampus of Young Targeted Replacement ApoE4 Mice. Mol. Neurodegener. 2013, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Yamada, K.; Liddelow, S.A.; Smith, S.T.; Zhao, L.; Luo, W.; Tsai, R.M.; Spina, S.; Grinberg, L.T.; Rojas, J.C.; et al. ApoE4 Markedly Exacerbates Tau-Mediated Neurodegeneration in a Mouse Model of Tauopathy. Nature 2017, 549, 523–527. [Google Scholar] [CrossRef]
- Kang, S.S.; Kurti, A.; Baker, K.E.; Liu, C.-C.; Colonna, M.; Ulrich, J.D.; Holtzman, D.M.; Bu, G.; Fryer, J.D. Behavioral and Transcriptomic Analysis of Trem2-Null Mice: Not All Knockout Mice Are Created Equal. Hum. Mol. Genet. 2018, 27, 211–223. [Google Scholar] [CrossRef]
- Kotredes, K.P.; Oblak, A.; Pandey, R.S.; Lin, P.B.-C.; Garceau, D.; Williams, H.; Uyar, A.; O’Rourke, R.; O’Rourke, S.; Ingraham, C.; et al. Uncovering Disease Mechanisms in a Novel Mouse Model Expressing Humanized APOEε4 and Trem2*R47H. Front. Aging Neurosci. 2021, 13, 735524. [Google Scholar] [CrossRef]
- Kotredes, K.P.; Pandey, R.S.; Persohn, S.; Elderidge, K.; Burton, C.P.; Miner, E.W.; Haynes, K.A.; Santos, D.F.S.; Williams, S.; Heaton, N.; et al. Characterizing Molecular and Synaptic Signatures in Mouse Models of Late-onset Alzheimer’s Disease Independent of Amyloid and Tau Pathology. Alzheimer’s Dement. 2024, 20, 4126–4146. [Google Scholar] [CrossRef]
- Sasner, M.; Preuss, C.; Pandey, R.S.; Uyar, A.; Garceau, D.; Kotredes, K.P.; Williams, H.; Oblak, A.L.; Lin, P.B.; Perkins, B.; et al. In Vivo Validation of Late-onset Alzheimer’s Disease Genetic Risk Factors. Alzheimer’s Dement. 2024, 20, 4970–4984. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.-M.; Kokjohn, T.A.; Beach, T.G.; Sue, L.I.; Brune, D.; Lopez, J.C.; Kalback, W.M.; Abramowski, D.; Sturchler-Pierrat, C.; Staufenbiel, M.; et al. Comparative Analysis of Amyloid-β Chemical Structure and Amyloid Plaque Morphology of Transgenic Mouse and Alzheimer’s Disease Brains. J. Biol. Chem. 2001, 276, 12991–12998. [Google Scholar] [CrossRef] [PubMed]
- Jacob, H.J.; Kwitek, A.E. Rat Genetics: Attachign Physiology and Pharmacology to the Genome. Nat. Rev. Genet. 2002, 3, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, R.A.; Weinstock, G.M.; Metzker, M.L.; Muzny, D.M.; Sodergren, E.J.; Scherer, S.; Scott, G.; Steffen, D.; Worley, K.C.; Burch, P.E.; et al. Genome Sequence of the Brown Norway Rat Yields Insights into Mammalian Evolution. Nature 2004, 428, 493–521. [Google Scholar] [CrossRef]
- Tesson, L.; Cozzi, J.; Ménoret, S.; Rémy, S.; Usal, C.; Fraichard, A.; Anegon, I. Transgenic Modifications of the Rat Genome. Transgenic Res. 2005, 14, 531–546. [Google Scholar] [CrossRef]
- Do Carmo, S.; Cuello, A.C. Modeling Alzheimer’s Disease in Transgenic Rats. Mol. Neurodegener. 2013, 8, 37. [Google Scholar] [CrossRef]
- Flood, D.G.; Lin, Y.G.; Lang, D.M.; Trusko, S.P.; Hirsch, J.D.; Savage, M.J.; Scott, R.W.; Howland, D.S. A Transgenic Rat Model of Alzheimer’s Disease with Extracellular Aβ Deposition. Neurobiol. Aging 2009, 30, 1078–1090. [Google Scholar] [CrossRef]
- Liu, L.; Orozco, I.J.; Planel, E.; Wen, Y.; Bretteville, A.; Krishnamurthy, P.; Wang, L.; Herman, M.; Figueroa, H.; Yu, W.H.; et al. A Transgenic Rat That Develops Alzheimer’s Disease-like Amyloid Pathology, Deficits in Synaptic Plasticity and Cognitive Impairment. Neurobiol. Dis. 2008, 31, 46–57. [Google Scholar] [CrossRef]
- Leon, W.C.; Canneva, F.; Partridge, V.; Allard, S.; Ferretti, M.T.; Dewilde, A.; Vercauteren, F.; Atifeh, R.; Ducatenzeiler, A.; Klein, W.; et al. A Novel Transgenic Rat Model with a Full Alzheimer’s—Like Amyloid Pathology Displays Pre—Plaque Intracellular Amyloid -β- Associated Cognitive Impairment. J. Alzheimer’s Dis. 2010, 20, 113–126. [Google Scholar] [CrossRef]
- Cohen, R.M.; Rezai-Zadeh, K.; Weitz, T.M.; Rentsendorj, A.; Gate, D.; Spivak, I.; Bholat, Y.; Vasilevko, V.; Glabe, C.G.; Breunig, J.J.; et al. A Transgenic Alzheimer Rat with Plaques, Tau Pathology, Behavioral Impairment, Oligomeric Aβ, and Frank Neuronal Loss. J. Neurosci. 2013, 33, 6245–6256. [Google Scholar] [CrossRef]
- Tambini, M.D.; Yao, W.; D’Adamio, L. Facilitation of Glutamate, but Not GABA, Release in Familial Alzheimer’s APP Mutant Knock-in Rats with Increased β-Cleavage of APP. Aging Cell 2019, 18, e13033. [Google Scholar] [CrossRef] [PubMed]
- Serneels, L.; T’Syen, D.; Perez-Benito, L.; Theys, T.; Holt, M.G.; De Strooper, B. Modeling the β-Secretase Cleavage Site and Humanizing Amyloid-Beta Precursor Protein in Rat and Mouse to Study Alzheimer’s Disease. Mol. Neurodegener. 2020, 15, 60. [Google Scholar] [CrossRef] [PubMed]
- McKean, N.E.; Handley, R.R.; Snell, R.G. A Review of the Current Mammalian Models of Alzheimer’s Disease and Challenges That Need to Be Overcome. Int. J. Mol. Sci. 2021, 22, 13168. [Google Scholar] [CrossRef] [PubMed]
- Kimura, N.; Nakamura, S.; Goto, N.; Narushima, E.; Hara, I.; Shichiri, S.; Saitou, K.; Nose, M.; Hayashi, T.; Kawamura, S.; et al. Senile Plaques in an Aged Western Lowland Gorilla. Exp. Anim. 2001, 50, 77–81. [Google Scholar] [CrossRef]
- Perez, S.E.; Raghanti, M.A.; Hof, P.R.; Kramer, L.; Ikonomovic, M.D.; Lacor, P.N.; Erwin, J.M.; Sherwood, C.C.; Mufson, E.J. Alzheimer’s Disease Pathology in the Neocortex and Hippocampus of the Western Lowland Gorilla (Gorilla Gorilla Gorilla). J. Comp. Neurol. 2013, 521, 4318–4338. [Google Scholar] [CrossRef]
- Perez, S.E.; Sherwood, C.C.; Cranfield, M.R.; Erwin, J.M.; Mudakikwa, A.; Hof, P.R.; Mufson, E.J. Early Alzheimer’s Disease-Type Pathology in the Frontal Cortex of Wild Mountain Gorillas (Gorilla Beringei Beringei). Neurobiol. Aging 2016, 39, 195–201. [Google Scholar] [CrossRef]
- Rosen, R.F.; Farberg, A.S.; Gearing, M.; Dooyema, J.; Long, P.M.; Anderson, D.C.; Davis-Turak, J.; Coppola, G.; Geschwind, D.H.; Paré, J.F.; et al. Tauopathy with Paired Helical Filaments in an Aged Chimpanzee. J. Comp. Neurol. 2008, 509, 259–270. [Google Scholar] [CrossRef]
- Holzer, M.; Craxton, M.; Jakes, R.; Arendt, T.; Goedert, M. Tau Gene (MAPT) Sequence Variation among Primates. Gene 2004, 341, 313–322. [Google Scholar] [CrossRef]
- Aguilera, B.; Perez Gomez, J.; DeGrazia, D. Should Biomedical Research with Great Apes Be Restricted? A Systematic Review of Reasons. BMC Med. Ethics 2021, 22, 15. [Google Scholar] [CrossRef]
- Sani, S.; Traul, D.; Klink, A.; Niaraki, N.; Gonzalo-Ruiz, A.; Wu, C.K.; Geula, C. Distribution, Progression and Chemical Composition of Cortical Amyloid-β Deposits in Aged Rhesus Monkeys: Similarities to the Human. Acta Neuropathol. 2003, 105, 145–156. [Google Scholar] [CrossRef]
- Shah, P.; Lal, N.; Leung, E.; Traul, D.E.; Gonzalo-Ruiz, A.; Geula, C. Neuronal and Axonal Loss Are Selectively Linked to Fibrillar Amyloid-β within Plaques of the Aged Primate Cerebral Cortex. Am. J. Pathol. 2010, 177, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Rosen, R.F.; Walker, L.C.; LeVine, H. PIB Binding in Aged Primate Brain: Enrichment of High-Affinity Sites in Humans with Alzheimer’s Disease. Neurobiol. Aging 2011, 32, 223–234. [Google Scholar] [CrossRef]
- Okano, H. Current Status of and Perspectives on the Application of Marmosets in Neurobiology. Annu. Rev. Neurosci. 2021, 44, 27–48. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Sasaki, E. Assisted Reproductive Techniques and Genetic Manipulation in the Common Marmoset. ILAR J. 2020, 61, 286–303. [Google Scholar] [CrossRef]
- Rodriguez-Callejas, J.D.; Fuchs, E.; Perez-Cruz, C. Evidence of Tau Hyperphosphorylation and Dystrophic Microglia in the Common Marmoset. Front. Aging Neurosci. 2016, 8, 315. [Google Scholar] [CrossRef]
- Paspalas, C.D.; Carlyle, B.C.; Leslie, S.; Preuss, T.M.; Crimins, J.L.; Huttner, A.J.; van Dyck, C.H.; Rosene, D.L.; Nairn, A.C.; Arnsten, A.F.T. The Aged Rhesus Macaque Manifests Braak Stage III/IV Alzheimer’s-like Pathology. Alzheimer’s Dement. 2018, 14, 680–691. [Google Scholar] [CrossRef]
- Schultz, C.; Hubbard, G.B.; Rüb, U.; Braak, E.; Braak, H. Age-Related Progression of Tau Pathology in Brains of Baboons. Neurobiol. Aging 2000, 21, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Beckman, D.; Morrison, J.H. Towards Developing a Rhesus Monkey Model of Early Alzheimer’s Disease Focusing on Women’s Health. Am. J. Primatol. 2021, 83, e23289. [Google Scholar] [CrossRef]
- Latimer, C.S.; Shively, C.A.; Keene, C.D.; Jorgensen, M.J.; Andrews, R.N.; Register, T.C.; Montine, T.J.; Wilson, A.M.; Neth, B.J.; Mintz, A.; et al. A Nonhuman Primate Model of Early Alzheimer’s Disease Pathologic Change: Implications for Disease Pathogenesis. Alzheimer’s Dement. 2019, 15, 93–105. [Google Scholar] [CrossRef]
- Seita, Y.; Morimura, T.; Watanabe, N.; Iwatani, C.; Tsuchiya, H.; Nakamura, S.; Suzuki, T.; Yanagisawa, D.; Tsukiyama, T.; Nakaya, M.; et al. Generation of Transgenic Cynomolgus Monkeys Overexpressing the Gene for Amyloid-β Precursor Protein. J. Alzheimer’s Dis. 2020, 75, 45–60. [Google Scholar] [CrossRef]
- Homanics, G.E.; Park, J.E.; Bailey, L.; Schaeffer, D.J.; Schaeffer, L.; He, J.; Li, S.; Zhang, T.; Haber, A.; Spruce, C.; et al. Early Molecular Events of Autosomal-Dominant Alzheimer’s Disease in Marmosets with PSEN1 Mutations. Alzheimer’s Dement. 2024, 20, 3455–3471. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, J.E.; Johansen, M.G.; Schmidt, M.; Liu, Y.; Li, R.; Callesen, H.; Melnikova, M.; Habekost, M.; Matrone, C.; Bouter, Y.; et al. Expression of the Alzheimer’s Disease Mutations AβPP695sw and PSEN1M146I in Double-Transgenic Göttingen Minipigs. J. Alzheimer’s Dis. 2016, 53, 1617–1630. [Google Scholar] [CrossRef] [PubMed]
- Alexander, A.G.; Marfil, V.; Li, C. Use of Caenorhabditis Elegans as a Model to Study Alzheimer’s Disease and Other Neurodegenerative Diseases. Front. Genet. 2014, 5, 279. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Funez, P.; de Mena, L.; Rincon-Limas, D.E. Modeling the Complex Pathology of Alzheimer’s Disease in Drosophila. Exp. Neurol. 2015, 274, 58–71. [Google Scholar] [CrossRef]
- Newman, M.; Verdile, G.; Martins, R.N.; Lardelli, M. Zebrafish as a Tool in Alzheimer’s Disease Research. Biochim. Biophys. Acta-Mol. Basis Dis. 2011, 1812, 346–352. [Google Scholar] [CrossRef]
- Abou-Dahech, M.S.; Williams, F.E. Aging, Age-Related Diseases, and the Zebrafish Model. J. Dement. Alzheimer’s Dis. 2024, 1, 48–71. [Google Scholar] [CrossRef]
- Gerhard, G.S.; Cheng, K.C. A Call to Fins! Zebrafish as a Gerontological Model. Aging Cell 2002, 1, 104–111. [Google Scholar] [CrossRef]
- Shenoy, A.; Banerjee, M.; Upadhya, A.; Bagwe-Parab, S.; Kaur, G. The Brilliance of the Zebrafish Model: Perception on Behavior and Alzheimer’s Disease. Front. Behav. Neurosci. 2022, 16, 861155. [Google Scholar] [CrossRef]
- Audira, G.; Siregar, P.; Strungaru, S.A.; Huang, J.C.; Hsiao, C.D. Which Zebrafish Strains Are More Suitable to Perform Behavioral Studies? A Comprehensive Comparison by Phenomic Approach. Biology 2020, 9, 200. [Google Scholar] [CrossRef]
- Teame, T.; Zhang, Z.; Ran, C.; Zhang, H.; Yang, Y.; Ding, Q.; Xie, M.; Gao, C.; Ye, Y.; Duan, M.; et al. The Use of Zebrafish (Danio Rerio) as Biomedical Models. Anim. Front. 2019, 9, 68–77. [Google Scholar] [CrossRef]
- Leimer, U.; Lun, K.; Romig, H.; Walter, J.; Grünberg, J.; Brand, M.; Haass, C. Zebrafish (Danio Rerio) Presenilin Promotes Aberrant Amyloid β-Peptide Production and Requires a Critical Aspartate Residue for Its Function in Amyloidogenesis. Biochemistry 1999, 38, 13602–13609. [Google Scholar] [CrossRef] [PubMed]
- Nornes, S.; Groth, C.; Camp, E.; Ey, P.; Lardelli, M. Developmental Control of Presenilin1 Expression, Endoproteolysis, and Interaction in Zebrafish Embryos. Exp. Cell Res. 2003, 289, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Groth, C.; Nornes, S.; McCarty, R.; Tamme, R.; Lardelli, M. Identification of a Second Presenilin Gene in Zebrafish with Similarity to the Human Alzheimer’s Disease Presenilin2. Dev. Genes Evol. 2002, 212, 486–490. [Google Scholar] [CrossRef]
- Musa, A.; Lehrach, H.; Russo, V.E.A. Distinct Expression Patterns of Two Zebrafish Homologues of the Human APP Gene during Embryonic Development. Dev. Genes Evol. 2001, 211, 563–567. [Google Scholar] [CrossRef]
- Chen, M.; Martins, R.N.; Lardelli, M. Complex Splicing and Neural Expression of Duplicated Tau Genes in Zebrafish Embryos. J. Alzheimer’s Dis. 2009, 18, 305–317. [Google Scholar] [CrossRef]
- Van Dam, D.; De Deyn, P.P. Animal Models in the Drug Discovery Pipeline for Alzheimer’s Disease. Br. J. Pharmacol. 2011, 164, 1285–1300. [Google Scholar] [CrossRef] [PubMed]
- Bill, B.R.; Petzold, A.M.; Clark, K.J.; Schimmenti, L.A.; Ekker, S.C. A Primer for Morpholino Use in Zebrafish. Zebrafish 2009, 6, 69–77. [Google Scholar] [CrossRef]
- Joshi, P.; Liang, J.O.; DiMonte, K.; Sullivan, J.; Pimplikar, S.W. Amyloid Precursor Protein Is Required for Convergent-Extension Movements during Zebrafish Development. Dev. Biol. 2009, 335, 1–11. [Google Scholar] [CrossRef]
- Nornes, S.; Newman, M.; Verdile, G.; Wells, S.; Stoick-Cooper, C.L.; Tucker, B.; Frederich-Sleptsova, I.; Martins, R.; Lardelli, M. Interference with Splicing of Presenilin Transcripts Has Potent Dominant Negative Effects on Presenilin Activity. Hum. Mol. Genet. 2008, 17, 402–412. [Google Scholar] [CrossRef]
- Saleem, S.; Kannan, R.R. Zebrafish: An Emerging Real-Time Model System to Study Alzheimer’s Disease and Neurospecific Drug Discovery. Cell Death Discov. 2018, 4, 45. [Google Scholar] [CrossRef]
- Tomasiewicz, H.G.; Flaherty, D.B.; Soria, J.P.; Wood, J.G. Transgenic Zebrafish Model of Neurodegeneration. J. Neurosci. Res. 2002, 70, 734–745. [Google Scholar] [CrossRef]
- Paquet, D.; Bhat, R.; Sydow, A.; Mandelkow, E.M.; Berg, S.; Hellberg, S.; Fälting, J.; Distel, M.; Köster, R.W.; Schmid, B.; et al. A Zebrafish Model of Tauopathy Allows in Vivo Imaging of Neuronal Cell Death and Drug Evaluation. J. Clin. Investig. 2009, 119, 1382–1395. [Google Scholar] [CrossRef] [PubMed]
- Nada, S.E.; Williams, F.E.; Shah, Z.A. Development of a Novel and Robust Pharmacological Model of Okadaic Acid-Induced Alzheimer’s Disease in Zebrafish. CNS Neurol. Disord.-Drug Targets 2016, 15, 86–94. [Google Scholar] [CrossRef]
- Koehler, D.; Williams, F.E. Utilizing Zebrafish and Okadaic Acid to Study Alzheimer’s Disease. Neural Regen. Res. 2018, 13, 1538–1541. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, P.; Thomas, A.K.; Zhang, Y.; Kizil, C. The Effects of Aging on Amyloid-Β42-Induced Neurodegeneration and Regeneration in Adult Zebrafish Brain. Neurogenesis 2017, 4, e1322666. [Google Scholar] [CrossRef] [PubMed]
- Nadiga, A.P.R.; Suman; Krishna, K.L. A Novel Zebrafish Model of Alzheimer’s Disease by Aluminium Chloride; Involving Nitro-Oxidative Stress, Neuroinflammation and Cholinergic Pathway. Eur. J. Pharmacol. 2024, 965, 176332. [Google Scholar] [CrossRef]
- Koehler, D.; Shah, Z.A.; Hensley, K.; Williams, F.E. Lanthionine Ketimine-5-Ethyl Ester Provides Neuroprotection in a Zebrafish Model of Okadaic Acid-Induced Alzheimer’s Disease. Neurochem. Int. 2018, 115, 61–68. [Google Scholar] [CrossRef]
- Arslanova, D.; Yang, T.; Xu, X.; Wong, S.T.; Augelli-Szafran, C.E.; Xia, W. Phenotypic Analysis of Images of Zebrafish Treated with Alzheimer’s γ-Secretase Inhibitors. BMC Biotechnol. 2010, 10, 24. [Google Scholar] [CrossRef]
- Bobrovskikh, A.V.; Zubairova, U.S.; Naumenko, L.G.; Doroshkov, A.V. Catching the Big Fish in Big Data: A Meta-Analysis of Zebrafish Kidney ScRNA-Seq Datasets Highlights Conserved Molecular Profiles of Macrophages and Neutrophils in Vertebrates. Biology 2024, 13, 773. [Google Scholar] [CrossRef]
- Bobrovskikh, A.V.; Zubairova, U.S.; Bondar, E.I.; Doroshkov, A.V.; Lavrekha, V.V. Transcriptomic Data Meta-Analysis Sheds Light on High Light Response in Arabidopsis thaliana L. Int. J. Mol. Sci. 2022, 23, 4455. [Google Scholar] [CrossRef]
- Moloney, A.; Sattelle, D.B.; Lomas, D.A.; Crowther, D.C. Alzheimer’s Disease: Insights from Drosophila Melanogaster Models. Trends Biochem. Sci. 2010, 35, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Iijima, K.; Liu, H.P.; Chiang, A.S.; Hearn, S.A.; Konsolaki, M.; Zhong, Y. Dissecting the Pathological Effects of Human Aβ40 and Aβ42 in Drosophila: A Potential Model for Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2004, 101, 6623–6628. [Google Scholar] [CrossRef] [PubMed]
- Shulman, J.M.; Imboywa, S.; Giagtzoglou, N.; Powers, M.P.; Hu, Y.; Devenport, D.; Chipendo, P.; Chibnik, L.B.; Diamond, A.; Perrimon, N.; et al. Functional Screening in Drosophila Identifies Alzheimer’s Disease Susceptibility Genes and Implicates Tau-Mediated Mechanisms. Hum. Mol. Genet. 2014, 23, 870–877. [Google Scholar] [CrossRef]
- Rosen, D.R.; Martin-Morris, L.; Luo, L.; White, K. A Drosophila Gene Encoding a Protein Resembling the Human β-Amyloid Protein Precursor. Proc. Natl. Acad. Sci. USA 1989, 86, 2478–2482. [Google Scholar] [CrossRef] [PubMed]
- Greeve, I.; Kretzschmar, D.; Tschäpe, J.A.; Beyn, A.; Brellinger, C.; Schweizer, M.; Nitsch, R.M.; Reifegerste, R. Age-Dependent Neurodegeneration and Alzheimer-Amyloid Plaque Formation in Transgenic Drosophila. J. Neurosci. 2004, 24, 3899–3906. [Google Scholar] [CrossRef]
- Jackson, G.R.; Wiedau-Pazos, M.; Sang, T.K.; Wagle, N.; Brown, C.A.; Massachi, S.; Geschwind, D.H. Human Wild-Type Tau Interacts with Wingless Pathway Components and Produces Neurofibrillary Pathology in Drosophila. Neuron 2002, 34, 509–519. [Google Scholar] [CrossRef]
- Lee, D.; O’Dowd, D.K. Fast Excitatory Synaptic Transmission Mediated by Nicotinic Acetylcholine Receptors in Drosophila Neurons. J. Neurosci. 1999, 19, 5311–5321. [Google Scholar] [CrossRef]
- Van Pelt, K.M.; Truttmann, M.C. Caenorhabditis Elegans as a Model System for Studying Aging-Associated Neurodegenerative Diseases. Transl. Med. Aging 2020, 4, 60–72. [Google Scholar] [CrossRef]
- Alvarez, J.; Alvarez-Illera, P.; Santo-Domingo, J.; Fonteriz, R.I.; Montero, M. Modeling Alzheimer’s Disease in Caenorhabditis Elegans. Biomedicines 2022, 10, 288. [Google Scholar] [CrossRef]
- Griffin, E.F.; Caldwell, K.A.; Caldwell, G.A. Genetic and Pharmacological Discovery for Alzheimer’s Disease Using Caenorhabditis Elegans. ACS Chem. Neurosci. 2017, 8, 2596–2606. [Google Scholar] [CrossRef]
- Yi, B.; Sahn, J.J.; Ardestani, P.M.; Evans, A.K.; Scott, L.L.; Chan, J.Z.; Iyer, S.; Crisp, A.; Zuniga, G.; Pierce, J.T.; et al. Small Molecule Modulator of Sigma 2 Receptor Is Neuroprotective and Reduces Cognitive Deficits and Neuroinflammation in Experimental Models of Alzheimer’s Disease. J. Neurochem. 2017, 140, 561–575. [Google Scholar] [CrossRef] [PubMed]
- Link, C.D. Expression of Human β-Amyloid Peptide in Transgenic Caenorhabditis Elegans. Proc. Natl. Acad. Sci. USA 1995, 92, 9368–9372. [Google Scholar] [CrossRef]
- Quartey, M.O.; Nyarko, J.N.K.; Maley, J.M.; Barnes, J.R.; Bolanos, M.A.C.; Heistad, R.M.; Knudsen, K.J.; Pennington, P.R.; Buttigieg, J.; De Carvalho, C.E.; et al. The Aβ(1–38) Peptide Is a Negative Regulator of the Aβ(1–42) Peptide Implicated in Alzheimer Disease Progression. Sci. Rep. 2021, 11, 431. [Google Scholar] [CrossRef] [PubMed]
- Hsien, L.C.; Kaur, P.; Teo, E.; Lam, V.Y.M.; Zhu, F.; Kibat, C.; Gruber, J.; Mathuru, A.; Tolwinski, N.S. Application of Optogenetic Amyloid-β Distinguishes between Metabolic and Physical Damage in Neurodegeneration. Elife 2020, 9, 836593. [Google Scholar] [CrossRef]
- Aprile, F.A.; Sormanni, P.; Podpolny, M.; Chhangur, S.; Needham, L.M.; Ruggeri, F.S.; Perni, M.; Limbocker, R.; Heller, G.T.; Sneideris, T.; et al. Rational Design of a Conformation-Specific Antibody for the Quantification of Aβ Oligomers. Proc. Natl. Acad. Sci. USA 2020, 117, 13509–13518. [Google Scholar] [CrossRef]
- Gallrein, C.; Iburg, M.; Michelberger, T.; Koçak, A.; Puchkov, D.; Liu, F.; Ayala Mariscal, S.M.; Nayak, T.; Kaminski Schierle, G.S.; Kirstein, J. Novel Amyloid-Beta Pathology C. Elegans Model Reveals Distinct Neurons as Seeds of Pathogenicity. Prog. Neurobiol. 2021, 198, 101907. [Google Scholar] [CrossRef]
- Pir, G.J.; Choudhary, B.; Mandelkow, E. Caenorhabditis Elegans Models of Tauopathy. FASEB J. 2017, 31, 5137–5148. [Google Scholar] [CrossRef]
- Liang, J.J.H.; McKinnon, I.A.; Rankin, C.H. The Contribution of C. Elegans Neurogenetics to Understanding Neurodegenerative Diseases. J. Neurogenet. 2020, 34, 527–548. [Google Scholar] [CrossRef]
- Chew, Y.L.; Fan, X.; Götz, J.; Nicholas, H.R. PTL-1 Regulates Neuronal Integrity and Lifespan in C. Elegans. J. Cell Sci. 2013, 126, 2079–2091. [Google Scholar] [CrossRef]
- Kraemer, B.C.; Burgess, J.K.; Chen, J.H.; Thomas, J.H.; Schellenberg, G.D. Molecular Pathways That Influence Human Tau-Induced Pathology in Caenorhabditis Elegans. Hum. Mol. Genet. 2006, 15, 1483–1496. [Google Scholar] [CrossRef]
- Ayyadevara, S.; Balasubramaniam, M.; Parcon, P.A.; Barger, S.W.; Griffin, W.S.T.; Alla, R.; Tackett, A.J.; Mackintosh, S.G.; Petricoin, E.; Zhou, W.; et al. Proteins That Mediate Protein Aggregation and Cytotoxicity Distinguish Alzheimer’s Hippocampus from Normal Controls. Aging Cell 2016, 15, 924–939. [Google Scholar] [CrossRef] [PubMed]
- Pir, G.J.; Choudhary, B.; Mandelkow, E.; Mandelkow, E.M. Tau Mutant A152T, a Risk Factor for FTD/PSP, Induces Neuronal Dysfunction and Reduced Lifespan Independently of Aggregation in a C. Elegans Tauopathy Model. Mol. Neurodegener. 2016, 11, 33. [Google Scholar] [CrossRef]
- Wang, C.; Saar, V.; Leung, K.L.; Chen, L.; Wong, G. Human Amyloid β Peptide and Tau Co-Expression Impairs Behavior and Causes Specific Gene Expression Changes in Caenorhabditis Elegans. Neurobiol. Dis. 2018, 109, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy Inhibits Amyloid-β and Tau Pathology and Reverses Cognitive Deficits in Models of Alzheimer’s Disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, A.; Gupta, S.M.; Dwivedi, S.; Kumar, D.; Shaikh, M.F.; Negi, A. Preclinical Models for Alzheimer’s Disease: Past, Present, and Future Approaches. ACS Omega 2022, 7, 47504–47517. [Google Scholar] [CrossRef] [PubMed]
- Tapia, R.; Peña, F.; Arias, C. Neurotoxic and Synaptic Effects of Okadaic Acid, an Inhibitor of Protein Phosphatases. Neurochem. Res. 1999, 24, 1423–1430. [Google Scholar] [CrossRef]
- Sontag, J.M.; Sontag, E. Protein Phosphatase 2A Dysfunction in Alzheimer’s Disease. Front. Mol. Neurosci. 2014, 7, 16. [Google Scholar] [CrossRef]
- Cohen, P.T.W.; Brewis, N.D.; Hughes, V.; Mann, D.J. Protein Serine/Threonine Phosphatases; an Expanding Family. FEBS Lett. 1990, 268, 355–359. [Google Scholar] [CrossRef]
- Arendt, T.; Holzer, M.; Fruth, R.; Brückner, M.K.; Gärtner, U. Phosphorylation of Tau, Aβ-Formation, and Apoptosis after in Vivo Inhibition of PP-1 and PP-2A. Neurobiol. Aging 1998, 19, 3–13. [Google Scholar] [CrossRef]
- Kamat, P.K.; Tota, S.; Rai, S.; Swarnkar, S.; Shukla, R.; Nath, C. A Study on Neuroinflammatory Marker in Brain Areas of Okadaic Acid (ICV) Induced Memory Impaired Rats. Life Sci. 2012, 90, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.Y.; Zhang, Y. Animal Models of Alzheimer’s Disease: Applications, Evaluation, and Perspectives. Zool. Res. 2022, 43, 1026–1040. [Google Scholar] [CrossRef]
- Tilson, H.A.; Rogers, B.C.; Grimes, L.; Harry, G.J.; Peterson, N.J.; Hong, J.S.; Dyer, R.S. Time-Dependent Neurobiological Effects of Colchicine Administered Directly into the Hippocampus of Rats. Brain Res. 1987, 408, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Geddes, J.W.; Bondada, V.; Keller, J.N. Effects of Intrahippocampal Colchicine Administration on the Levels and Localization of Microtubule-Associated Proteins, Tau and MAP2. Brain Res. 1994, 633, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Merrick, S.E.; Demoise, D.C.; Lee, V.M.Y. Site-Specific Dephosphorylation of Tau Protein at Ser202/Thr205 in Response to Microtubule Depolymerization in Cultured Human Neurons Involves Protein Phosphatase 2A. J. Biol. Chem. 1996, 271, 5589–5594. [Google Scholar] [CrossRef] [PubMed]
- Riedel, G.; Kang, S.H.; Choi, D.Y.; Platt, B. Scopolamine-Induced Deficits in Social Memory in Mice: Reversal by Donepezil. Behav. Brain Res. 2009, 204, 217–225. [Google Scholar] [CrossRef]
- Schliebs, R.; Arendt, T. The Significance of the Cholinergic System in the Brain during Aging and in Alzheimer’s Disease. J. Neural Transm. 2006, 113, 1625–1644. [Google Scholar] [CrossRef]
- Safar, M.M.; Arab, H.H.; Rizk, S.M.; El-Maraghy, S.A. Bone Marrow-Derived Endothelial Progenitor Cells Protect Against Scopolamine-Induced Alzheimer-Like Pathological Aberrations. Mol. Neurobiol. 2016, 53, 1403–1418. [Google Scholar] [CrossRef]
- Mostafa, D.K.; Ismail, C.A.; Ghareeb, D.A. Differential Metformin Dose-Dependent Effects on Cognition in Rats: Role of Akt. Psychopharmacology 2016, 233, 2513–2524. [Google Scholar] [CrossRef]
- Yeo, H.G.; Lee, Y.; Jeon, C.Y.; Jeong, K.J.; Jin, Y.B.; Kang, P.; Kim, S.U.; Kim, J.S.; Huh, J.W.; Kim, Y.H.; et al. Characterization of Cerebral Damage in a Monkey Model of Alzheimer’s Disease Induced by Intracerebroventricular Injection of Streptozotocin. J. Alzheimer’s Dis. 2015, 46, 989–1005. [Google Scholar] [CrossRef]
- Neha; Sodhi, R. K.; Jaggi, A.S.; Singh, N. Animal Models of Dementia and Cognitive Dysfunction. Life Sci. 2014, 109, 73–86. [Google Scholar] [CrossRef]
- Han, S.; Yang, E.M.; Hur, E.-M. A Brief Guide for Gene Delivery to the Brain Using Adeno-Associated Viral Vectors. Mol. Cells 2025, 48, 100189. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C.R.R.; Emson, P.C. AChE-Stained Horizontal Sections of the Rat Brain in Stereotaxic Coordinates. J. Neurosci. Methods 1980, 3, 129–149. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://labs.gaidi.ca/mouse-brain-atlas/ (accessed on 30 April 2025).
- Götz, J.; Chen, F.; Van Dorpe, J.; Nitsch, R.M. Formation of Neurofibrillary Tangles in P301L Tau Transgenic Mice Induced by Aβ42 Fibrils. Science 2001, 293, 1491–1495. [Google Scholar] [CrossRef]
- Stöhr, J.; Watts, J.C.; Mensinger, Z.L.; Oehler, A.; Grillo, S.K.; DeArmond, S.J.; Prusiner, S.B.; Giles, K. Purified and Synthetic Alzheimer’s Amyloid Beta (Aβ) Prions. Proc. Natl. Acad. Sci. USA 2012, 109, 11025–11030. [Google Scholar] [CrossRef]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and Spreading of Tauopathy in Transgenic Mouse Brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Z.; Cooper, J.; Murray, T.K.; Garn, K.; McNaughton, E.; Clarke, H.; Parhizkar, S.; Ward, M.A.; Cavallini, A.; Jackson, S.; et al. A Novel in Vivo Model of Tau Propagation with Rapid and Progressive Neurofibrillary Tangle Pathology: The Pattern of Spread Is Determined by Connectivity, Not Proximity. Acta Neuropathol. 2014, 127, 667–683. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s Disease Is a Synaptic Failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef]
- Selkoe, D.J. Soluble Oligomers of the Amyloid β-Protein Impair Synaptic Plasticity and Behavior. Behav. Brain Res. 2008, 192, 106–113. [Google Scholar] [CrossRef]
- Shankar, G.M.; Walsh, D.M. Alzheimer’s Disease: Synaptic Dysfunction and Aβ. Mol. Neurodegener. 2009, 4, 48. [Google Scholar] [CrossRef]
- Fá, M.; Puzzo, D.; Piacentini, R.; Staniszewski, A.; Zhang, H.; Baltrons, M.A.; Li Puma, D.D.; Chatterjee, I.; Li, J.; Saeed, F.; et al. Extracellular Tau Oligomers Produce An Immediate Impairment of LTP and Memory. Sci. Rep. 2016, 6, 19393. [Google Scholar] [CrossRef]
- Hayden, E.Y.; Teplow, D.B. Amyloid β-Protein Oligomers and Alzheimer’s Disease. Alzheimer’s Res. Ther. 2013, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Balducci, C.; Beeg, M.; Stravalaci, M.; Bastone, A.; Sclip, A.; Biasini, E.; Tapella, L.; Colombo, L.; Manzoni, C.; Borsello, T.; et al. Synthetic Amyloid-β Oligomers Impair Long-Term Memory Independently of Cellular Prion Protein. Proc. Natl. Acad. Sci. USA 2010, 107, 2295–2300. [Google Scholar] [CrossRef] [PubMed]
- Zussy, C.; Brureau, A.; Delair, B.; Marchal, S.; Keller, E.; Ixart, G.; Naert, G.; Meunier, J.; Chevallier, N.; Maurice, T.; et al. Time-Course and Regional Analyses of the Physiopathological Changes Induced after Cerebral Injection of an Amyloid β Fragment in Rats. Am. J. Pathol. 2011, 179, 315–334. [Google Scholar] [CrossRef]
- Kim, H.Y.; Lee, D.K.; Chung, B.R.; Kim, H.V.; Kim, Y. Intracerebroventricular Injection of Amyloid-# Peptides in Normal Mice to Acutely Induce Alzheimer-like Cognitive Deficits. J. Vis. Exp. 2016, 2016, 53308. [Google Scholar] [CrossRef]
- Kasza, Á.; Penke, B.; Frank, Z.; Bozsó, Z.; Szegedi, V.; Hunya, Á.; Németh, K.; Kozma, G.; Fülöp, L. Studies for Improving a Rat Model of Alzheimer’s Disease: ICV Administration of Well-Characterized β-Amyloid 1-42 Oligomers Induce Dysfunction in Spatial Memory. Molecules 2017, 22, 2007. [Google Scholar] [CrossRef] [PubMed]
- He, F.Q.; Qiu, B.Y.; Zhang, X.H.; Li, T.K.; Xie, Q.; Cui, D.J.; Huang, X.L.; Gan, H.T. Tetrandrine Attenuates Spatial Memory Impairment and Hippocampal Neuroinflammation via Inhibiting NF-ΚB Activation in a Rat Model of Alzheimer’s Disease Induced by Amyloid-β(1-42). Brain Res. 2011, 1384, 89–96. [Google Scholar] [CrossRef]
- Chacón, M.A.; Barría, M.I.; Soto, C.; Inestrosa, N.C. β-Sheet Breaker Peptide Prevents Aβ-Induced Spatial Memory Impairments with Partial Reduction of Amyloid Deposits. Mol. Psychiatry 2004, 9, 953–961. [Google Scholar] [CrossRef]
- Chen, Z.; Mengel, D.; Keshavan, A.; Rissman, R.A.; Billinton, A.; Perkinton, M.; Percival-Alwyn, J.; Schultz, A.; Properzi, M.; Johnson, K.; et al. Learnings about the Complexity of Extracellular Tau Aid Development of a Blood-Based Screen for Alzheimer’s Disease. Alzheimer’s Dement. 2019, 15, 487–496. [Google Scholar] [CrossRef]
- Lasagna-Reeves, C.; Castillo-Carranza, D.L.; Jackson, G.R.; Kayed, R. Tau Oligomers as Potential Targets for Immunotherapy for Alzheimers Disease and Tauopathies. Curr. Alzheimer Res. 2011, 8, 659–665. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Kopeikina, K.J.; Koffie, R.M.; De Calignon, A.; Hyman, B.T. Are Tangles as Toxic as They Look? J. Mol. Neurosci. 2011, 45, 438–444. [Google Scholar] [CrossRef]
- Ondrejcak, T.; Klyubin, I.; Hu, N.W.; O’Malley, T.T.; Corbett, G.T.; Winters, R.; Perkinton, M.S.; Billinton, A.; Prenderville, J.A.; Walsh, D.M.; et al. Tau and Amyloid b Protein in Patient-Derived Aqueous Brain Extracts Act Concomitantly to Disrupt Long-Term Potentiation in Vivo. J. Neurosci. 2023, 43, 5870–5879. [Google Scholar] [CrossRef]
- Ondrejcak, T.; Klyubin, I.; Corbett, G.T.; Fraser, G.; Hong, W.; Mably, A.J.; Gardener, M.; Hammersley, J.; Perkinton, M.S.; Billinton, A.; et al. Cellular Prion Protein Mediates the Disruption of Hippocampal Synaptic Plasticity by Soluble Tau in Vivo. J. Neurosci. 2018, 38, 10595–10606. [Google Scholar] [CrossRef]
- Puzzo, D.; Piacentini, R.; Fá, M.; Gulisano, W.; Li Puma, D.D.; Staniszewski, A.; Zhang, H.; Tropea, M.R.; Cocco, S.; Palmeri, A.; et al. LTP and Memory Impairment Caused by Extracellular Aβ and Tau Oligomers Is APP-Dependent. Elife 2017, 6, e26991. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Muñoz, M.J.; Gerson, J.; Castillo-Carranza, D.L. Tau Oligomers: The Toxic Player at Synapses in Alzheimer’s Disease. Front. Cell. Neurosci. 2015, 9, 464. [Google Scholar] [CrossRef]
- Zhang, J.; Ke, K.F.; Liu, Z.; Qiu, Y.H.; Peng, Y.P. Th17 Cell-Mediated Neuroinflammation Is Involved in Neurodegeneration of Aβ1-42-Induced Alzheimer’s Disease Model Rats. PLoS ONE 2013, 8, e75786. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Haam, J.; Walker, M.; Scappini, E.; Naughton, J.; Martin, N.P. Recombinant Viral Vectors as Neuroscience Tools. Curr. Protoc. Neurosci. 2019, 87, e67. [Google Scholar] [CrossRef] [PubMed]
- Penaud-Budloo, M.; Le Guiner, C.; Nowrouzi, A.; Toromanoff, A.; Chérel, Y.; Chenuaud, P.; Schmidt, M.; von Kalle, C.; Rolling, F.; Moullier, P.; et al. Adeno-Associated Virus Vector Genomes Persist as Episomal Chromatin in Primate Muscle. J. Virol. 2008, 82, 7875–7885. [Google Scholar] [CrossRef]
- Wu, Z.; Yang, H.; Colosi, P. Effect of Genome Size on AAV Vector Packaging. Mol. Ther. 2010, 18, 80–86. [Google Scholar] [CrossRef]
- Sakuma, T.; Barry, M.A.; Ikeda, Y. Lentiviral Vectors: Basic to Translational. Biochem. J. 2012, 443, 603–618. [Google Scholar] [CrossRef]
- Sakurai, K.; Zhao, S.; Takatoh, J.; Rodriguez, E.; Lu, J.; Leavitt, A.D.; Fu, M.; Han, B.X.; Wang, F. Capturing and Manipulating Activated Neuronal Ensembles with CANE Delineates a Hypothalamic Social-Fear Circuit. Neuron 2016, 92, 739–753. [Google Scholar] [CrossRef]
- Lawlor, P.A.; Bland, R.J.; Das, P.; Price, R.W.; Holloway, V.; Smithson, L.; Dicker, B.L.; During, M.J.; Young, D.; Golde, T.E. Novel Rat Alzheimer’s Disease Models Based on AAV-Mediated Gene Transfer to Selectively Increase Hippocampal Aβ Levels. Mol. Neurodegener. 2007, 2, 11. [Google Scholar] [CrossRef] [PubMed]
- Koukouli, F.; Rooy, M.; Maskos, U. Early and Progressive Deficit of Neuronal Activity Patterns in a Model of Local Amyloid Pathology in Mouse Prefrontal Cortex. Aging (Albany N. Y.) 2016, 8, 3430–3449. [Google Scholar] [CrossRef] [PubMed]
- Parsi, S.; Pandamooz, S.; Heidari, S.; Naji, M.; Morfini, G.; Ahmadiani, A.; Dargahi, L. A Novel Rat Model of Alzheimer’s Disease Based on Lentiviral-Mediated Expression of Mutant APP. Neuroscience 2015, 284, 99–106. [Google Scholar] [CrossRef]
- Caillierez, R.; Bégard, S.; Lécolle, K.; Deramecourt, V.; Zommer, N.; Dujardin, S.; Loyens, A.; Dufour, N.; Aurégan, G.; Winderickx, J.; et al. Lentiviral Delivery of the Human Wild-Type Tau Protein Mediates a Slow and Progressive Neurodegenerative Tau Pathology in the Rat Brain. Mol. Ther. 2013, 21, 1358–1368. [Google Scholar] [CrossRef] [PubMed]
- Osinde, M.; Clavaguera, F.; May-Nass, R.; Tolnay, M.; Dev, K.K. Lentivirus Tau (P301S) Expression in Adult Amyloid Precursor Protein (APP)-Transgenic Mice Leads to Tangle Formation. Neuropathol. Appl. Neurobiol. 2008, 34, 523–531. [Google Scholar] [CrossRef]
- Cook, C.; Kang, S.S.; Carlomagno, Y.; Lin, W.L.; Yue, M.; Kurti, A.; Shinohara, M.; Jansen-West, K.; Perkerson, E.; Castanedes-Casey, M.; et al. Tau Deposition Drives Neuropathological, Inflammatory and Behavioral Abnormalities Independently of Neuronal Loss in a Novel Mouse Model. Hum. Mol. Genet. 2015, 24, 6198–6212. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Zouboulis, C.C.; Makrantonaki, E.; Hossini, A.M. Skin Mirrors Brain: A Chance for Alzheimer’s Disease Research. Adv. Exp. Med. Biol. 2021, 1339, 371–380. [Google Scholar] [CrossRef]
- Sharipova, D.V.; Kovalenko, V.R.; Bairamova, E.M.; Vartanova, V.A.; Grigor’eva, E.V.; Vyatkin, Y.V.; Khabarova, E.A.; Rzaev, D.A.; Zakian, S.M.; Medvedev, S.P. Generation of Two IPSC Lines, (ICGi015-A and ICGi015-B), by Reprogramming Peripheral Blood Mononuclear Cells from a Patient with Parkinson’s Disease. Stem Cell Res. 2019, 41, 101652. [Google Scholar] [CrossRef]
- Grigor’eva, E.V.; Drozdova, E.S.; Sorogina, D.A.; Malakhova, A.A.; Pavlova, S.V.; Vyatkin, Y.V.; Khabarova, E.A.; Rzaev, J.A.; Medvedev, S.P.; Zakian, S.M. Generation of Induced Pluripotent Stem Cell Line, ICGi034-A, by Reprogramming Peripheral Blood Mononuclear Cells from a Patient with Parkinson’s Disease Associated with GBA Mutation. Stem Cell Res. 2022, 59, 102651. [Google Scholar] [CrossRef]
- Soldner, F.; Hockemeyer, D.; Beard, C.; Gao, Q.; Bell, G.W.; Cook, E.G.; Hargus, G.; Blak, A.; Cooper, O.; Mitalipova, M.; et al. Parkinson’s Disease Patient-Derived Induced Pluripotent Stem Cells Free of Viral Reprogramming Factors. Cell 2009, 136, 964–977. [Google Scholar] [CrossRef] [PubMed]
- Mitne-Neto, M.; Machado-Costa, M.; Marchetto, M.C.N.; Bengtson, M.H.; Joazeiro, C.A.; Tsuda, H.; Bellen, H.J.; Silva, H.C.A.; Oliveira, A.S.B.; Lazar, M.; et al. Downregulation of VAPB Expression in Motor Neurons Derived from Induced Pluripotent Stem Cells of ALS8 Patients. Hum. Mol. Genet. 2011, 20, 3642–3652. [Google Scholar] [CrossRef] [PubMed]
- Ustyantseva, E.I.; Medvedev, S.P.; Zakian, S.M. Studying ALS: Current Approaches, Effect on Potential Treatment Strategy. Adv. Exp. Med. Biol. 2020, 1241, 195–217. [Google Scholar] [CrossRef]
- Ustyantseva, E.I.; Medvedev, S.P.; Vetchinova, A.S.; Illarioshkin, S.N.; Leonov, S.V.; Zakian, S.M. Generation of an Induced Pluripotent Stem Cell Line, ICGi014-A, by Reprogramming Peripheral Blood Mononuclear Cells from a Patient with Homozygous D90A Mutation in SOD1 Causing Amyotrophic Lateral Sclerosis. Stem Cell Res. 2020, 42, 101675. [Google Scholar] [CrossRef]
- Ebert, A.D.; Yu, J.; Rose, F.F.; Mattis, V.B.; Lorson, C.L.; Thomson, J.A.; Svendsen, C.N. Induced Pluripotent Stem Cells from a Spinal Muscular Atrophy Patient. Nature 2009, 457, 277–280. [Google Scholar] [CrossRef]
- Valetdinova, K.R.; Maretina, M.A.; Kuranova, M.L.; Grigor’eva, E.V.; Minina, Y.M.; Kizilova, E.A.; Kiselev, A.V.; Medvedev, S.P.; Baranov, V.S.; Zakian, S.M. Generation of Two Spinal Muscular Atrophy (SMA) Type I Patient-Derived Induced Pluripotent Stem Cell (IPSC) Lines and Two SMA Type II Patient-Derived IPSC Lines. Stem Cell Res. 2019, 34, 101376. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Papapetrou, E.P.; Kim, H.; Chambers, S.M.; Tomishima, M.J.; Fasano, C.A.; Ganat, Y.M.; Menon, J.; Shimizu, F.; Viale, A.; et al. Modelling Pathogenesis and Treatment of Familial Dysautonomia Using Patient-Specific IPSCs. Nature 2009, 461, 402–406. [Google Scholar] [CrossRef]
- Marchetto, M.C.N.; Carromeu, C.; Acab, A.; Yu, D.; Yeo, G.W.; Mu, Y.; Chen, G.; Gage, F.H.; Muotri, A.R. A Model for Neural Development and Treatment of Rett Syndrome Using Human Induced Pluripotent Stem Cells. Cell 2010, 143, 527–539. [Google Scholar] [CrossRef]
- Brennand, K.J.; Simone, A.; Jou, J.; Gelboin-Burkhart, C.; Tran, N.; Sangar, S.; Li, Y.; Mu, Y.; Chen, G.; Yu, D.; et al. Modelling Schizophrenia Using Human Induced Pluripotent Stem Cells. Nature 2011, 473, 221–225. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Hossini, A.M.; Megges, M.; Prigione, A.; Lichtner, B.; Toliat, M.R.; Wruck, W.; Schröter, F.; Nuernberg, P.; Kroll, H.; Makrantonaki, E.; et al. Induced Pluripotent Stem Cell-Derived Neuronal Cells from a Sporadic Alzheimer’s Disease Donor as a Model for Investigating AD-Associated Gene Regulatory Networks. BMC Genom. 2015, 16, 84. [Google Scholar] [CrossRef]
- Yagi, T.; Ito, D.; Okada, Y.; Akamatsu, W.; Nihei, Y.; Yoshizaki, T.; Yamanaka, S.; Okano, H.; Suzuki, N. Modeling Familial Alzheimer’s Disease with Induced Pluripotent Stem Cells. Hum. Mol. Genet. 2011, 20, 4530–4539. [Google Scholar] [CrossRef]
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C.; Hefferan, M.P.; Van Gorp, S.; Nazor, K.L.; Boscolo, F.S.; et al. Probing Sporadic and Familial Alzheimer’s Disease Using Induced Pluripotent Stem Cells. Nature 2012, 482, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Grigor’eva, E.V.; Malankhanova, T.B.; Ustyantseva, E.I.; Minina, J.M.; Redina, O.E.; Morozov, V.V.; Shevela, A.I.; Zakian, S.M.; Medvedev, S.P. Generation of Two IPSC Lines (ICGi008-A and ICGi008-B) from Skin Fibroblasts of a Patient with Early-Onset Alzheimer’s Disease Caused by London Familial APP Mutation (V717I). Stem Cell Res. 2019, 36, 101415. [Google Scholar] [CrossRef]
- Lunn, J.S.; Sakowski, S.A.; Federici, T.; Glass, J.D.; Boulis, N.M.; Feldman, E.L. Stem Cell Technology for the Study and Treatment of Motor Neuron Diseases. Regen. Med. 2011, 6, 201–213. [Google Scholar] [CrossRef]
- Amemori, T.; Romanyuk, N.; Jendelova, P.; Herynek, V.; Turnovcova, K.; Prochazka, P.; Kapcalova, M.; Cocks, G.; Price, J.; Sykova, E. Human Conditionally Immortalized Neural Stem Cells Improve Locomotor Function after Spinal Cord Injury in the Rat. Stem Cell Res. Ther. 2013, 4, 68. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Madhu, L.N.; Babu, R.S.; Shankar, G.; Kotian, S.; Nagarajan, A.; Upadhya, R.; Narvekar, E.; Cai, J.J.; Shetty, A.K. Extracellular Vesicles from HiPSC-Derived NSCs Protect Human Neurons against Aβ-42 Oligomers Induced Neurodegeneration, Mitochondrial Dysfunction and Tau Phosphorylation. Stem Cell Res. Ther. 2025, 16, 191. [Google Scholar] [CrossRef]
- Kondo, T.; Asai, M.; Tsukita, K.; Kutoku, Y.; Ohsawa, Y.; Sunada, Y.; Imamura, K.; Egawa, N.; Yahata, N.; Okita, K.; et al. Modeling Alzheimer’s Disease with IPSCs Reveals Stress Phenotypes Associated with Intracellular Aβ and Differential Drug Responsiveness. Cell Stem Cell 2013, 12, 487–496. [Google Scholar] [CrossRef]
- Muratore, C.R.; Rice, H.C.; Srikanth, P.; Callahan, D.G.; Shin, T.; Benjamin, L.N.P.; Walsh, D.M.; Selkoe, D.J.; Young-Pearse, T.L. The Familial Alzheimer’s Disease APPV717I Mutation Alters APP Processing and Tau Expression in IPSC-Derived Neurons. Hum. Mol. Genet. 2014, 23, 3523–3536. [Google Scholar] [CrossRef]
- Sproul, A.A.; Jacob, S.; Pre, D.; Kim, S.H.; Nestor, M.W.; Navarro-Sobrino, M.; Santa-Maria, I.; Zimmer, M.; Aubry, S.; Steele, J.W.; et al. Characterization and Molecular Profiling of PSEN1 Familial Alzheimer’s Disease IPSC-Derived Neural Progenitors. PLoS ONE 2014, 9, e84547. [Google Scholar] [CrossRef]
- Koch, P.; Tamboli, I.Y.; Mertens, J.; Wunderlich, P.; Ladewig, J.; Stüber, K.; Esselmann, H.; Wiltfang, J.; Brüstle, O.; Walter, J. Presenilin-1 L166P Mutant Human Pluripotent Stem Cell-Derived Neurons Exhibit Partial Loss of γ-Secretase Activity in Endogenous Amyloid-β Generation. Am. J. Pathol. 2012, 180, 2404–2416. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Virumbrales, M.; Moreno, C.L.; Kruglikov, I.; Marazuela, P.; Sproul, A.; Jacob, S.; Zimmer, M.; Paull, D.; Zhang, B.; Schadt, E.E.; et al. CRISPR/Cas9-Correctable Mutation-Related Molecular and Physiological Phenotypes in IPSC-Derived Alzheimer’s PSEN2N141I Neurons. Acta Neuropathol. Commun. 2017, 5, 77. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, M.; Petersen, A.J.; Naumenko, N.; Puttonen, K.; Lehtonen, Š.; Gubert Olivé, M.; Shakirzyanova, A.; Leskelä, S.; Sarajärvi, T.; Viitanen, M.; et al. PSEN1 Mutant IPSC-Derived Model Reveals Severe Astrocyte Pathology in Alzheimer’s Disease. Stem Cell Rep. 2017, 9, 1885–1897. [Google Scholar] [CrossRef]
- Konttinen, H.; Cabral-da-Silva, M.e.C.; Ohtonen, S.; Wojciechowski, S.; Shakirzyanova, A.; Caligola, S.; Giugno, R.; Ishchenko, Y.; Hernández, D.; Fazaludeen, M.F.; et al. PSEN1ΔE9, APPswe, and APOE4 Confer Disparate Phenotypes in Human IPSC-Derived Microglia. Stem Cell Rep. 2019, 13, 669–683. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; Malhotra, M.; Curtin, C.M.; O’Brien, F.J.; O’Driscoll, C.M. Life in 3D Is Never Flat: 3D Models to Optimise Drug Delivery. J. Control. Release 2015, 215, 39–54. [Google Scholar] [CrossRef]
- Slanzi, A.; Iannoto, G.; Rossi, B.; Zenaro, E.; Constantin, G. In Vitro Models of Neurodegenerative Diseases. Front. Cell Dev. Biol. 2020, 8, 328. [Google Scholar] [CrossRef]
- Bosi, S.; Rauti, R.; Laishram, J.; Turco, A.; Lonardoni, D.; Nieus, T.; Prato, M.; Scaini, D.; Ballerini, L. From 2D to 3D: Novel Nanostructured Scaffolds to Investigate Signalling in Reconstructed Neuronal Networks. Sci. Rep. 2015, 5, 9562. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Kim, Y.H.; Quinti, L.; Tanzi, R.E.; Kim, D.Y. 3D Culture Models of Alzheimer’s Disease: A Road Map to a “Cure-in-a-Dish”. Mol. Neurodegener. 2016, 11, 75. [Google Scholar] [CrossRef] [PubMed]
- Guttikonda, S.R.; Sikkema, L.; Tchieu, J.; Saurat, N.; Walsh, R.M.; Harschnitz, O.; Ciceri, G.; Sneeboer, M.; Mazutis, L.; Setty, M.; et al. Fully Defined Human Pluripotent Stem Cell-Derived Microglia and Tri-Culture System Model C3 Production in Alzheimer’s Disease. Nat. Neurosci. 2021, 24, 343–354. [Google Scholar] [CrossRef]
- Flanagan, L.A.; Rebaza, L.M.; Derzic, S.; Schwartz, P.H.; Monuki, E.S. Regulation of Human Neural Precursor Cells by Laminin and Integrins. J. Neurosci. Res. 2006, 83, 845–856. [Google Scholar] [CrossRef]
- Park, H.J.; Kim, J.; Ryou, C. A Three-Dimensional Spheroid Co-Culture System of Neurons and Astrocytes Derived from Alzheimer’s Disease Patients for Drug Efficacy Testing. Cell Prolif. 2023, 56, e13399. [Google Scholar] [CrossRef] [PubMed]
- Louit, A.; Galbraith, T.; Berthod, F. In Vitro 3D Modeling of Neurodegenerative Diseases. Bioengineering 2023, 10, 93. [Google Scholar] [CrossRef] [PubMed]
- Boder, E.J.; Banerjee, I.A. Alzheimer’s Disease: Current Perspectives and Advances in Physiological Modeling. Bioengineering 2021, 8, 211. [Google Scholar] [CrossRef] [PubMed]
- Bédard, P.; Gauvin, S.; Ferland, K.; Caneparo, C.; Pellerin, È.; Chabaud, S.; Bolduc, S. Innovative Human Three-Dimensional Tissue-Engineered Models as an Alternative to Animal Testing. Bioengineering 2020, 7, 115. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, Y.H.; Hebisch, M.; Sliwinski, C.; Lee, S.; D’Avanzo, C.; Chen, H.; Hooli, B.; Asselin, C.; Muffat, J.; et al. A Three-Dimensional Human Neural Cell Culture Model of Alzheimer’s Disease. Nature 2014, 515, 274–278. [Google Scholar] [CrossRef]
- Simpson, L.W.; Szeto, G.L.; Boukari, H.; Good, T.A.; Leach, J.B. Impact of Four Common Hydrogels on Amyloid-β (Aβ) Aggregation and Cytotoxicity: Implications for 3d Models of Alzheimer’s Disease. ACS Omega 2020, 5, 20250–20260. [Google Scholar] [CrossRef]
- Castillo, G.M.; Lukito, W.; Peskind, E.; Raskind, M.; Kirschner, D.A.; Yee, A.G.; Snow, A.D. Laminin Inhibition of Beta-Amyloid Protein (Abeta) Fibrillogenesis and Identification of an Abeta Binding Site Localized to the Globular Domain Repeats on the Laminin a Chain. J. Neurosci. Res. 2000, 62, 451–462. [Google Scholar] [CrossRef]
- De Souza, N. Organoids. Nat. Methods 2018, 15, 23. [Google Scholar] [CrossRef]
- Lee, H.K.; Velazquez Sanchez, C.; Chen, M.; Morin, P.J.; Wells, J.M.; Hanlon, E.B.; Xia, W. Three Dimensional Human Neuro-Spheroid Model of Alzheimer’s Disease Based on Differentiated Induced Pluripotent Stem Cells. PLoS ONE 2016, 11, e0163072. [Google Scholar] [CrossRef]
- Pomeshchik, Y.; Klementieva, O.; Gil, J.; Martinsson, I.; Hansen, M.G.; de Vries, T.; Sancho-Balsells, A.; Russ, K.; Savchenko, E.; Collin, A.; et al. Human IPSC-Derived Hippocampal Spheroids: An Innovative Tool for Stratifying Alzheimer Disease Patient-Specific Cellular Phenotypes and Developing Therapies. Stem Cell Rep. 2020, 15, 256–273. [Google Scholar] [CrossRef]
- Bi, F.C.; Yang, X.H.; Cheng, X.Y.; Deng, W.B.; Guo, X.L.; Yang, H.; Wang, Y.; Li, J.; Yao, Y. Optimization of Cerebral Organoids: A More Qualified Model for Alzheimer’s Disease Research. Transl. Neurodegener. 2021, 10, 27. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Knoblich, J.A. Generation of Cerebral Organoids from Human Pluripotent Stem Cells. Nat. Protoc. 2014, 9, 2329–2340. [Google Scholar] [CrossRef]
- Birtele, M.; Lancaster, M.; Quadrato, G. Modelling Human Brain Development and Disease with Organoids. Nat. Rev. Mol. Cell Biol. 2024, 26, 389–412. [Google Scholar] [CrossRef]
- Raja, W.K.; Mungenast, A.E.; Lin, Y.T.; Ko, T.; Abdurrob, F.; Seo, J.; Tsai, L.H. Self-Organizing 3D Human Neural Tissue Derived from Induced Pluripotent Stem Cells Recapitulate Alzheimer’s Disease Phenotypes. PLoS ONE 2016, 11, e0161969. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.; Armijo, E.; Bravo-Alegria, J.; Becerra-Calixto, A.; Mays, C.E.; Soto, C. Modeling Amyloid Beta and Tau Pathology in Human Cerebral Organoids. Mol. Psychiatry 2018, 23, 2363–2374. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Song, L.; Bejoy, J.; Zhao, J.; Kanekiyo, T.; Bu, G.; Zhou, Y.; Li, Y. Modeling Neurodegenerative Microenvironment Using Cortical Organoids Derived from Human Stem Cells. Tissue Eng. Part A 2018, 24, 1125–1137. [Google Scholar] [CrossRef]
- Vanova, T.; Sedmik, J.; Raska, J.; Amruz Cerna, K.; Taus, P.; Pospisilova, V.; Nezvedova, M.; Fedorova, V.; Kadakova, S.; Klimova, H.; et al. Cerebral Organoids Derived from Patients with Alzheimer’s Disease with PSEN1/2 Mutations Have Defective Tissue Patterning and Altered Development. Cell Rep. 2023, 42, 113310. [Google Scholar] [CrossRef]
- Kuehner, J.N.; Chen, J.; Bruggeman, E.C.; Wang, F.; Li, Y.; Xu, C.; McEachin, Z.T.; Li, Z.; Chen, L.; Hales, C.M.; et al. 5-Hydroxymethylcytosine Is Dynamically Regulated during Forebrain Organoid Development and Aberrantly Altered in Alzheimer’s Disease. Cell Rep. 2021, 35, 109042. [Google Scholar] [CrossRef]
- Zhao, J.; Fu, Y.; Yamazaki, Y.; Ren, Y.; Davis, M.D.; Liu, C.-C.; Lu, W.; Wang, X.; Chen, K.; Cherukuri, Y.; et al. APOE4 Exacerbates Synapse Loss and Neurodegeneration in Alzheimer’s Disease Patient IPSC-Derived Cerebral Organoids. Nat. Commun. 2020, 11, 5540. [Google Scholar] [CrossRef]
- Hernández, D.; Rooney, L.A.; Daniszewski, M.; Gulluyan, L.; Liang, H.H.; Cook, A.L.; Hewitt, A.W.; Pébay, A. Culture Variabilities of Human IPSC-Derived Cerebral Organoids Are a Major Issue for the Modelling of Phenotypes Observed in Alzheimer’s Disease. Stem Cell Rev. Rep. 2022, 18, 718–731. [Google Scholar] [CrossRef]
- Kistemaker, L.; van Bodegraven, E.J.; de Vries, H.E.; Hol, E.M. Vascularized Human Brain Organoids: Current Possibilities and Prospects. Trends Biotechnol. 2025. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.; Rhee, S.W.; Tu, C.H.; Cribbs, D.H.; Cotman, C.W.; Jeon, N.L. Microfluidic Multicompartment Device for Neuroscience Research. Langmuir 2003, 19, 1551–1556. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Wetzel, I.; Marriott, I.; Dréau, D.; D’Avanzo, C.; Kim, D.Y.; Tanzi, R.E.; Cho, H. A 3D Human Triculture System Modeling Neurodegeneration and Neuroinflammation in Alzheimer’s Disease. Nat. Neurosci. 2018, 21, 941–951. [Google Scholar] [CrossRef]
- Jorfi, M.; Park, J.; Hall, C.K.; Lin, C.C.J.; Chen, M.; von Maydell, D.; Kruskop, J.M.; Kang, B.; Choi, Y.; Prokopenko, D.; et al. Infiltrating CD8+ T Cells Exacerbate Alzheimer’s Disease Pathology in a 3D Human Neuroimmune Axis Model. Nat. Neurosci. 2023, 26, 1489–1504. [Google Scholar] [CrossRef] [PubMed]
- Holloway, P.M.; Hallinan, G.I.; Hegde, M.; Lane, S.I.R.; Deinhardt, K.; West, J. Asymmetric Confinement for Defining Outgrowth Directionality. Lab Chip 2019, 19, 1484–1489. [Google Scholar] [CrossRef]
- Pediaditakis, I.; Kodella, K.R.; Manatakis, D.V.; Le, C.Y.; Hinojosa, C.D.; Tien-Street, W.; Manolakos, E.S.; Vekrellis, K.; Hamilton, G.A.; Ewart, L.; et al. Modeling Alpha-Synuclein Pathology in a Human Brain-Chip to Assess Blood-Brain Barrier Disruption. Nat. Commun. 2021, 12, 5907. [Google Scholar] [CrossRef]
- Park, T.E.; Mustafaoglu, N.; Herland, A.; Hasselkus, R.; Mannix, R.; FitzGerald, E.A.; Prantil-Baun, R.; Watters, A.; Henry, O.; Benz, M.; et al. Hypoxia-Enhanced Blood-Brain Barrier Chip Recapitulates Human Barrier Function and Shuttling of Drugs and Antibodies. Nat. Commun. 2019, 10, 2621. [Google Scholar] [CrossRef]
- Brown, J.A.; Codreanu, S.G.; Shi, M.; Sherrod, S.D.; Markov, D.A.; Neely, M.D.; Britt, C.M.; Hoilett, O.S.; Reiserer, R.S.; Samson, P.C.; et al. Metabolic Consequences of Inflammatory Disruption of the Blood-Brain Barrier in an Organ-on-Chip Model of the Human Neurovascular Unit. J. Neuroinflamm. 2016, 13, 306. [Google Scholar] [CrossRef]
- Vatine, G.D.; Barrile, R.; Workman, M.J.; Sances, S.; Barriga, B.K.; Rahnama, M.; Barthakur, S.; Kasendra, M.; Lucchesi, C.; Kerns, J.; et al. Human IPSC-Derived Blood-Brain Barrier Chips Enable Disease Modeling and Personalized Medicine Applications. Cell Stem Cell 2019, 24, 995–1005.e6. [Google Scholar] [CrossRef]
- Ko, E.C.; Spitz, S.; Pramotton, F.M.; Barr, O.M.; Xu, C.; Pavlou, G.; Zhang, S.; Tsai, A.; Maaser-Hecker, A.; Jorfi, M.; et al. Accelerating the in Vitro Emulation of Alzheimer’s Disease-Associated Phenotypes Using a Novel 3D Blood-Brain Barrier Neurosphere Co-Culture Model. Front. Bioeng. Biotechnol. 2023, 11, 1251195. [Google Scholar] [CrossRef]
- Ekins, S.; Mestres, J.; Testa, B. In Silico Pharmacology for Drug Discovery: Methods for Virtual Ligand Screening and Profiling. Br. J. Pharmacol. 2007, 152, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Abbas, Q.; Seo, S.Y.; Shahzadi, S.; Al Ashwal, H.; Zaki, N.; Iqbal, Z.; Moustafa, A.A. Computational Modeling and Biomarker Studies of Pharmacological Treatment of Alzheimer’s Disease (Review). Mol. Med. Rep. 2018, 18, 639–655. [Google Scholar] [CrossRef] [PubMed]
- Caligiore, D.; Silvetti, M.; D’Amelio, M.; Puglisi-Allegra, S.; Baldassarre, G.; Anbarjafari, G. Computational Modeling of Catecholamines Dysfunction in Alzheimer’s Disease at Pre-Plaque Stage. J. Alzheimer’s Dis. 2020, 77, 275–290. [Google Scholar] [CrossRef]
- Leonard, C.; Phillips, C.; McCarty, J. Insight Into Seeded Tau Fibril Growth From Molecular Dynamics Simulation of the Alzheimer’s Disease Protofibril Core. Front. Mol. Biosci. 2021, 8, 624302. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Gong, Y.; Xu, Z.; Dong, X.; Wei, G.; Zhang, Q. Molecular Dynamics Simulations Reveal the Destabilization Mechanism of Alzheimer’s Disease-Related Tau R3-R4 Protofilament by Norepinephrine. Biophys. Chem. 2021, 271, 106541. [Google Scholar] [CrossRef]
- Fan, H.M.; Gu, R.X.; Wang, Y.J.; Pi, Y.L.; Zhang, Y.H.; Xu, Q.; Wei, D.Q. Destabilization of Alzheimer’s Aβ42 Protofibrils with a Novel Drug Candidate Wgx-50 by Molecular Dynamics Simulations. J. Phys. Chem. B 2015, 119, 11196–11202. [Google Scholar] [CrossRef]
- Brodland, G.W. How Computational Models Can Help Unlock Biological Systems. Semin. Cell Dev. Biol. 2015, 47, 62–73. [Google Scholar] [CrossRef]
- Carpanini, S.M.; Torvell, M.; Morgan, B.P. Therapeutic Inhibition of the Complement System in Diseases of the Central Nervous System. Front. Immunol. 2019, 10, 362. [Google Scholar] [CrossRef]
- Schadt, E.E. Molecular Networks as Sensors and Drivers of Common Human Diseases. Nature 2009, 461, 218–223. [Google Scholar] [CrossRef]
- Eisen, M.B.; Spellman, P.T.; Brown, P.O.; Botstein, D. Cluster Analysis and Display of Genome-Wide Expression Patterns. Proc. Natl. Acad. Sci. USA 1998, 95, 14863–14868. [Google Scholar] [CrossRef]
- Berardini, T.Z.; Li, D.; Huala, E.; Bridges, S.; Burgess, S.; McCarthy, F.; Carbon, S.; Lewis, S.E.; Mungall, C.J.; Abdulla, A.; et al. The Gene Ontology in 2010: Extensions and Refinements. Nucleic Acids Res. 2010, 38, D331–D335. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-P.; Wang, Y.; Zhang, X.-S.; Chen, L. Identifying Dysfunctional Crosstalk of Pathways in Various Regions of Alzheimer’s Disease Brains. BMC Syst. Biol. 2010, 4, S11. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.P.; Wang, Y.; Zhang, X.S.; Xia, W.; Chen, L. Detecting and Analyzing Differentially Activated Pathways in Brain Regions of Alzheimer’s Disease Patients. Mol. Biosyst. 2011, 7, 1441–1452. [Google Scholar] [CrossRef]
- Liang, D.; Han, G.; Feng, X.; Sun, J.; Duan, Y.; Lei, H. Concerted Perturbation Observed in a Hub Network in Alzheimer’s Disease. PLoS ONE 2012, 7, e40498. [Google Scholar] [CrossRef]
- Hallock, P.; Thomas, M.A. Integrating the Alzheimer’s Disease Proteome and Transcriptome: A Comprehensive Network Model of a Complex Disease. Omi. A J. Integr. Biol. 2012, 16, 37–49. [Google Scholar] [CrossRef]
- Keshava Prasad, T.S.; Goel, R.; Kandasamy, K.; Keerthikumar, S.; Kumar, S.; Mathivanan, S.; Telikicherla, D.; Raju, R.; Shafreen, B.; Venugopal, A.; et al. Human Protein Reference Database—2009 Update. Nucleic Acids Res. 2009, 37, D767–D772. [Google Scholar] [CrossRef]
- Chen, J.Y.; Shen, C.; Sivachenko, A.Y. Mining Alzheimer Disease Relevant Proteins from Integrated Protein Interactome Data. In Biocomputing 2006—Proceedings of the Pacific Symposium; World Scientific Press: London, UK, 2006; pp. 367–378. [Google Scholar] [CrossRef]
- Neff, R.A.; Wang, M.; Vatansever, S.; Guo, L.; Ming, C.; Wang, Q.; Wang, E.; Horgusluoglu-Moloch, E.; Song, W.M.; Li, A.; et al. Molecular Subtyping of Alzheimer’s Disease Using RNA Sequencing Data Reveals Novel Mechanisms and Targets. Sci. Adv. 2021, 7, eabb5398. [Google Scholar] [CrossRef]
- Guennewig, B.; Lim, J.; Marshall, L.; McCorkindale, A.N.; Paasila, P.J.; Patrick, E.; Kril, J.J.; Halliday, G.M.; Cooper, A.A.; Sutherland, G.T. Defining Early Changes in Alzheimer’s Disease from RNA Sequencing of Brain Regions Differentially Affected by Pathology. Sci. Rep. 2021, 11, 4865. [Google Scholar] [CrossRef]
- Alsema, A.M.; Jiang, Q.; Kracht, L.; Gerrits, E.; Dubbelaar, M.L.; Miedema, A.; Brouwer, N.; Hol, E.M.; Middeldorp, J.; van Dijk, R.; et al. Profiling Microglia From Alzheimer’s Disease Donors and Non-Demented Elderly in Acute Human Postmortem Cortical Tissue. Front. Mol. Neurosci. 2020, 13, 134. [Google Scholar] [CrossRef]
- Trapnell, C. Defining Cell Types and States with Single-Cell Genomics. Genome Res. 2015, 25, 1491–1498. [Google Scholar] [CrossRef] [PubMed]
- Luecken, M.D.; Büttner, M.; Chaichoompu, K.; Danese, A.; Interlandi, M.; Mueller, M.F.; Strobl, D.C.; Zappia, L.; Dugas, M.; Colomé-Tatché, M.; et al. Benchmarking Atlas-Level Data Integration in Single-Cell Genomics. Nat. Methods 2022, 19, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Lähnemann, D.; Köster, J.; Szczurek, E.; McCarthy, D.J.; Hicks, S.C.; Robinson, M.D.; Vallejos, C.A.; Campbell, K.R.; Beerenwinkel, N.; Mahfouz, A.; et al. Eleven Grand Challenges in Single-Cell Data Science. Genome Biol. 2020, 21, 31. [Google Scholar] [CrossRef]
- Bobrovskikh, A.; Doroshkov, A.; Mazzoleni, S.; Cartenì, F.; Giannino, F.; Zubairova, U. A Sight on Single-Cell Transcriptomics in Plants Through the Prism of Cell-Based Computational Modeling Approaches: Benefits and Challenges for Data Analysis. Front. Genet. 2021, 12, 652974. [Google Scholar] [CrossRef] [PubMed]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-Cell Transcriptomic Analysis of Alzheimer’s Disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Kale, M.; Wankhede, N.; Pawar, R.; Ballal, S.; Kumawat, R.; Goswami, M.; Khalid, M.; Taksande, B.; Upaganlawar, A.; Umekar, M.; et al. AI-Driven Innovations in Alzheimer’s Disease: Integrating Early Diagnosis, Personalized Treatment, and Prognostic Modelling. Ageing Res. Rev. 2024, 101, 102497. [Google Scholar] [CrossRef]
- Saleem, T.J.; Zahra, S.R.; Wu, F.; Alwakeel, A.; Alwakeel, M.; Jeribi, F.; Hijji, M. Deep Learning-Based Diagnosis of Alzheimer’s Disease. J. Pers. Med. 2022, 12, 815. [Google Scholar] [CrossRef]
- Bhatti, U.A.; Tang, H.; Wu, G.; Marjan, S.; Hussain, A. Deep Learning with Graph Convolutional Networks: An Overview and Latest Applications in Computational Intelligence. Int. J. Intell. Syst. 2023, 2023, 8342104. [Google Scholar] [CrossRef]
- Vieira, S.; Pinaya, W.H.L.; Mechelli, A. Using Deep Learning to Investigate the Neuroimaging Correlates of Psychiatric and Neurological Disorders: Methods and Applications. Neurosci. Biobehav. Rev. 2017, 74, 58–75. [Google Scholar] [CrossRef]
- Hojjati, S.H.; Ebrahimzadeh, A.; Khazaee, A.; Babajani-Feremi, A. Predicting Conversion from MCI to AD Using Resting-State FMRI, Graph Theoretical Approach and SVM. J. Neurosci. Methods 2017, 282, 69–80. [Google Scholar] [CrossRef]
- Zhang, L.; Ni, H.; Yu, Z.; Wang, J.; Qin, J.; Hou, F.; Yang, A. Investigation on the Alteration of Brain Functional Network and Its Role in the Identification of Mild Cognitive Impairment. Front. Neurosci. 2020, 14, 558434. [Google Scholar] [CrossRef] [PubMed]
- Aghdam, M.A.; Bozdag, S.; Saeed, F. Machine-Learning Models for Alzheimer’s Disease Diagnosis Using Neuroimaging Data: Survey, Reproducibility, and Generalizability Evaluation. Brain Inform. 2025, 12, 8. [Google Scholar] [CrossRef]
- Moradi, E.; Pepe, A.; Gaser, C.; Huttunen, H.; Tohka, J. Machine Learning Framework for Early MRI-Based Alzheimer’s Conversion Prediction in MCI Subjects. Neuroimage 2015, 104, 398–412. [Google Scholar] [CrossRef] [PubMed]
- Plis, S.M.; Hjelm, D.R.; Salakhutdinov, R.; Allen, E.A.; Bockholt, H.J.; Long, J.D.; Johnson, H.J.; Paulsen, J.S.; Turner, J.A.; Calhoun, V.D. Deep Learning for Neuroimaging: A Validation Study. Front. Neurosci. 2014, 8, 229. [Google Scholar] [CrossRef]
- Koga, S.; Ikeda, A.; Dickson, D.W. Deep Learning-based Model for Diagnosing Alzheimer’s Disease and Tauopathies. Neuropathol. Appl. Neurobiol. 2022, 48, e12759. [Google Scholar] [CrossRef]
- Qiu, S.; Miller, M.I.; Joshi, P.S.; Lee, J.C.; Xue, C.; Ni, Y.; Wang, Y.; De Anda-Duran, I.; Hwang, P.H.; Cramer, J.A.; et al. Multimodal Deep Learning for Alzheimer’s Disease Dementia Assessment. Nat. Commun. 2022, 13, 3404. [Google Scholar] [CrossRef] [PubMed]
- Alhudhaif, A.; Polat, K. Residual Block Fully Connected DCNN with Categorical Generalized Focal Dice Loss and Its Application to Alzheimer’s Disease Severity Detection. PeerJ Comput. Sci. 2023, 9, e1599. [Google Scholar] [CrossRef]
- Basaia, S.; Agosta, F.; Wagner, L.; Canu, E.; Magnani, G.; Santangelo, R.; Filippi, M. Automated Classification of Alzheimer’s Disease and Mild Cognitive Impairment Using a Single MRI and Deep Neural Networks. NeuroImage Clin. 2019, 21, 101645. [Google Scholar] [CrossRef]
- Cheng, F.; Wang, F.; Tang, J.; Zhou, Y.; Fu, Z.; Zhang, P.; Haines, J.L.; Leverenz, J.B.; Gan, L.; Hu, J.; et al. Artificial Intelligence and Open Science in Discovery of Disease-Modifying Medicines for Alzheimer’s Disease. Cell Rep. Med. 2024, 5, 101379. [Google Scholar] [CrossRef]
- Wahler, F.; Neubert, M. A Scientific Definition of Explainable Artificial Intelligence for Decision Making. Int. J. Teach. Case Stud. 2023, 14, 88–116. [Google Scholar] [CrossRef]
- Dwivedi, R.; Dave, D.; Naik, H.; Singhal, S.; Omer, R.; Patel, P.; Qian, B.; Wen, Z.; Shah, T.; Morgan, G.; et al. Explainable AI (XAI): Core Ideas, Techniques, and Solutions. ACM Comput. Surv. 2023, 55, 194. [Google Scholar] [CrossRef]
- Bordin, V.; Coluzzi, D.; Rivolta, M.W.; Baselli, G. Explainable AI Points to White Matter Hyperintensities for Alzheimer’s Disease Identification: A Preliminary Study. In Proceedings of the 2022 44th Annual International Conference of the IEEE Engineering in Medicine & Biology Society (EMBC), Glasgow, UK, 11 July 2022; pp. 484–487. [Google Scholar]
- Tekkesinoglu, S.; Pudas, S. Explaining Graph Convolutional Network Predictions for Clinicians—An Explainable AI Approach to Alzheimer’s Disease Classification. Front. Artif. Intell. 2024, 6, 1334613. [Google Scholar] [CrossRef] [PubMed]
- Hopfield, J.J. Neural Networks and Physical Systems with Emergent Collective Computational Abilities. Proc. Natl. Acad. Sci. USA 1982, 79, 2554–2558. [Google Scholar] [CrossRef]
- Pinheiro, R.F.; Colón, D.; Fonseca-Pinto, R. A Memory Failure Computational Model in Alzheimer-like Disease via Continuous Delayed Hopfield Network with Lurie Control System Based Healing. Neurocomputing 2025, 636, 129967. [Google Scholar] [CrossRef]
- Eyigoz, E.; Mathur, S.; Santamaria, M.; Cecchi, G.; Naylor, M. Linguistic Markers Predict Onset of Alzheimer’s Disease. EClinicalMedicine 2020, 28, 100583. [Google Scholar] [CrossRef] [PubMed]
- Haraldsen, I.H.; Hatlestad-Hall, C.; Marra, C.; Renvall, H.; Maestú, F.; Acosta-Hernández, J.; Alfonsin, S.; Andersson, V.; Anand, A.; Ayllón, V.; et al. Intelligent Digital Tools for Screening of Brain Connectivity and Dementia Risk Estimation in People Affected by Mild Cognitive Impairment: The AI-Mind Clinical Study Protocol. Front. Neurorobot. 2024, 17, 1289406. [Google Scholar] [CrossRef]
- Duff, K. Transgenic Mouse Models of Alzheimer’s Disease: How Useful Have They Been for Therapeutic Development? Brief. Funct. Genom. Proteom. 2004, 3, 47–59. [Google Scholar] [CrossRef]
- Tsolaki, M.; Makri, M.; Tsatali, M.; Teichmann, Β. European Projects for Patients with Dementia and Their Caregivers. In Worldwide Congress on “Genetics, Geriatrics and Neurodegenerative Diseases Research”; Springer International Publishing: Cham, Germany, 2023; pp. 609–618. [Google Scholar]
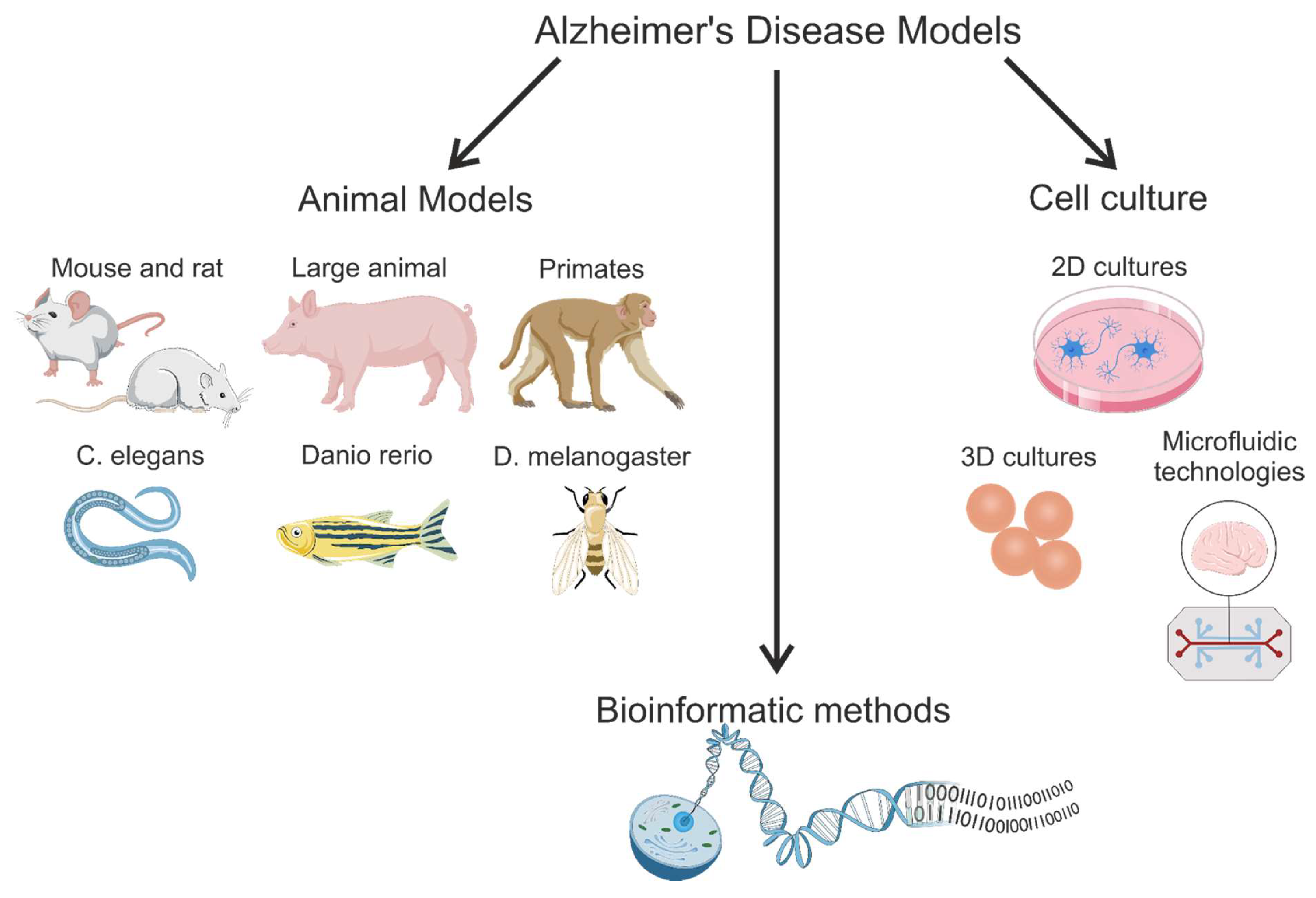
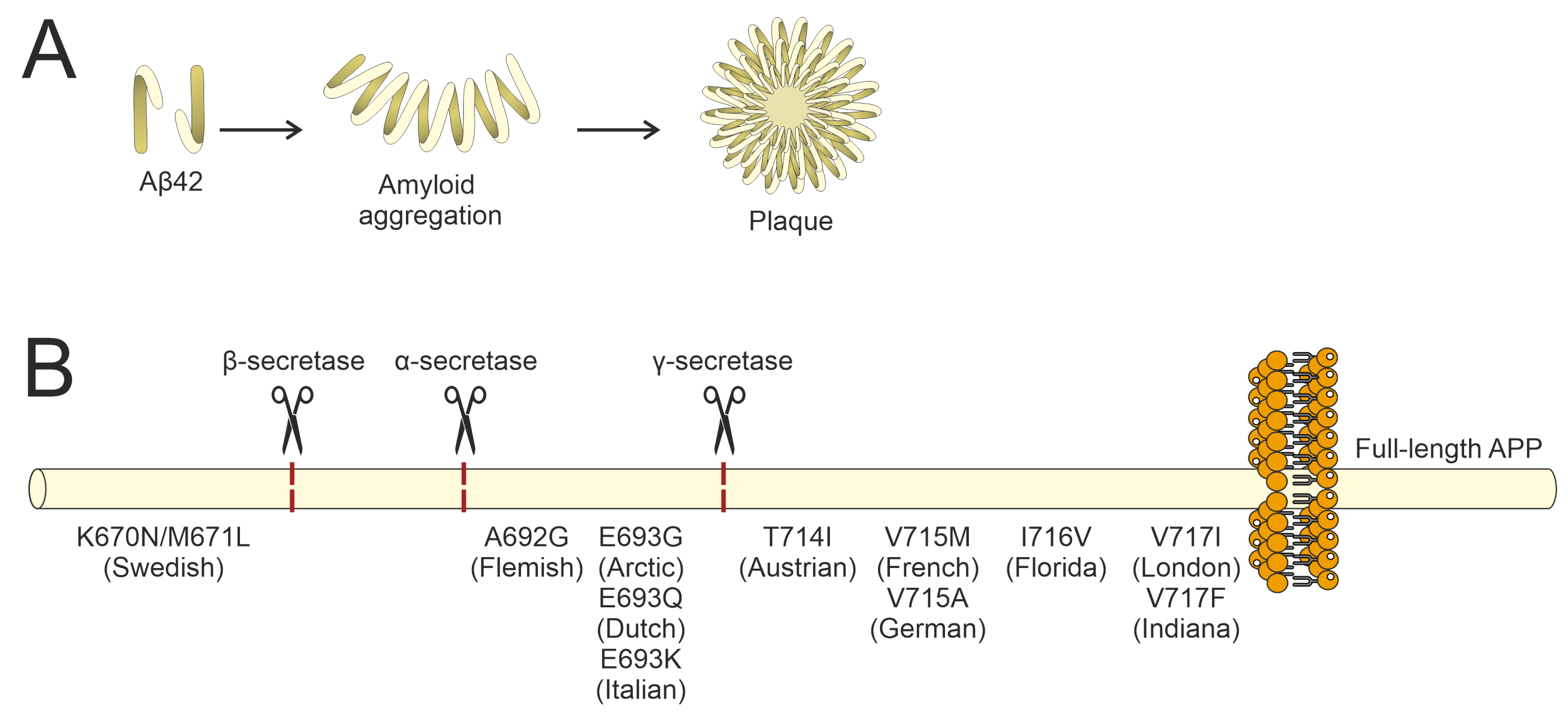

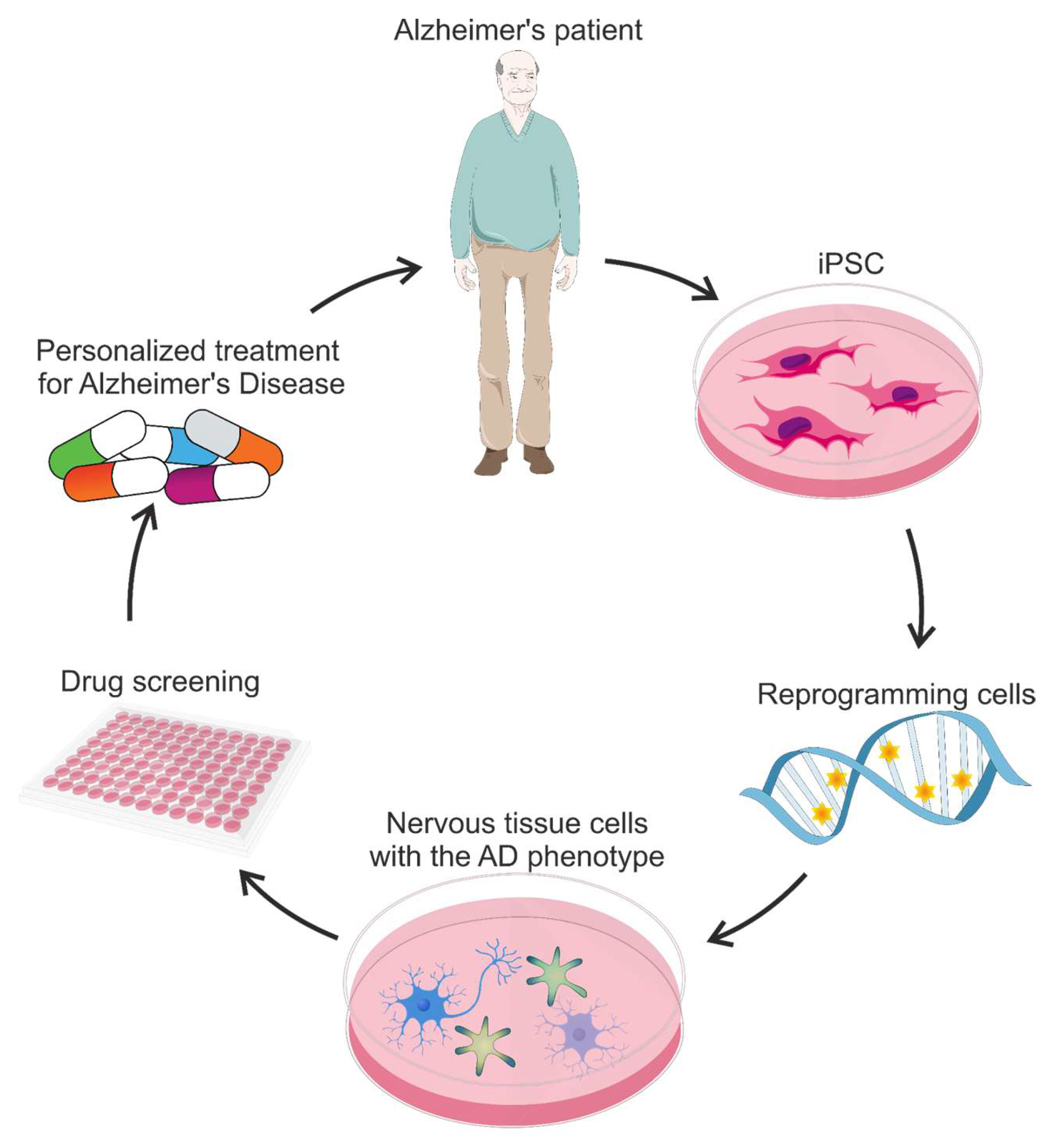
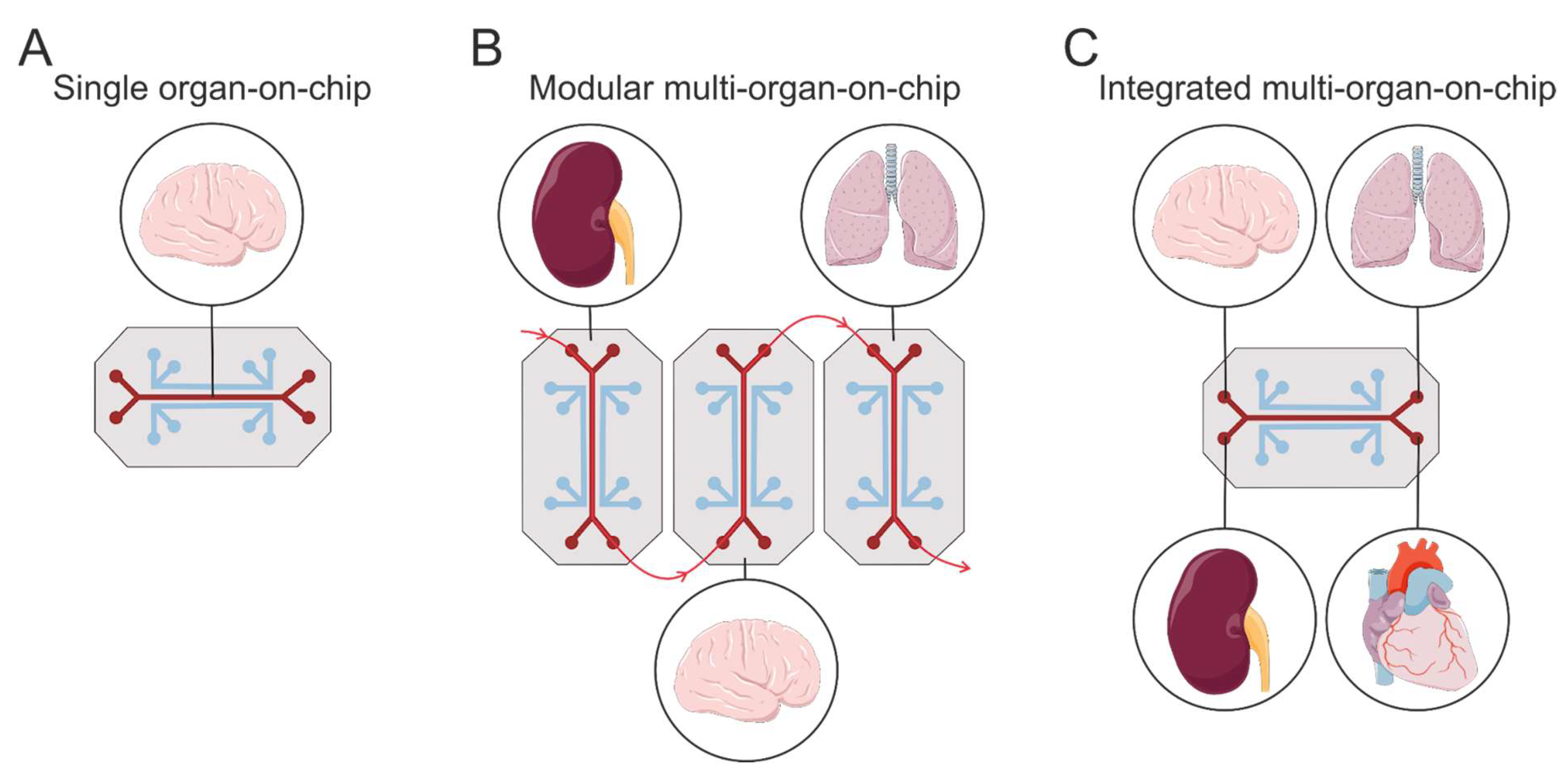
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Timofeeva, A.M.; Aulova, K.S.; Nevinsky, G.A. Modeling Alzheimer’s Disease: A Review of Gene-Modified and Induced Animal Models, Complex Cell Culture Models, and Computational Modeling. Brain Sci. 2025, 15, 486. https://doi.org/10.3390/brainsci15050486
Timofeeva AM, Aulova KS, Nevinsky GA. Modeling Alzheimer’s Disease: A Review of Gene-Modified and Induced Animal Models, Complex Cell Culture Models, and Computational Modeling. Brain Sciences. 2025; 15(5):486. https://doi.org/10.3390/brainsci15050486
Chicago/Turabian StyleTimofeeva, Anna M., Kseniya S. Aulova, and Georgy A. Nevinsky. 2025. "Modeling Alzheimer’s Disease: A Review of Gene-Modified and Induced Animal Models, Complex Cell Culture Models, and Computational Modeling" Brain Sciences 15, no. 5: 486. https://doi.org/10.3390/brainsci15050486
APA StyleTimofeeva, A. M., Aulova, K. S., & Nevinsky, G. A. (2025). Modeling Alzheimer’s Disease: A Review of Gene-Modified and Induced Animal Models, Complex Cell Culture Models, and Computational Modeling. Brain Sciences, 15(5), 486. https://doi.org/10.3390/brainsci15050486