
Mitochondrial Chronic Progressive External Ophthalmoplegia


Abstract
1. Introduction
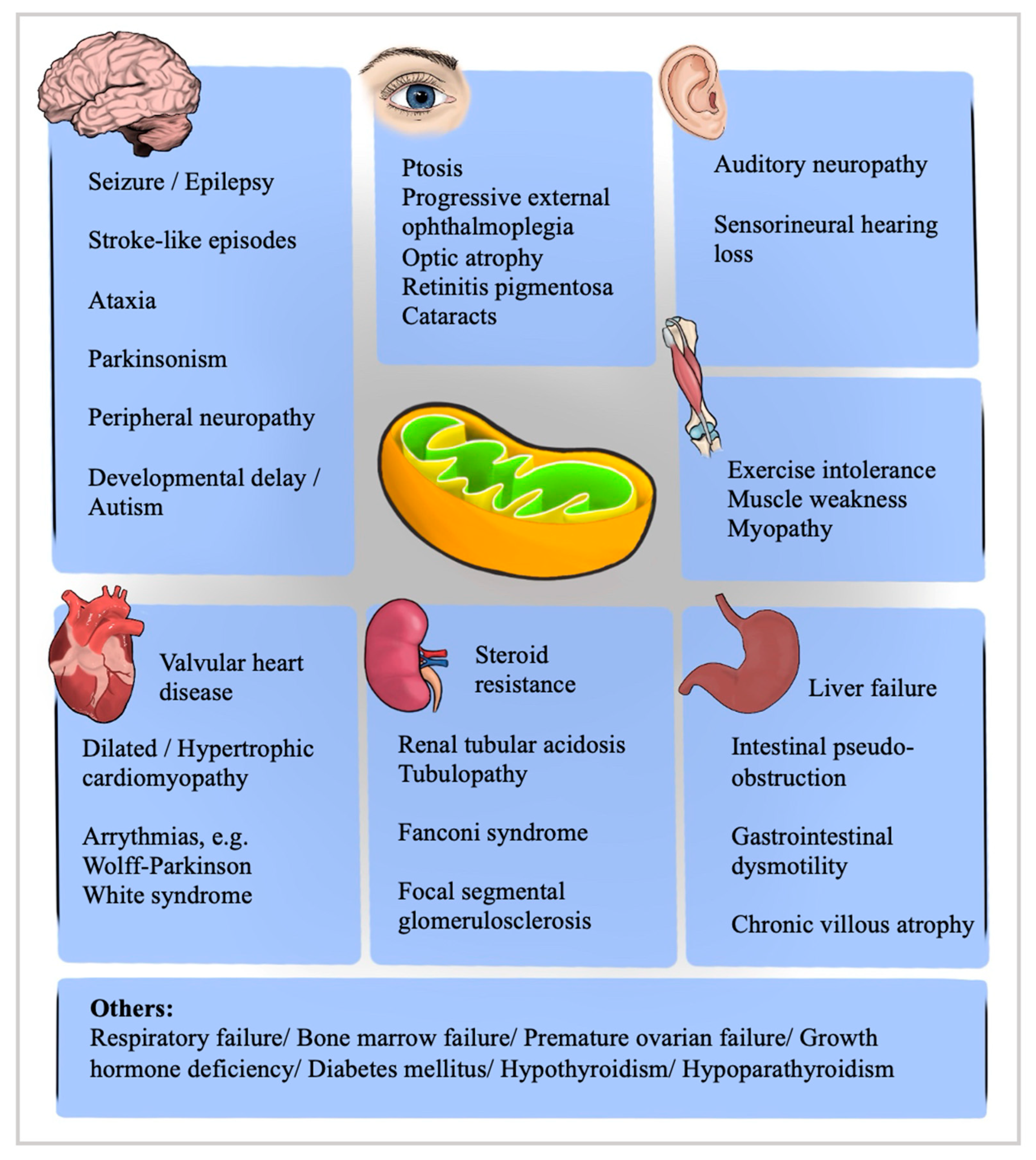
2. Pathophysiology, Genetics and Classification
3. Mitochondrial DNA Deletion/Depletion Disorders Causing CPEO
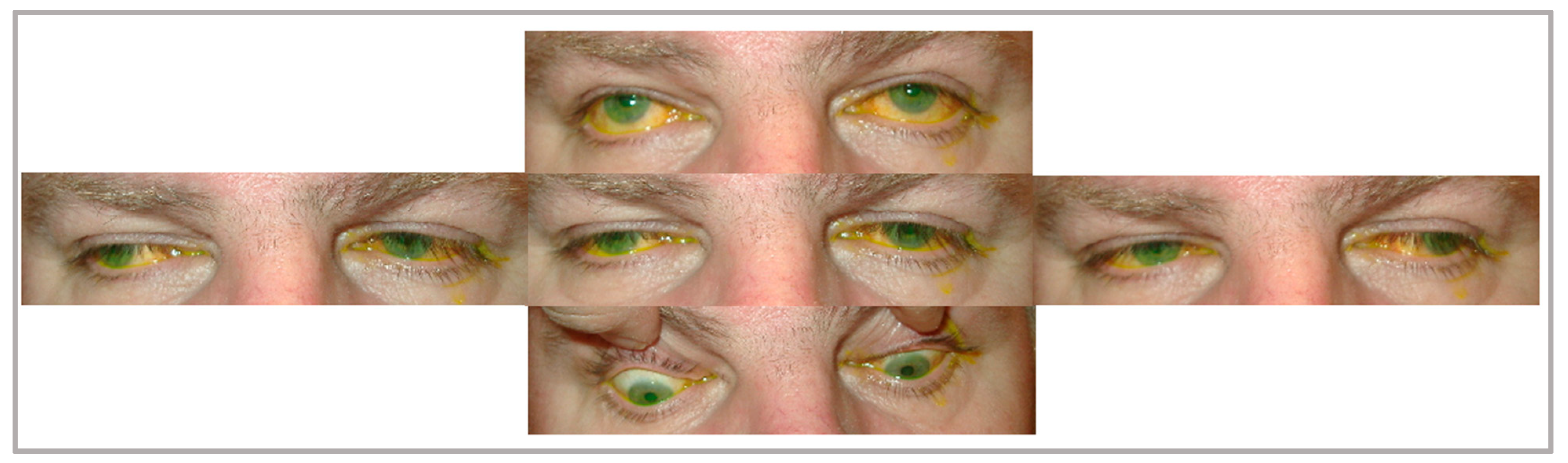
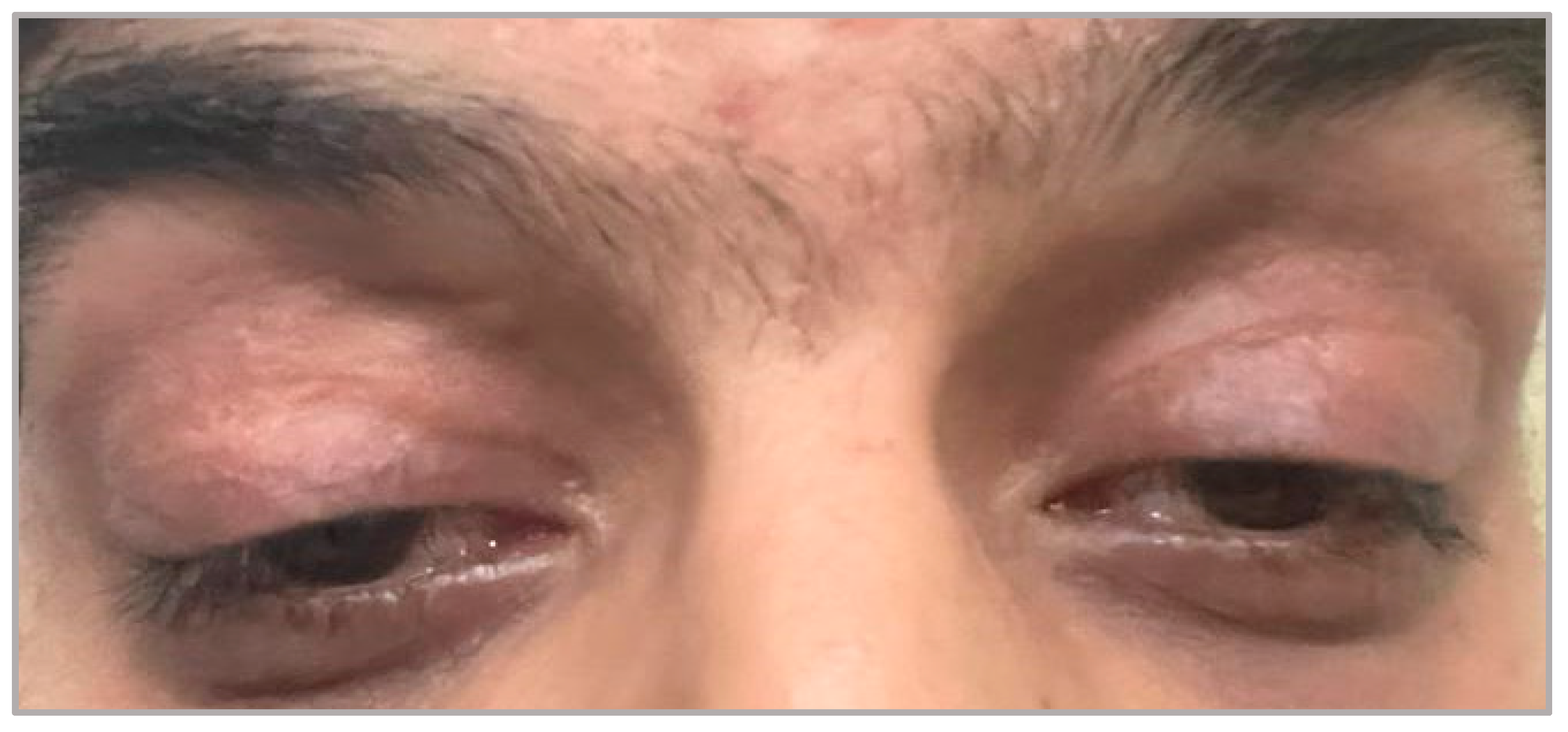
3.1. CPEO
3.2. Kearns–Sayre Syndrome
3.3. Pearson Syndrome
3.4. Leigh Syndrome
3.5. MELAS
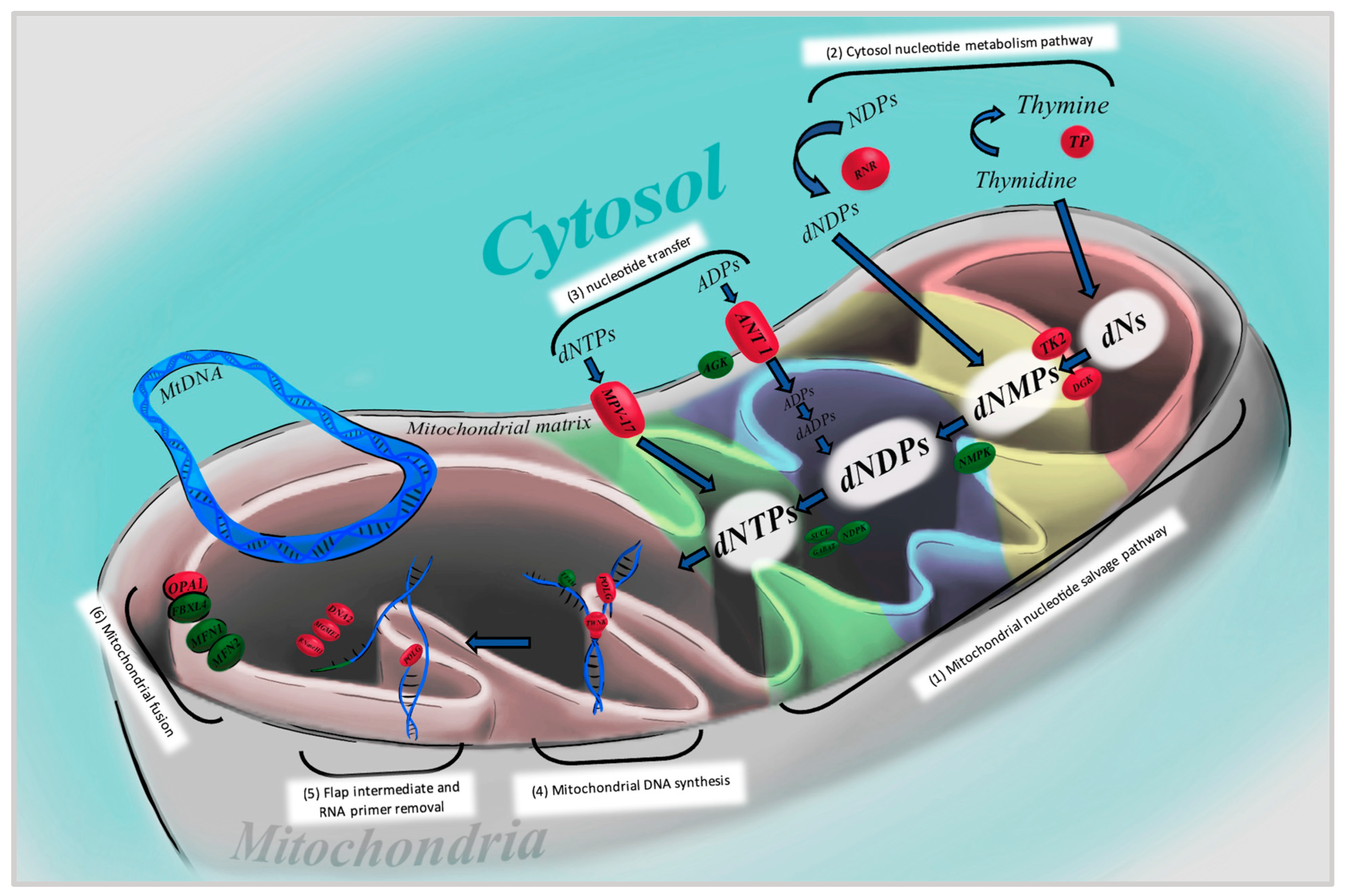
4. Nuclear DNA Gene Mutations/Protein Dysfunction Causing CPEO
4.1. DNA Polymerase Subunit Gamma (POLG)
4.2. Twinkle mtDNA Helicase (TWNK)
4.3. Thymidine Phosphorylase
4.4. Ribonucleoside–Diphosphate Reductase Subunit M2 B (RRM2B)
4.5. Optic Atrophy 1 (OPA-1)
4.6. Thymidine Kinase 2 (TK2)
4.7. Deoxyguanosine Kinase
4.8. Ribonuclease H1 (RNase H1)
4.9. Mitochondrial Genome Maintenance Exonuclease 1 (MGME1)
4.10. Adenine Nucleotide Translocator 1 (ANT1)
4.11. Mitochondrial Inner Membrane Protein MPV17
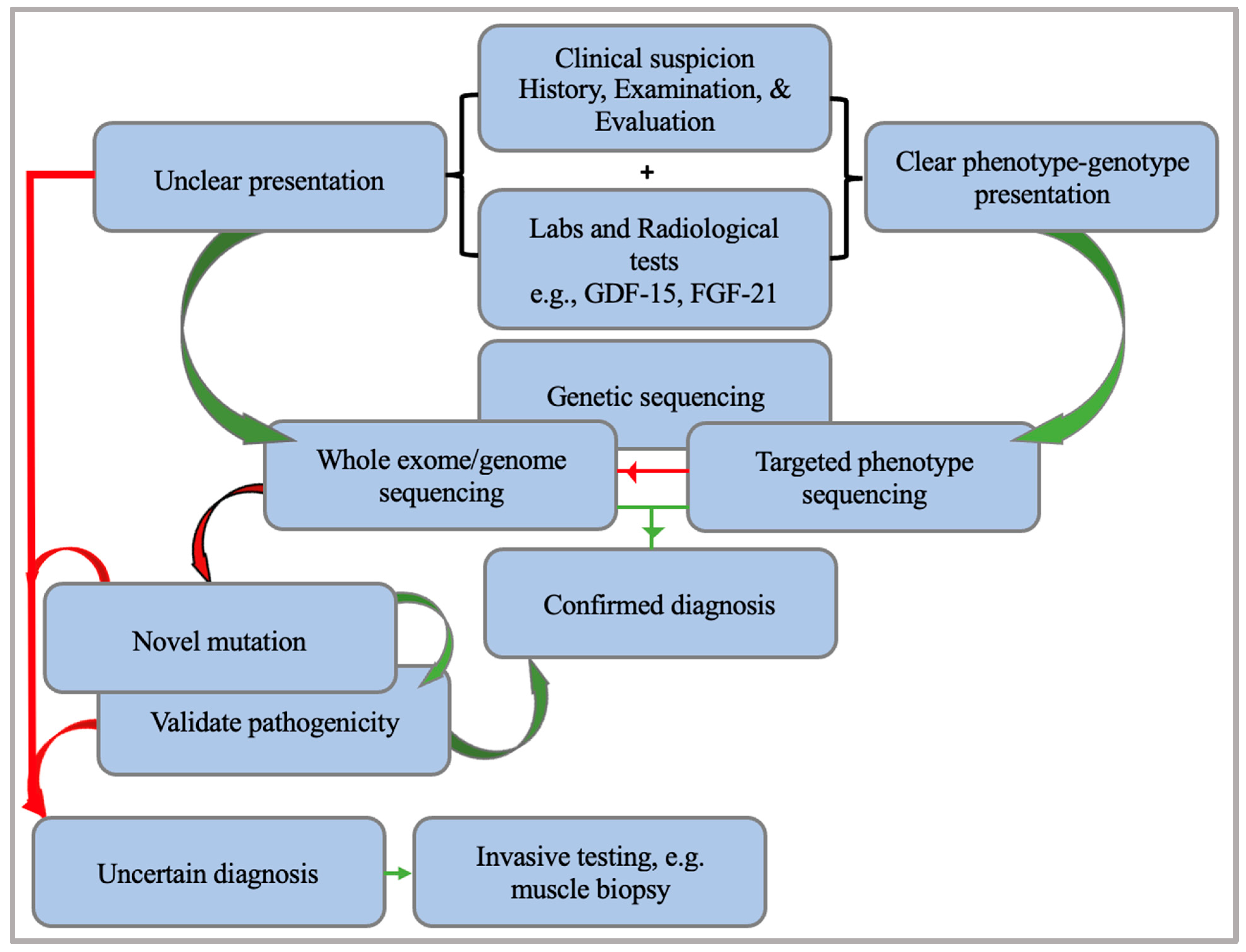
5. A Diagnostic Approach to Mitochondrial Encephalomyopathies
6. Treatment of Mitochondrial Diseases
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mitochondrial DNA Disorder | Acronym | Possible Clinical Features | Reference |
|---|---|---|---|
| Chronic progressive external ophthalmoplegia | CPEO | Progressive bilateral ptosis, ophthalmoplegia, diplopia | [117] |
| CPEO “plus”: muscle weakness, exercise intolerance, short stature, pharyngeal muscle weakness, cognitive impairment, decreased vision, cardiac conduction block | |||
| Kearns–Sayre syndrome | KSS | Criterion: CPEO and pigmentary retinopathy (onset < 20 years), cerebrospinal fluid protein >1 g/L. Plus one of the following: Cerebellar ataxia, myopathy, dysphagia, sensorineural hearing loss, heart block, diabetes mellitus, endocrine dysfunction | [118] |
| Pearson syndrome | PS | Sideroblastic anemia, pancytopenia, exocrine pancreatic failure, renal tubular defects | [119] |
| Mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes | MELAS | Criterion: Stroke-like episodes (SLEs) (age < 40 years), seizures and/or dementia, signs of myopathy, e.g., ragged red fibers and/or lactic acidosis. Pigmentary retinopathy, diabetes mellitus, cardiomyopathy, bilateral sensorineural deafness | [120,121,122] |
| Leigh syndrome * | LS | Developmental delay, hypotonia, respiratory dysfunction, epilepsy, reduced feeding, weakness. Cardiomyopathy, cardiac conduction defects. Ptosis, ophthalmoplegia, nystagmus, strabismus, pigmentary retinopathy, optic atrophy | [34,35,36,37] |
| Maternally Inherited Leigh Syndrome | MILS | Intractable epileptic seizures, chorea, hyporeflexia, psychomotor retardation, hypoacusis, dystonia, hypotonia, hypertrophic cardiomyopathy, hepatopathy, lactic acidosis | [123] |
| Neuropathy, Ataxia, Retinitis Pigmentosa | NARP | Axonal neuropathy, cerebellar ataxia, proximal muscle weakness, retinitis pigmentosa. Epilepsy, cerebellar or cerebral atrophy, dementia, hypoacusis, optic atrophy, sleep apnea syndrome, renal impairment, diabetes mellitus | [124] |
| Leber’s hereditary optic neuropathy | LHON | Bilateral vision loss, central/centro cecal scotoma, mild disc pseudo edema, dyschromatopsia, peripheral neuropathy, clonus, dystonia, postural tremor, myopathy, Wolff Parkinson White syndrome | [125] |
| Maternally Inherited Diabetes and Deafness | MIDD | Diabetes mellitus, progressive sensorineural deafness, thin and short stature, macular dystrophy, proliferative retinopathy, myopathy, left ventricular hypertrophy, Wolff Parkinson White syndrome, atrial fibrillation, constipation, diarrhea, intestinal pseudo-obstruction | [126] |
| Myoclonic Epilepsy and Ragged Red Fibers | MERRF | Myoclonus, epilepsy, ataxia, migraine, dementia, SLEs, myopathy, myalgia, dysphagia, dysmotility, polyneuropathy, hearing loss, optic atrophy, ptosis, ophthalmoparesis, cardiomyopathy, arrhythmias, lipomatosis | [127] |
| Sensory ataxic neuropathy with dysarthria and opthalmoparesis * | SANDO | Ataxia, ptosis, dysarthria, sensory neuropathy, dysphagia, myalgia, seizures, diabetes mellitus | [128,129] |
| Nuclear DNA gene | Protein | Possible clinical manifestations | Reference |
| POLG | DNA polymerase subunit gamma | Seizures, ataxia, SLEs, peripheral neuropathy, migraine-like headache, hypotonia, liver involvement, anemia, ptosis, PEO | [55] |
| TWNK | Twinkle | Ataxia, neuropathy, myopathy, epileptic encephalopathy, CPEO, cataracts, mild cardiac abnormalities, parkinsonism | [58] |
| TYMP | Thymidine phosphorylase | Mitochondrial neurogastrointestinal encephalopathy (MNGIE): CPEO, cachexia, severe gastrointestinal dysmotility, sensorineural hearing loss, peripheral neuropathy, leukoencephalopathy | [59] |
| RRM2B | P-53 subunit of ribonucleotide reductase | Autosomal dominant PEO: Bulbar dysfunction, hearing loss, gastrointestinal dysmotility | [64] |
| Autosomal recessive PEO: Retinopathy, myopathy, mood disorders | [66] | ||
| Encephalomyopathy phenotype: Hypotonia, failure to thrive, sensorineural hearing loss, retinopathy, renal tubular necrosis, respiratory failure | [67] | ||
| MNGIE-like phenotype: cachexia, gastrointestinal dysmotility, peripheral neuropathy | [67] | ||
| RNASEH1 | Ribonuclease H1 | CPEO, cerebellar ataxia, dysphagia Proximal muscle weakness, peripheral neuropathy, pyramidal signs | [87,88] |
| TK2 | Thymidine kinase 2 | Early-onset (≤1 year): Esophageal reflux, vomiting, intestinal dysmotility, failure to thrive, severe myopathy, seizures, cognitive impairment, rigid spine, multiple fractures, nephropathy, cardiomyopathy, bilateral optic atrophy Childhood-onset (>1 to ≤12 years): Gowers sign, dropped head, CPEO, facial diplegia, dysphagia, restrictive lung disease, encephalopathy, hearing loss, cognitive decline, multiple fractures, arrhythmias, renal tubulopathy Late-onset (>12 years): Proximal muscle wasting, facial and axial neck flexor muscle weakness, CPEO, bulbar and diaphragmatic weakness | [76] |
| DGOUK | Deoxyguanosine kinase | Psychomotor delay, hypotonia, nystagmus, Optic disc dysplasia, renal involvement, jaundice, cholestasis, hepatomegaly, progressive hepatic disease | [82,130] |
| myopathy, parkinsonism, CPEO, rigidity, bradykinesia, progressive hepatic disease | [84] | ||
| OPA1 | GTPase mitochondrial fusion | Progressive bilateral optic neuropathy, optic atrophy. Dominant optic atrophy-plus: CPEO, myopathy; ataxia; peripheral neuropathy sensorineural hearing loss | [70] |
| MGME1 | Mitochondrial genome maintenance exonuclease 1 | PEO, muscle wasting, emaciation, respiratory failure, skeletal malformations, atrioventricular block, cerebellar atrophy | [92,93] |
| ANT1 | Adenine nucleotide translocator 1 | Exercise intolerance, muscle weakness, ptosis, cardiomyopathy | [97] |
| MPV17 | MPV17 protein | Early-onset hepatocerebral phenotype: hypoglycemia, metabolic acidosis, failure to thrive, liver failure, dysmotility, corneal scarring | [131] |
| neuromyopathic phenotype | [132] | ||
| DNA2 | DNA replication ATP-dependent | CPEO, limb-gridle weakness, Gowers sign, progressive muscle weakness, myalgia, and dyspnea | [133] |
| POLG2 | DNA polymerase subunit gamma 2 | Cerebellar ataxia, CPEO, neuropathy, seizures, parkinsonism, and exercise intolerance | [134] |
| AFG3L2 | AFG3-like protein 2 | Spinocerebellar ataxia type28, CPEO, optic atrophy, nystagmus, parkinsonism | [135,136,137] |
| SPG7 | Paraplegin | Spastic paraplegia 7, Proximal myopathy, CPEO, optic atrophy, dysphagia, spasticity, ataxia, cerebellar atrophy | [138] |
| TOP3A | DNA topoisomerase 3 alpha | Cerebellar ataxia, neuropathy, sensorineural hearing loss, CPEO, myopathy, cardiac conduction defects | [139,140] |
| LIG3 | DNA ligase 3 | MNGIE and MELAS-like phenotype, headache, neurogenic bladder, cataracts, macular degeneration | [63] |
| RRM1 | Ribonucleotide reductase catalytic subunit M1 | Proximal myopathy, dysphagia, ptosis, ophthalmoparesis, peripheral neuropathy, nausea, vomiting, cachexia, intestinal dysmotility | [141] |
| C1QBP | Compliment C1q binding protein | Exercise intolerance, muscle weakness, CPEO, left ventricular hypertrophy | [142,143] |
| GMPR | Guanosine monophosphate reductase | CPEO | [144] |
| Treatment | Mechanism of Action | Conditions | Results | Clinical Trial/Reference |
|---|---|---|---|---|
| Mitochondrial cocktail | ||||
| Coenzyme Q10 (ubiquinone) | Antioxidant/ electron flow restoration in the respiratory chain complex | Mitochondrial diseases | Phase 3 clinical trial (completed) No statistical difference in measured outcomes. | NCT00432744 |
| Primary and Secondary Coenzyme Q10 deficiency | Variable outcomes. | [145] | ||
| L-carnitine | Fatty acid metabolism, detoxification, cell membrane stabilization/control of ketogenesis/gluconeogenesis/Improves metabolic flexibility | Mitochondrial myopathy/CPEO | Improved exercise tolerance and aerobic capacity. | [146] |
| Carnitine deficiency-related mitochondrial dysfunction | Maintenance of metabolic flexibility. | [147] | ||
| Riboflavin (Vitamin B2) | Electron carrier component | Primary and secondary flavoenzyme defects with mitochondrial dysfunction, e.g., complex 1 and 2 deficiencies | Dramatic improvements in supplementation for riboflavin-associated deficiencies. | [148] |
| Thiamine (Vitamin B1) | Coenzyme in the maintenance of carbohydrate metabolism | Genetic dysfunction of thiamine metabolism and transport. | Supplementation improves symptoms, clinical outcomes, and survival. | [149] |
| Thiamine-deficient leigh syndrome | Significant reduction in morbidity and mortality. | [150] | ||
| Alpha lipoic acid (ALA) | Antioxidant | Age-associated cognitive and mitochondrial dysfunction | Improves age-associated memory loss, mitochondrial function, and structure. | [151] |
| ALA and L-acetyl carnitine for supranuclear palsy | Phase 2 clinical trial (completed). Results posted and updated in 2017. | NCT01537549 | ||
| Parkinson’s disease | Slows cognitive decline effectively. | [152] | ||
| Improved mitochondrial function and autophagy. | [153] | |||
| Folinic acid | Increases levels of 5MTHF levels in the brain, Folate is believed to play a role in mitochondrial nucleotide biosynthesis, and mtDNA replication | Cerebral folate deficiency in KSS | Reversal of leukoencephalopathy and normalization of 5MTHF CSF levels. | [154] |
| Early, high dose treatment seems advisable in KSS. | [155] | |||
| Cerebral folate deficiency and mitochondrial complex 1 deficiency | Improvement in hypotonia, ataxia, and cerebral hypomyelination. | [156] | ||
| Mito Q | Antioxidant targeting mitochondria | Mitochondrial diseases, e.g., Friedreich ataxia | Targeted antioxidants are more effective than non-targeting ones. | [157] |
| Benefits including protection from ischemia/ reperfusion injury and endothelial damage. | [158] | |||
| Therapy tackling mitochondrial disease outcomes | ||||
| Idebenone (RAXONE) | Electron carrier, antioxidant | LHON | (RODOS study) No statistically significant difference in recovery of visual acuity except in discordant visual acuity Secondary endpoints were all statistically significant. | NCT00747487 |
| RODOS OFU study Beneficial effects persisted for a median of 30 months post discontinuation. | NCT01421381 | |||
| Phase 4 CT LOROS study Long term treatment results in prolonged clinical benefit in the subacute phase. | NCT02774005 | |||
| MELAS | Phase 2 clinical trial Non statistically significant difference in primary outcomes. | NCT00887562 | ||
| Vatiquinone (EPI-743) | Inhibitor of 15-lipooxygenase, a regulator of oxidative stress pathways Augmentation of glutathione synthesis | Friedreich’s ataxia | Phase 2/3 clinical trial Non statistical difference in primary endpoints. However, there was a statistically significant impact in fatigue scores and disease progression of upright stability and bulbar subscales in a 72-week period. | NCT04577352 |
| Parkinson’s disease | Phase 2 clinical trial Statistically significant in retinal function and unified Parkinson’s disease rating scale. Decrease in oxidative stress markers in the basal ganglia with MRS. | NCT01923584 | ||
| Leigh syndrome | Phase 2 CT Fewer adverse events and hospitalizations in treatment group compared to placebo. | NCT02352896 | ||
| Nicotinamide Riboside | Increasing intracellular levels of NAD, the crucial cofactor for mitochondrial energy production | Obesity and the related metabolic complications | Better systemic NAD metabolism, composition of gut microbiota, myoblast differentiation and mitochondrial number in the muscles. | [159] |
| Healthy subjects | Increased NAD pools without apparent side effects. | [160] | ||
| Parkinson’s disease | NADPARK study Phase 1 clinical trial Increased intracellular levels of NAD, lysosomal and proteasomal function of genes related to the mitochondria of blood and muscles. Decreased serum and CSF inflammatory cytokines. | NCT03816020 | ||
| ANT1-deficient mice | Increased exercise capacity and mitochondrial respiration. | [100] | ||
| L-arginine | Nitric oxide precursor, regulates respiratory chain and oxidative stress in the mitochondria | MELAS | IV arginine improves symptoms during acute MELAS attacks. arginine supplementation increases endothelial function, preventing further stroke-like episodes. | [51] |
| Sickle cell disease, vaso-occlusive pain | Phase 2 clinical trial increases mitochondrial activity and reduces oxidative stress. | NCT02536170 | ||
| L-citrulline | Precursor of arginine | MELAS | Increases the production of NO, as well as concentrations and fluxes of arginine and citrulline. | [161] |
| deoxythymidine and deoxycytidine substrates | Supplies the nucleoside salvage pathway | TK2d | Improve muscle weakness and ambulation. Discontinuing gastrostomy and mechanical ventilation. | [78] |
| elamipretide | improves coupling of electron transport chain by Targeting cardiolipin, a phospholipid in the inner mitochondrial membrane | Heart failure | Improvement of cardiac mitochondrial function, and increased efficiency of complex 1 and 4. | [162] |
| Barth syndrome cardiomyopathy | Improves mitochondrial bioenergetics and morphology rapidly in induced pluripotent stem cells, normalizes mitochondrial ultrastructure and dynamics | [163] | ||
| Increased left ventricular mass and stroke volume | [164] | |||
| Primary mitochondrial myopathy | MMPOWER-3 clinical trial Primary endpoints were not statistically significant, Class I evidence that elamipretide does not improve the 6-min walk test or fatigue at 24 weeks compared with placebo. | NCT03323749 | ||
| LHON | Phase 2 clinical trial Did not achieve primary BCVA outcomes. Improvements in color discrimination, contrast sensitivity and central visual field. | NCT02693119 | ||
| Age-related macular degeneration with non-central geographic atrophy | Phase 2 clinical trial Did not reach primary outcomes. Ameliorated mitochondrial-rich ellipsoid zone progressive decline. Improvement of >2 lines visual improvement in low luminance visual acuity. | NCT03891875 | ||
| Sonlicromanol (KH176) | Antioxidant and redox modulator | Mitochondrial m.3243A>G Spectrum Disorders, e.g., MELAS, MIDD. | KHENERGY Study Phase 2a clinical trial Positive effect on mood and alertness with no other significant parameters. | NCT02909400 |
| Mammalian model of Leigh syndrome | Improved abnormal gait, motor coordination, learning, and decreased the loss of retinal ganglion cells. | [165] | ||
| KL1333 | Increase in intracellular NAD | MELAS fibroblasts | Increased ATP decreases ROS and lactate. Improved mitochondrial biogenesis and function. | [166] |
| PMD | Phase 1 completed (no posted results) | NCT03888716 | ||
| Phase 2 active (no posted results) | NCT05650229 | |||
| edaravone | antioxidant | MELAS | Scavenges ROS, and inhibits inflammation in cerebrovascular disease, improving vascular function. | [42] |
| hyperosmolarity-induced oxidative stress and apoptosis in primary human corneal epithelial cells | Partially attenuated low ATP production induced by hyperosmolarity. | [167] | ||
| Bocidelpar (ASP0367) | Modulation of peroxisome proliferator-activated receptor delta, a modulator of cellular energy consumption | PMM | MOUNTAINSIDE study Phase2/3 clinical trial (no results posted) | NCT04641962 |
| Genetic therapy | ||||
| GS010 | recombinant adeno-associated viral vector serotype 2 containing human wildtype mitochondrial NADH dehydrogenase 4 (ND4) gene | LHON | RESCUE Trial and REVERSE trial Significant improvement of visual acuity in both eyes despite injecting one eye with treatment and the other with a sham injection. | NCT02652767 NCT02652780 |
| REFLECT Trial Subjects treated bilaterally had better average visual acuity than those treated unilaterally. | NCT03293524 | |||
| Mitochondrial Augmentation Therapy | Replacing dysfunctional mitochondria with healthy donor mitochondria | Pearson syndrome | Phase1/2 clinical trial | NCT03384420 |
| Improved mitochondrial function, heteroplasmy, and respiratory capacity. Improved quality of life, aerobic ability, and fine motor functions. | [168] | |||
| Mesoangioblasts (MABs) | Lowering the percentage of mtDNA mutational load | m.3243A>G Mutation Carriers | Phase 2 clinical trial Active, recruiting. | NCT05962333 |
| mtDNA mutated myotubes, m.3271T>C and m.3291T>C mutation | Proportional reduction in mtDNA mutational load in vitro after fusion of wild type MABs. | [169] | ||
| mtDNA point mutation or large-scale deletions | Half the mtDNA carriers have nearly mutation free MABs. | [170] | ||
| Allogeneic hematopoietic stem cell transplant | Restoring thymidine phosphorylase enzyme function | MNGIE | Short- and long-term outcomes are influenced by a diagnosis earlier than irreversible gastrointestinal symptoms, a fit matched HLA-donor, and a busulfan-based conditioning regimen. | [171] |
| Unfavorable overall outcome pertaining to mortality. Significant improvement in progression and clinical manifestations over time. | [172] | |||
References
- Yu-Wai-Man, P.; Clements Al Nesbitt, V.; Griffiths, P.G.; Gorman, G.S.; Schaefer, A.M.; Turnbull, D.M.; Taylor, R.W.; McFarland, R. A national epidemiological study of chronic progressive external ophthalmoplegia in the United Kingdom—Molecular genetic features and neurological burden. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5109. [Google Scholar]
- Kearns, T.P.; Sayre, G.P. Retinitis pigmentosa, external ophthalmophegia, and complete heart block: Unusual syndrome with histologic study in one of two cases. AMA Arch. Ophthalmol. 1958, 60, 280–289. [Google Scholar] [CrossRef]
- Olson, W.; Engel, W.K.; Walsh, G.O.; Einaugler, R. Oculocraniosomatic neuromuscular disease with “ragged-red” fibers. Arch. Neurol. 1972, 26, 193–211. [Google Scholar] [CrossRef]
- Zintz, R.; Villiger, W. Electron microscopic findings in 3 cases of chronic progressive ocular muscular dystrophy. Ophthalmologica 1967, 153, 439–459. [Google Scholar] [CrossRef]
- Reske-Nielsen, E.; Lou, H.C. Lowes, MProgressive external ophthalmoplegia. Evidence for a generalised mitochondrial disease with a defect in pyruvate metabolism. Acta Ophthalmol. 1976, 54, 553–573. [Google Scholar] [CrossRef]
- Holt, I.J.; Harding, A.E.; Morgan-Hughes, J.A. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 1988, 331, 717–719. [Google Scholar] [CrossRef] [PubMed]
- Zeviani, M.; Servidei, S.; Gellera, C.; Bertini, E.; DiMauro, S.; DiDonato, S. An autosomal dominant disorder with multiple deletions of mitochondrial DNA starting at the D-loop region. Nature 1989, 339, 309–311. [Google Scholar] [CrossRef] [PubMed]
- Kaukonen, J.; Juselius, J.K.; Tiranti, V.; Kyttälä, A.; Zeviani, M.; Comi, G.P.; Keränen, S.; Peltonen, L.; Suomalainen, A. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science 2000, 289, 782–785. [Google Scholar] [CrossRef]
- Thangaraj, K.; Khan, N.A.; Govindaraj, P.; Meena, A.K. Mitochondrial disorders: Challenges in diagnosis & treatment. Indian J. Med. Res. 2015, 141, 13–26. [Google Scholar]
- Greaves, L.C.; Yu-Wai-Man, P.; Blakely, E.L.; Krishnan, K.J.; Beadle, N.E.; Kerin, J.; Barron, M.J.; Griffiths, P.G.; Dickinson, A.J.; Turnbull, D.M.; et al. Mitochondrial DNA Defects and Selective Extraocular Muscle Involvement in CPEO. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3340–3346. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Burr, S.P.; Chinnery, P.F. The mitochondrial DNA genetic bottleneck: Inheritance and beyond. Essays Biochem. 2018, 62, 225–234. [Google Scholar]
- Zaidi, A.A.; Wilton, P.R.; Su, M.S.-W.; Paul, I.M.; Arbeithuber, B.; Anthony, K.; Nekrutenko, A.; Nielsen, R.; Makova, K.D. Bottleneck and selection in the germline and maternal age influence transmission of mitochondrial DNA in human pedigrees. Proc. Natl. Acad. Sci. USA 2019, 116, 25172–25178. [Google Scholar] [CrossRef]
- Rusecka, J.; Kaliszewska, M.; Bartnik, E.; Tońska, K. Nuclear genes involved in mitochondrial diseases caused by instability of mitochondrial DNA. J. Appl. Genet. 2018, 59, 43–57. [Google Scholar] [CrossRef]
- Chatzistefanou, K.I.; Brouzas, D.; Asproudis, I.; Tsina, E.; Droutsas, K.D.; Koutsandrea, C. Strabismus surgery for diplopia in chronic progressive external ophthalmoplegia. Int. Ophthalmol. 2019, 39, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Richardson, C.; Smith, T.; Schaefer, A.; Turnbull, D.; Griffiths, P. Ocular motility findings in chronic progressive external ophthalmoplegia. Eye 2005, 19, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, G.; Sirrs, S.; Wade, N.K.; Mezei, M.M. Multisystem Disorder in Late-Onset Chronic Progressive External Ophthalmoplegia. Can. J. Neurol. Sci. 2011, 38, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.G.; Rojdamrongratana, D.; Rosenblatt, M.I.; Aakalu, V.K.; Yu, C.Q. 3D printing for low cost, rapid prototyping of eyelid crutches. Orbit 2019, 38, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Kim, N.J.; Choung, H.K.; Hwang, S.W.; Sung, M.; Lee, M.J.; Khwarg, S.I. Frontalis sling operation using silicone rod for the correction of ptosis in chronic progressive external ophthalmoplegia. Br. J. Ophthalmol. 2008, 92, 1685–1688. [Google Scholar] [CrossRef]
- Rajabi, M.T.; Tabatabaie, S.Z.; Rajabi, M.B.; Abrishami, Y.; Hosseini, S.S.; Oestreicher, J. Management of myogenic ptosis in chronic progressive external ophtalmoplegia. Iran. J. Neurol. 2014, 13, 185–187. [Google Scholar]
- Shemesh, A.; Margolin, E. Kearns Sayre Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Ortiz, A.; Arias, J.; Cárdenas, P.; Villamil, J.; Peralta, M.; Escaf, L.C.; Ortiz, J. Macular findings in Spectral Domain Optical Coherence Tomography and OCT Angiography in a patient with Kearns–Sayre syndrome. Int. J. Retin. Vitr. 2017, 3, 24. [Google Scholar] [CrossRef]
- Kang, Y.X.; Wang, Y.J.; Zhang, Q.; Pang, X.H.; Gu, W. A case of hypopituitarism accompanying Kearns–Sayre syndrome treated with human chorionic gonadotropin: A case report and literature review. Andrologia 2017, 49, e12711. [Google Scholar] [CrossRef]
- Ng, Y.S.; Lim, A.Z.; Panagiotou, G.; Turnbull, D.M.; Walker, M. Endocrine Manifestations and New Developments in Mitochondrial Disease. Endocr. Rev. 2021, 43, 583–609. [Google Scholar] [CrossRef]
- Katsanos, K.H.; Nastos, D.; Noussias, V.; Christodoulou, D.; Kappas, A.; Tsianos, E.V. Manometric study in Kearns–Sayre syndrome. Dis. Esophagus 2001, 14, 63–66. [Google Scholar] [CrossRef]
- Vadhul, R.; Halbach, C.S.; Areaux, R.G., Jr.; Berry, S.; Hou, J.H. Endothelial dysfunction in a child with Pearson marrow-pancreas syndrome managed with Descemet stripping automated endothelial keratoplasty using a suture pull-through technique. Digit. J. Ophthalmol. 2019, 25, 59–64. [Google Scholar] [CrossRef]
- Shoeleh, C.; Donato, U.M.; Galligan, A.; Vitko, J. A Case Report on Pearson Syndrome with Emphasis on Genetic Screening in Patients Presenting with Sideroblastic Anemia and Lactic Acidosis. Cureus 2023, 15, e33963. [Google Scholar] [CrossRef]
- Jennifer, M.S.; Cortez, D. Pearson marrow-pancreas syndrome with cardiac conduction abnormality necessitating prophylactic pacemaker implantation. Ann. Noninvasive Electrocardiol. 2020, 25, e12681. [Google Scholar] [CrossRef]
- Reddy, J.; Jose, J.; Prakash, A.; Devi, S. Pearson syndrome: A rare inborn error of metabolism with bone marrow morphology providing a clue to diagnosis. Sudan. J. Paediatr. 2019, 19, 161–164. [Google Scholar] [CrossRef]
- Broomfield, A.; Sweeney, M.G.; Woodward, C.E.; Fratter, C.; Morris, A.M.; Leonard, J.V.; Abulhoul, L.; Grunewald, S.; Clayton, P.T.; Hanna, M.G.; et al. Paediatric single mitochondrial DNA deletion disorders: An overlapping spectrum of disease. J. Inherit. Metab. Dis. 2015, 38, 445–457. [Google Scholar] [CrossRef]
- Faraci, M.; Cuzzubbo, D.; Micalizzi, C.; Lanino, E.; Morreale, G.; Dallorso, S.; Castagnola, E.; Schiaffino, M.C.; Bruno, C.; Rossi, A.; et al. Allogeneic bone marrow transplantation for Pearson’s syndrome. Bone Marrow Transplant. 2007, 39, 563–565. [Google Scholar] [CrossRef]
- Saini, A.G.; Chatterjee, D.; Bhagwat, C.; Vyas, S.; Attri, S.V. Leigh syndrome in an infant: Autopsy and histopathology findings. Autops. Case Rep. 2021, 11, e2021334. [Google Scholar] [CrossRef]
- Lake, N.J.; Compton, A.G.; Rahman, S.; Thorburn, D.R. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann. Neurol. 2016, 79, 190–203. [Google Scholar] [CrossRef]
- Kistol, D.; Tsygankova, P.; Krylova, T.; Bychkov, I.; Itkis, Y.; Nikolaeva, E.; Mikhailova, S.; Sumina, M.; Pechatnikova, N.; Kurbatov, S.; et al. Leigh Syndrome: Spectrum of Molecular Defects and Clinical Features in Russia. Int. J. Mol. Sci. 2023, 24, 1597. [Google Scholar] [CrossRef]
- Chang, X.; Wu, Y.; Zhou, J.; Meng, H.; Zhang, W.; Guo, J. A meta-analysis and systematic review of Leigh syndrome: Clinical manifestations, respiratory chain enzyme complex deficiency, and gene mutations. Medicine 2020, 99, e18634. [Google Scholar] [CrossRef]
- Han, J.; Lee, Y.-M.; Kim, S.M.; Han, S.Y.; Lee, J.B. Ophthalmological manifestations in patients with Leigh syndrome. Br. J. Ophthalmol. 2015, 99, 528–535. [Google Scholar] [CrossRef]
- Sofou, K.; de Coo, I.F.M.; Ostergaard, E.; Isohanni, P.; Naess, K.; De Meirleir, L.; Tzoulis, C.; Uusimaa, J.; Lönnqvist, T.; Bindoff, L.A.; et al. Phenotype-genotype correlations in Leigh syndrome: New insights from a multicentre study of 96 patients. J. Med. Genet. 2018, 55, 21–27. [Google Scholar] [CrossRef]
- Brecht, M.; Richardson, M.; Taranath, A.; Grist, S.; Thorburn, D.; Bratkovic, D. Leigh Syndrome Caused by the MT-ND5 m.13513G>A Mutation: A Case Presenting with WPW-Like Conduction Defect, Cardiomyopathy, Hypertension and Hyponatraemia. JIMD Rep. 2015, 19, 95–100. [Google Scholar]
- Ardissone, A.; Bruno, C.; Diodato, D.; Donati, A.; Ghezzi, D.; Lamantea, E.; Lamperti, C.; Mancuso, M.; Martinelli, D.; Primiano, G.; et al. Clinical, imaging, biochemical and molecular features in Leigh syndrome: A study from the Italian network of mitochondrial diseases. Orphanet J. Rare Dis. 2021, 16, 413. [Google Scholar] [CrossRef]
- Alves, C.A.P.F.; Teixeira, S.R.; Martin-Saavedra, J.S.; Gonçalves, F.G.; Russo, F.L.; Muraresku, C.; McCormick, E.M.; Falk, M.J.; Zolkipli-Cunningham, Z.; Ganetzky, R.; et al. Pediatric Leigh Syndrome: Neuroimaging Features and Genetic Correlations. Ann. Neurol. 2020, 88, 218–232. [Google Scholar] [CrossRef]
- Baertling, F.; Rodenburg, R.J.; Schaper, J.; Smeitink, J.A.; Koopman, W.J.H.; Mayatepek, E.; Morava, E.; Distelmaier, F. A guide to diagnosis and treatment of Leigh syndrome. J. Neurol. Neurosurg. Psychiatry 2014, 85, 257–265. [Google Scholar] [CrossRef]
- Ogawa, E.; Shimura, M.; Fushimi, T.; Tajika, M.; Ichimoto, K.; Matsunaga, A.; Tsuruoka, T.; Ishige, M.; Fuchigami, T.; Yamazaki, T.; et al. Clinical validity of biochemical and molecular analysis in diagnosing Leigh syndrome: A study of 106 Japanese patients. J. Inherit. Metab. Dis. 2017, 40, 685–693. [Google Scholar] [CrossRef]
- Pek, N.M.Q.; Phua, Q.H.; Ho, B.X.; Pang, J.K.S.; Hor, J.-H.; An, O.; Yang, H.H.; Yu, Y.; Fan, Y.; Ng, S.-Y.; et al. Mitochondrial 3243A>G mutation confers pro-atherogenic and pro-inflammatory properties in MELAS iPS derived endothelial cells. Cell Death Dis. 2019, 10, 802. [Google Scholar] [CrossRef]
- Kaufmann, P.; Engelstad, K.; Wei, Y.; Kulikova, R.; Oskoui, M.; Sproule, D.; Battista, V.; Koenigsberger, D.; Pascual, J.; Shanske, S.; et al. Natural history of MELAS associated with mitochondrial DNA m.3243A>G genotype. Neurology 2011, 77, 1965–1971. [Google Scholar] [CrossRef]
- Seed, L.M.; Dean, A.; Krishnakumar, D.; Phyu, P.; Horvath, R.; Harijan, P.D. Molecular and neurological features of MELAS syndrome in paediatric patients: A case series and review of the literature. Mol. Genet. Genom. Med. 2022, 10, e1955. [Google Scholar] [CrossRef]
- Al-Enezi, M.; Al-Saleh, H.; Nasser, M. Mitochondrial disorders with significant ophthalmic manifestations. Middle East Afr. J. Ophthalmol. 2008, 15, 81–86. [Google Scholar] [CrossRef]
- Weiduschat, N.; Kaufmann, P.; Mao, X.; Engelstad, K.M.; Hinton, V.; DiMauro, S.; De Vivo, D.; Shungu, D. Cerebral metabolic abnormalities in A3243G mitochondrial DNA mutation carriers. Neurology 2014, 82, 798–805. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Emrick, L.T.; Craigen, W.J.; Scaglia, F. Citrulline and arginine utility in treating nitric oxide deficiency in mitochondrial disorders. Mol. Genet. Metab. 2012, 107, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Ouchi, A.; Miura, D.; Shimoji, K.; Kinjo, K.; Sueyoshi, T.; Jonosono, M.; Rajput, V. MELAS and reversible vasoconstriction of the major cerebral arteries. Intern. Med. 2013, 52, 1389–1392. [Google Scholar] [CrossRef][Green Version]
- Cheng, W.; Zhang, Y.; He, L. MRI Features of Stroke-Like Episodes in Mitochondrial Encephalomyopathy with Lactic Acidosis and Stroke-Like Episodes. Front. Neurol. 2022, 13, 843386. [Google Scholar] [CrossRef]
- Ng, Y.S.; Bindoff, L.A.; Gorman, G.S.; Horvath, R.; Klopstock, T.; Mancuso, M.; Martikainen, M.H.; Mcfarland, R.; Nesbitt, V.; Pitceathly, R.D.S.; et al. Consensus-based statements for the management of mitochondrial stroke-like episodes. Wellcome Open Res. 2019, 4, 201. [Google Scholar] [CrossRef] [PubMed]
- Argudo, J.M.; Moncayo, O.M.A.; Insuasti, W.; Garofalo, G.; Aguirre, A.S.; Encalada, S.; Villamarin, J.; Oña, S.; Tenemaza, M.G.; Eissa-Garcés, A.; et al. Arginine for the Treatment of Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-Like Episodes: A Systematic Review. Cureus 2022, 14, e32709. [Google Scholar] [CrossRef]
- Pérez-Cruz, E.; González-Rivera, C.; Valencia-Olvera, L.d.C.G. Immunonutrition for the acute treatment of MELAS syndrome. Endocrinología. Diabetes Nutr. 2022, 69, 144–148. [Google Scholar]
- Li, J.; Zhang, W.; Cui, Z.; Li, Z.; Jiang, T.; Meng, H. Epilepsy Associated with Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-Like Episodes. Front. Neurol. 2021, 12, 675816. [Google Scholar] [CrossRef]
- Rahman, S.; Copeland, W.C. POLG-related disorders and their neurological manifestations. Nat. Rev. Neurol. 2019, 15, 40–52. [Google Scholar] [CrossRef]
- Hikmat, O.; Naess, K.; Engvall, M.; Klingenberg, C.; Rasmussen, M.; Tallaksen, C.M.; Brodtkorb, E.; Ostergaard, E.; de Coo, I.F.M.; Pias-Peleteiro, L.; et al. Simplifying the clinical classification of polymerase gamma (POLG) disease based on age of onset; studies using a cohort of 155 cases. J. Inherit. Metab. Dis. 2020, 43, 726–736. [Google Scholar] [CrossRef]
- Ciesielski, G.L.; Rosado-Ruiz, F.A.; Kaguni, L.S. Purification and Comparative Assay of Human Mitochondrial Single-Stranded DNA-Binding Protein. Methods Mol. Biol. 2016, 1351, 211–222. [Google Scholar]
- Goffart, S.; Cooper, H.M.; Tyynismaa, H.; Wanrooij, S.; Suomalainen, A.; Spelbrink, J.N. Twinkle mutations associated with autosomal dominant progressive external ophthalmoplegia lead to impaired helicase function and in vivo mtDNA replication stalling. Hum. Mol. Genet. 2009, 18, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Bermejo-Guerrero, L.; de Fuenmayor-Fernández de la Hoz, C.P.; Serrano-Lorenzo, P.; Blázquez-Encinar, A.; Gutiérrez-Gutiérrez, G.; Martínez-Vicente, L.; Galán-Dávila, L.; García-García, J.; Arenas, J.; Muelas, N.; et al. Clinical, Histological, and Genetic Features of 25 Patients with Autosomal Dominant Progressive External Ophthalmoplegia (ad-PEO)/PEO-Plus Due to TWNK Mutations. J. Clin. Med. 2021, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Ronchi, D.; Caporali, L.; Manenti, G.F.; Meneri, M.; Mohamed, S.; Bordoni, A.; Tagliavini, F.; Contin, M.; Piga, D.; Sciacco, M.; et al. TYMP Variants Result in Late-Onset Mitochondrial Myopathy With Altered Muscle Mitochondrial DNA Homeostasis. Front. Genet. 2020, 11, 860. [Google Scholar] [CrossRef] [PubMed]
- Cardaioli, E.; Da Pozzo, P.; Malfatti, E.; Battisti, C.; Gallus, G.N.; Gaudiano, C.; Macucci, M.; Malandrini, A.; Margollicci, M.; Rubegni, A.; et al. A second MNGIE patient without typical mitochondrial skeletal muscle involvement. Neurol. Sci. 2010, 31, 491–494. [Google Scholar] [CrossRef]
- Martí, R.; Spinazzola, A.; Tadesse, S.; Nishino, I.; Nishigaki, Y.; Hirano, M. Definitive Diagnosis of Mitochondrial Neurogastrointestinal Encephalomyopathy by Biochemical Assays. Clin. Chem. 2004, 50, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Filosto, M.; Piccinelli, S.C.; Caria, F.; Cassarino, S.G.; Baldelli, E.; Galvagni, A.; Volonghi, I.; Scarpelli, M.; Padovani, A. Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE-MTDPS1). J. Clin. Med. 2018, 7, 389. [Google Scholar] [CrossRef] [PubMed]
- Bonora, E.; Chakrabarty, S.; Kellaris, G.; Tsutsumi, M.; Bianco, F.; Bergamini, C.; Ullah, F.; Isidori, F.; Liparulo, I.; Diquigiovanni, C.; et al. Biallelic variants in LIG3 cause a novel mitochondrial neurogastrointestinal encephalomyopathy. Brain 2021, 144, 1451–1466. [Google Scholar] [CrossRef] [PubMed]
- Pitceathly, R.D.S.; Smith, C.; Fratter, C.; Alston, C.L.; He, L.; Craig, K.; Blakely, E.L.; Evans, J.C.; Taylor, J.; Shabbir, Z.; et al. Adults with RRM2B-related mitochondrial disease have distinct clinical and molecular characteristics. Brain 2012, 135, 3392–3403. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Craigen, W.J.; Scaglia, F. Mitochondrial DNA maintenance defects. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 1539–1555. [Google Scholar] [CrossRef] [PubMed]
- Takata, A.; Kato, M.; Nakamura, M.; Yoshikawa, T.; Kanba, S.; Sano, A.; Kato, T. Exome sequencing identifies a novel missense variant in RRM2B associated with autosomal recessive progressive external ophthalmoplegia. Genome Biol. 2011, 12, R92. [Google Scholar] [CrossRef]
- Keshavan, N.; Abdenur, J.; Anderson, G.; Assouline, Z.; Barcia, G.; Bouhikbar, L.; Chakrapani, A.; Cleary, M.; Cohen, M.C.; Feillet, F.; et al. The natural history of infantile mitochondrial DNA depletion syndrome due to RRM2B deficiency. Genet. Med. 2020, 22, 199–209. [Google Scholar] [CrossRef]
- Shaibani, A.; Shchelochkov, O.A.; Zhang, S.; Katsonis, P.; Lichtarge, O.; Wong, L.J.; Shinawi, M. Mitochondrial neurogastrointestinal encephalopathy due to mutations in RRM2B. Arch. Neurol. 2009, 66, 1028–1032. [Google Scholar] [CrossRef]
- Lee, H.; Yoon, Y. Mitochondrial Membrane Dynamics-Functional Positioning of OPA1. Antioxidants 2018, 7, 186. [Google Scholar] [CrossRef]
- Zeng, T.; Liao, L.; Guo, Y.; Liu, X.; Xiong, X.; Zhang, Y.; Cen, S.; Li, H.; Wei, S. Concurrent OPA1 mutation and chromosome 3q deletion leading to Behr syndrome: A case report. BMC Pediatr. 2020, 20, 420. [Google Scholar] [CrossRef]
- Carelli, V.; Musumeci, O.; Caporali, L.; Zanna, C.; La Morgia, C.; Del Dotto, V.; Porcelli, A.M.; Rugolo, M.; Valentino, M.L.; Iommarini, L.; et al. Syndromic parkinsonism and dementia associated with OPA1 missense mutations. Ann. Neurol. 2015, 78, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Griffiths, P.G.; Gorman, G.S.; Lourenco, C.M.; Wright, A.F.; Auer-Grumbach, M.; Toscano, A.; Musumeci, O.; Valentino, M.L.; Caporali, L.; et al. Multi-system neurological disease is common in patients with OPA1 mutations. Brain 2010, 133, 771–786. [Google Scholar] [CrossRef]
- Romagnoli, M.; La Morgia, C.; Carbonelli, M.; Di Vito, L.; Amore, G.; Zenesini, C.; Cascavilla, M.L.; Barboni, P.; Carelli, V. Dominant optic atrophy. Orphanet J. Rare Dis. 2012, 7, 46. [Google Scholar]
- Gedikbasi, A.; Toksoy, G.; Karaca, M.; Gulec, C.; Balci, M.C.; Gunes, D.; Gunes, S.; Aslanger, A.D.; Unverengil, G.; Karaman, B.; et al. Idebenone increases chance of stabilization/recovery of visual acuity in OPA1-dominant optic atrophy. Ann. Clin. Transl. Neurol. 2020, 7, 590–594. [Google Scholar]
- Gedikbasi, A.; Toksoy, G.; Karaca, M.; Gulec, C.; Balci, M.C.; Gunes, D.; Gunes, S.; Aslanger, A.D.; Unverengil, G.; Karaman, B.; et al. Clinical and bi-genomic DNA findings of patients suspected to have mitochondrial diseases. Front. Genet. 2023, 14, 1191159. [Google Scholar] [CrossRef]
- Berardo, A.; Domínguez-González, C.; Engelstad, K.; Hirano, M. Advances in Thymidine Kinase 2 Deficiency: Clinical Aspects, Translational Progress, and Emerging Therapies. J. Neuromuscul. Dis. 2022, 9, 225–235. [Google Scholar] [CrossRef]
- Domínguez-González, C.; Fernández-Torrón, R.; Moore, U.; Hoz, C.P.d.F.-F.d.l.; Vélez-Gómez, B.; Cabezas, J.A.; Alonso-Pérez, J.; González-Mera, L.; Olivé, M.; García-García, J.; et al. Muscle MRI characteristic pattern for late-onset TK2 deficiency diagnosis. J. Neurol. 2022, 269, 3550–3562. [Google Scholar] [CrossRef] [PubMed]
- Domínguez-González, C.; Madruga-Garrido, M.; Mavillard, F.; Garone, C.; Aguirre-Rodríguez, F.J.; Donati, M.A.; Kleinsteuber, K.; Martí, I.; Martín-Hernández, E.; Morealejo-Aycinena, J.P.; et al. Deoxynucleoside Therapy for Thymidine Kinase 2–Deficient Myopathy. Ann. Neurol. 2019, 86, 293–303. [Google Scholar] [CrossRef]
- Montano, V.; Simoncini, C.; Calì, C.L.; Legati, A.; Siciliano, G.; Mancuso, M. CPEO and Mitochondrial Myopathy in a Patient with DGUOK Compound Heterozygous Pathogenetic Variant and mtDNA Multiple Deletions. Case Rep. Neurol. Med. 2019, 2019, 5918632. [Google Scholar] [CrossRef] [PubMed]
- Copeland, W.C. Defects in mitochondrial DNA replication and human disease. Crit. Rev. Biochem. Mol. Biol. 2011, 47, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Dimmock, D.P.; Dunn, J.K.; Feigenbaum, A.; Rupar, A.; Horvath, R.; Freisinger, P.; de Camaret, B.M.; Wong, L.-J.; Scaglia, F. Abnormal neurological features predict poor survival and should preclude liver transplantation in patients with deoxyguanosine kinase deficiency. Liver Transplant. 2008, 14, 1480–1485. [Google Scholar] [CrossRef] [PubMed]
- Dimmock, D.P.; Zhang, Q.; Dionisi-Vici, C.; Carrozzo, R.; Shieh, J.; Tang, L.-Y.; Truong, C.; Schmitt, E.; Sifry-Platt, M.; Lucioli, S.; et al. Clinical and molecular features of mitochondrial DNA depletion due to mutations in deoxyguanosine kinase. Hum. Mutat. 2008, 29, 330–331. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J.; Scorza, F. Renal manifestations of primary mitochondrial disorders (Review). Biomed. Rep. 2017, 6, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Caporali, L.; Bello, L.; Tagliavini, F.; La Morgia, C.; Maresca, A.; Di Vito, L.; Liguori, R.; Valentino, M.L.; Cecchin, D.; Pegoraro, E.; et al. DGUOK recessive mutations in patients with CPEO, mitochondrial myopathy, parkinsonism and mtDNA deletions. Brain 2017, 141, e3. [Google Scholar] [CrossRef]
- Grabhorn, E.; Tsiakas, K.; Herden, U.; Fischer, L.; Freisinger, P.; Marquardt, T.; Ganschow, R.; Briem-Richter, A.; Santer, R. Long-term outcomes after liver transplantation for deoxyguanosine kinase deficiency: A single-center experience and a review of the literature. Liver Transplant. 2014, 20, 464–472. [Google Scholar] [CrossRef]
- Misic, J.; Milenkovic, D.; Al-Behadili, A.; Xie, X.; Jiang, M.; Jiang, S.; Filograna, R.; Koolmeister, C.; Siira, S.J.; Jenninger, L.; et al. Mammalian RNase H1 directs RNA primer formation for mtDNA replication initiation and is also necessary for mtDNA replication completion. Nucleic Acids Res. 2022, 50, 8749–8766. [Google Scholar] [CrossRef] [PubMed]
- Manini, A.; Caporali, L.; Meneri, M.; Zanotti, S.; Piga, D.; Arena, I.G.; Corti, S.; Toscano, A.; Comi, G.P.; Musumeci, O.; et al. Case Report: Rare Homozygous RNASEH1 Mutations Associated with Adult-Onset Mitochondrial Encephalomyopathy and Multiple Mitochondrial DNA Deletions. Front. Genet. 2022, 13, 906667. [Google Scholar] [CrossRef]
- Bugiardini, E.; Poole, O.V.; Manole, A.; Pittman, A.M.; Horga, A.; Hargreaves, I.; Woodward, C.E.; Sweeney, M.G.; Holton, J.L.; Taanman, J.W.; et al. Clinicopathologic and molecular spectrum of RNASEH1-related mitochondrial disease. Neurol. Genet. 2017, 3, e149. [Google Scholar] [CrossRef]
- Uhler, J.P.; Thörn, C.; Nicholls, T.J.; Matic, S.; Milenkovic, D.; Gustafsson, C.M.; Falkenberg, M. MGME1 processes flaps into ligatable nicks in concert with DNA polymerase γ during mtDNA replication. Nucleic Acids Res. 2016, 44, 5861–5871. [Google Scholar] [CrossRef]
- Szczesny, R.J.; Hejnowicz, M.S.; Steczkiewicz, K.; Muszewska, A.; Borowski, L.S.; Ginalski, K.; Dziembowski, A. Identification of a novel human mitochondrial endo-/exonuclease Ddk1/c20orf72 necessary for maintenance of proper 7S DNA levels. Nucleic Acids Res. 2013, 41, 3144–3161. [Google Scholar] [CrossRef]
- Zhao, L. Mitochondrial DNA degradation: A quality control measure for mitochondrial genome maintenance and stress response. Enzymes 2019, 45, 311–341. [Google Scholar]
- Kornblum, C.; Nicholls, T.J.; Haack, T.B.; Schöler, S.; Peeva, V.; Danhauser, K.; Hallmann, K.; Zsurka, G.; Rorbach, J.; Iuso, A.; et al. Loss-of-function mutations in MGME1 impair mtDNA replication and cause multisystemic mitochondrial disease. Nat. Genet. 2013, 45, 214–219. [Google Scholar] [CrossRef]
- Rocha, E.B.d.S.; Rodrigues, K.d.L.; Montouro, L.A.M.; Coelho, É.N.; Kouyoumdjian, J.A.; Kok, F.; Nóbrega, P.R.; Graca, C.R.; Morita, M.d.P.A.; Estephan, E.d.P. A case of mitochondrial DNA depletion syndrome type 11—Expanding the genotype and phenotype. Neuromuscul. Disord. 2023, 33, 692–696. [Google Scholar] [CrossRef]
- Lee, J.; Schriner, S.E.; Wallace, D.C. Adenine nucleotide translocator 1 deficiency increases resistance of mouse brain and neurons to excitotoxic insults. Biochim. Biophys. Acta (BBA)-Bioenerg. 2009, 1787, 364–370. [Google Scholar] [CrossRef]
- Echaniz-Laguna, A.; Chassagne, M.; Ceresuela, J.; Rouvet, I.; Padet, S.; Acquaviva, C.; Nataf, S.; Vinzio, S.; Bozon, D.; de Camaret, B.M. Complete loss of expression of the ANT1 gene causing cardiomyopathy and myopathy. J. Med. Genet. 2012, 49, 146–150. [Google Scholar] [CrossRef]
- Klumpe, I.; Savvatis, K.; Westermann, D.; Tschöpe, C.; Rauch, U.; Landmesser, U.; Schultheiss, H.-P.; Dörner, A. Transgenic overexpression of adenine nucleotide translocase 1 protects ischemic hearts against oxidative stress. J. Mol. Med. 2016, 94, 645–653. [Google Scholar] [CrossRef]
- Kawamata, H.; Tiranti, V.; Magrané, J.; Chinopoulos, C.; Manfredi, G. adPEO mutations in ANT1 impair ADP-ATP translocation in muscle mitochondria. Hum. Mol. Genet. 2011, 20, 2964–2974. [Google Scholar] [CrossRef] [PubMed]
- Strauss, K.A.; DuBiner, L.; Simon, M.; Zaragoza, M.; Sengupta, P.P.; Li, P.; Narula, N.; Dreike, S.; Platt, J.; Procaccio, V.; et al. Severity of cardiomyopathy associated with adenine nucleotide translocator-1 deficiency correlates with mtDNA haplogroup. Proc. Natl. Acad. Sci. USA 2013, 110, 3453–3458. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Alder, N.N.; Wang, W.; Szeto, H.; Marcinek, D.J.; Rabinovitch, P.S. Reduction of elevated proton leak rejuvenates mitochondria in the aged cardiomyocyte. eLife 2020, 9, e60827. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, P.M.; Huang, J.; Butic, A.; Perry, C.; Yardeni, T.; Tan, W.; Morrow, R.; Baur, J.A.; Wallace, D.C. Nicotinamide riboside alleviates exercise intolerance in ANT1-deficient mice. Mol. Metab. 2022, 64, 101560. [Google Scholar] [CrossRef] [PubMed]
- Alonzo, J.R.; Venkataraman, C.; Field, M.S.; Stover, P.J. The mitochondrial inner membrane protein MPV17 prevents uracil accumulation in mitochondrial DNA. J. Biol. Chem. 2018, 293, 20285–20294. [Google Scholar] [CrossRef]
- Jacinto, S.; Guerreiro, P.; de Oliveira, R.M.; Cunha-Oliveira, T.; Santos, M.J.; Grazina, M.; Rego, A.C.; Outeiro, T.F. MPV17 Mutations Are Associated with a Quiescent Energetic Metabolic Profile. Front. Cell. Neurosci. 2021, 15, 641264. [Google Scholar] [CrossRef]
- Blakely, E.L.; Butterworth, A.; Hadden, R.D.; Bodi, I.; He, L.; McFarland, R.; Taylor, R.W. MPV17 mutation causes neuropathy and leukoencephalopathy with multiple mtDNA deletions in muscle. Neuromuscul. Disord. 2012, 22, 587–591. [Google Scholar] [CrossRef]
- Uusimaa, J.; Evans, J.; Smith, C.; Butterworth, A.; Craig, K.; Ashley, N.; Liao, C.; Carver, J.; Diot, A.; Macleod, L.; et al. Clinical, biochemical, cellular and molecular characterization of mitochondrial DNA depletion syndrome due to novel mutations in the MPV17 gene. Eur. J. Hum. Genet. 2014, 22, 184–191. [Google Scholar] [CrossRef]
- Tsygankova, P.G.; Itkis, Y.S.; Krylova, T.D.; Kurkina, M.V.; Bychkov, I.O.; Ilyushkina, A.A.; Zabnenkova, V.V.; Mikhaylova, S.V.; Pechatnikova, N.L.; Sheremet, N.L.; et al. Plasma FGF-21 and GDF-15 are elevated in different inherited metabolic diseases and are not diagnostic for mitochondrial disorders. J. Inherit. Metab. Dis. 2019, 42, 918–933. [Google Scholar] [CrossRef]
- Yatsuga, S.; Fujita, Y.; Ishii, A.; Fukumoto, Y.; Arahata, H.; Kakuma, T.; Kojima, T.; Ito, M.; Tanaka, M.; Saiki, R.; et al. Growth differentiation factor 15 as a useful biomarker for mitochondrial disorders. Ann. Neurol. 2015, 78, 814–823. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.L.; Liang, C.; Sue, C.M. A comparison of current serum biomarkers as diagnostic indicators of mitochondrial diseases. Neurology 2016, 86, 2010–2015. [Google Scholar] [CrossRef] [PubMed]
- Bernier, F.P.; Boneh, A.; Dennett, X.; Chow, C.W.; Cleary, M.A.; Thorburn, D.R. Diagnostic criteria for respiratory chain disorders in adults and children. Neurology 2002, 59, 1406–1411. [Google Scholar] [CrossRef] [PubMed]
- Eloise, W.; Ryan, D.; Carolyn, M.S. New diagnostic pathways for mitochondrial disease. J. Transl. Genet. Genom. 2020, 4, 188–202. [Google Scholar]
- Cejudo, P.; Bautista, J.; Montemayor, T.; Villagómez, R.; Jiménez, L.; Ortega, F.; Campos, Y.; Sánchez, H.; Arenas, J. Exercise training in mitochondrial myopathy: A randomized controlled trial. Muscle Nerve 2005, 32, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Jeppesen, T.D. Aerobic Exercise Training in Patients with mtDNA-Related Mitochondrial Myopathy. Front. Physiol. 2020, 11, 349. [Google Scholar] [CrossRef]
- Zweers, H.; van Wegberg, A.M.J.; Janssen, M.C.H.; Wortmann, S.B. Ketogenic diet for mitochondrial disease: A systematic review on efficacy and safety. Orphanet J. Rare Dis. 2021, 16, 295. [Google Scholar] [CrossRef]
- Hung, K.-M.; Chen, P.-C.; Hsieh, H.-C.; Calkins, M.J. Mitochondrial defects arise from nucleoside/nucleotide reverse transcriptase inhibitors in neurons: Potential contribution to HIV-associated neurocognitive disorders. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Orsucci, D.; Ienco, E.C.; Siciliano, G.; Mancuso, M. Mitochondrial disorders and drugs: What every physician should know. Drugs Context 2019, 8, 212588. [Google Scholar] [CrossRef] [PubMed]
- de Barcelos, I.P.; Emmanuele, V.; Hirano, M. Advances in primary mitochondrial myopathies. Curr. Opin. Neurol. 2019, 32, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Slone, J.; Gui, B.; Huang, T. The current landscape for the treatment of mitochondrial disorders. J. Genet. Genom. 2018, 45, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Hou, Y.; Zhao, X.; Liufu, T.; Yu, M.; Zhang, W.; Xie, Z.; Zhang, V.W.; Yuan, Y.; Wang, Z. The clinical, myopathological, and genetic analysis of 155 Chinese mitochondrial ophthalmoplegia patients with mitochondrial DNA single large deletions. Mol. Genet. Genom. Med. 2023, 12, e2328. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Yu, L.; Wang, Z.-X. Diagnosis and Management of Kearns-Sayre Syndrome Rely on Comprehensive Clinical Evaluation. Chin. Med. J. 2016, 129, 2519–2520. [Google Scholar] [CrossRef]
- Ying, Y.; Liang, Y.; Luo, X.; Wei, M. Case Report: Clinical and Genetic Characteristics of Pearson Syndrome in a Chinese Boy and 139 Patients. Front. Genet. 2022, 13, 802402. [Google Scholar] [CrossRef]
- Latvala, T.; Mustonen, E.; Uusitalo, R.; Majamaa, K. Pigmentary retinopathy in patients with the MELAS mutation 3243A→G in mitochondrial DNA. Graefe’s Arch. Clin. Exp. Ophthalmol. 2002, 240, 795–801. [Google Scholar] [CrossRef]
- Seitun, S.; Massobrio, L.; Rubegni, A.; Nesti, C.; Morelli, M.C.; Boccalini, S.; Pregliasco, A.G.; Budaj, I.; Deferrari, L.; Rosa, G.M.; et al. MELAS Syndrome with Cardiac Involvement: A Multimodality Imaging Approach. Case Rep. Cardiol. 2016, 2016, 1490181. [Google Scholar] [CrossRef]
- Wu, G.; Shen, Y.M.; Zhu, F.M.; Tao, W.M.; Zhou, Y.M.; Ke, S.M.; Jiang, H.M. Comprehensive Diagnostic Criteria for MELAS Syndrome; a Case Study Involving an Elderly Patient With MT-TWm.5541C>T Mutation. Neurologist 2023, 28, 190–194. [Google Scholar] [CrossRef]
- Finsterer, J. Intractable Epilepsy in Maternally Inherited Leigh Syndrome (MILS) Due to the Sporadic Variant m.8993T>G in MT-ATP6: A Case Report. Cureus 2022, 14, e22716. [Google Scholar] [CrossRef]
- Finsterer, J. Neuropathy, Ataxia, and Retinitis Pigmentosa Syndrome. J. Clin. Neuromuscul. Dis. 2023, 24, 140–146. [Google Scholar] [CrossRef]
- Esmaeil, A.; Ali, A.; Behbehani, R. Leber’s hereditary optic neuropathy: Update on current diagnosis and treatment. Front. Ophthalmol. 2023, 2, 1077395. [Google Scholar] [CrossRef]
- Yang, M.; Xu, L.; Xu, C.; Cui, Y.; Jiang, S.; Dong, J.; Liao, L. The Mutations and Clinical Variability in Maternally Inherited Diabetes and Deafness: An Analysis of 161 Patients. Front. Endocrinol. 2021, 12, 728043. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J.; Zarrouk-Mahjoub, S.; Shoffner, J.M. MERRF Classification: Implications for Diagnosis and Clinical Trials. Pediatr. Neurol. 2018, 80, 8–23. [Google Scholar] [CrossRef]
- Hanisch, F.; Kornhuber, M.; Alston, C.L.; Taylor, R.W.; Deschauer, M.; Zierz, S. SANDO syndrome in a cohort of 107 patients with CPEO and mitochondrial DNA deletions. J. Neurol. Neurosurg. Psychiatry 2015, 86, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Parada-Garza, J.D.; López-Valencia, G.; Miranda-García, L.A.; Pérez-García, G.; Ruiz-Sandoval, J.L. MRI findings in SANDO variety of the ataxia-neuropathy spectrum with a novel mutation in POLG (c.3287G>T): A case report. Neuromuscul. Disord. 2020, 30, 590–592. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, M.; Ferraris, S.; Pancrudo, J.; Feigenbaum, A.; Raiman, J.; Christodoulou, J.; Thorburn, D.R.; DiMauro, S. New DGK gene mutations in the hepatocerebral form of mitochondrial DNA depletion syndrome. Arch. Neurol. 2005, 62, 745–747. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Li, F.-Y.; Schmitt, E.; Zhang, S.; Craigen, W.J.; Wong, L.-J.C. MPV17-associated hepatocerebral mitochondrial DNA depletion syndrome: New patients and novel mutations. Mol. Genet. Metab. 2010, 99, 300–308. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Wang, J.; Dai, H.; Almannai, M.; Staufner, C.; Alfadhel, M.; Gambello, M.J.; Prasun, P.; Raza, S.; Lyons, H.J.; et al. MPV17-related mitochondrial DNA maintenance defect: New cases and review of clinical, biochemical, and molecular aspects. Hum. Mutat. 2018, 39, 461–470. [Google Scholar] [CrossRef]
- Ronchi, D.; Di Fonzo, A.; Lin, W.; Bordoni, A.; Liu, C.; Fassone, E.; Pagliarani, S.; Rizzuti, M.; Zheng, L.; Filosto, M.; et al. Mutations in DNA2 Link Progressive Myopathy to Mitochondrial DNA Instability. Am. J. Hum. Genet. 2013, 92, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Borsche, M.; Dulovic-Mahlow, M.; Baumann, H.; Tunc, S.; Lüth, T.; Schaake, S.; Özcakir, S.; Westenberger, A.; Münchau, A.; Knappe, E.; et al. POLG2-Linked Mitochondrial Disease: Functional Insights from New Mutation Carriers and Review of the Literature. Cerebellum, 2023; ahead of print. [Google Scholar]
- Wong, W.K.; Troedson, C.; Dale, R.C.; Roscioli, T.; Field, M.; Palmer, E.; Martin, E.M.; Kumar, K.R.; Mohammad, S.S. Levodopa Responsive Dystonia Parkinsonism, Intellectual Disability, and Optic Atrophy Due to a Heterozygous Missense Variant in AFG3L2. Mov. Disord. Clin. Pract. 2022, 9 (Suppl. S2), S32–S35. [Google Scholar] [CrossRef]
- Maricich, S.M.; Zoghbi, H.Y. Chapter 50—Dominantly Inherited Spinocerebellar Syndromes. In Neuromuscular Disorders of Infancy, Childhood, and Adolescence, 2nd ed.; Darras, B.T., Jones, H.R., Ryan, M.M., De Vivo, D.C., Eds.; Academic Press: San Diego, CA, USA, 2015; pp. 1003–1032. [Google Scholar]
- Gorman, G.S.; Pfeffer, G.; Griffin, H.; Blakely, E.L.; Kurzawa-Akanbi, M.; Gabriel, J.; Sitarz, K.; Roberts, M.; Schoser, B.; Pyle, A.; et al. Clonal Expansion of Secondary Mitochondrial DNA Deletions Associated With Spinocerebellar Ataxia Type 28. JAMA Neurol. 2015, 72, 106–111. [Google Scholar] [CrossRef]
- Pfeffer, G.; Gorman, G.S.; Griffin, H.; Kurzawa-Akanbi, M.; Blakely, E.L.; Wilson, I.; Sitarz, K.; Moore, D.; Murphy, J.L.; Alston, C.L.; et al. Mutations in the SPG7 gene cause chronic progressive external ophthalmoplegia through disordered mitochondrial DNA maintenance. Brain 2014, 137, 1323–1336. [Google Scholar] [CrossRef]
- Erdinc, D.; Rodríguez-Luis, A.; Fassad, M.R.; Mackenzie, S.; Watson, C.M.; Valenzuela, S.; Xie, X.; Menger, K.E.; Sergeant, K.; Craig, K.; et al. Pathological variants in TOP3A cause distinct disorders of mitochondrial and nuclear genome stability. EMBO Mol. Med. 2023, 15, e16775. [Google Scholar] [CrossRef] [PubMed]
- Llauradó, A.; Rovira-Moreno, E.; Codina-Solà, M.; Martínez-Saez, E.; Salvadó, M.; Sanchez-Tejerina, D.; Sotoca, J.; López-Diego, V.; Restrepo-Vera, J.L.; Garcia-Arumi, E.; et al. Chronic progressive external ophthalmoplegia plus syndrome due to homozygous missense variant in TOP3A gene. Clin. Genet. 2023, 103, 492–494. [Google Scholar] [CrossRef]
- Shintaku, J.; Pernice, W.M.; Eyaid, W.; Gc, J.B.; Brown, Z.P.; Juanola-Falgarona, M.; Torres-Torronteras, J.; Sommerville, E.W.; Hellebrekers, D.M.; Blakely, E.L.; et al. RRM1 variants cause a mitochondrial DNA maintenance disorder via impaired de novo nucleotide synthesis. J. Clin. Investig. 2022, 132, e145660. [Google Scholar] [CrossRef]
- Feichtinger, R.G.; Oláhová, M.; Kishita, Y.; Garone, C.; Kremer, L.S.; Yagi, M.; Uchiumi, T.; Jourdain, A.A.; Thompson, K.; D’souza, A.R.; et al. Biallelic C1QBP Mutations Cause Severe Neonatal-, Childhood-, or Later-Onset Cardiomyopathy Associated with Combined Respiratory-Chain Deficiencies. Am. J. Hum. Genet. 2017, 101, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Marchet, S.; Legati, A.; Nasca, A.; Di Meo, I.; Spagnolo, M.; Zanetti, N.; Lamantea, E.; Catania, A.; Lamperti, C.; Ghezzi, D. Homozygous mutations in C1QBP as cause of progressive external ophthalmoplegia (PEO) and mitochondrial myopathy with multiple mtDNA deletions. Hum. Mutat. 2020, 41, 1745–1750. [Google Scholar] [CrossRef]
- Sommerville, E.W.; Dalla Rosa, I.; Rosenberg, M.M.; Bruni, F.; Thompson, K.; Rocha, M.; Blakely, E.L.; He, L.; Falkous, G.; Schaefer, A.M.; et al. Identification of a novel heterozygous guanosine monophosphate reductase (GMPR) variant in a patient with a late-onset disorder of mitochondrial DNA maintenance. Clin. Genet. 2020, 97, 276–286. [Google Scholar] [CrossRef]
- Neergheen, V.; Chalasani, A.; Wainwright, L.; Yubero, D.; Montero, R.; Artuch, R.; Hargreaves, I. Coenzyme Q10 in the Treatment of Mitochondrial Disease. J. Inborn Errors Metab. Screen. 2017, 5, 2326409817707771. [Google Scholar] [CrossRef]
- Gimenes, A.; Bravo, D.; Nápolis, L.; Mello, M.; Oliveira, A.; Neder, J.; Nery, L. Effect of L-carnitine on exercise performance in patients with mitochondrial myopathy. Braz. J. Med. Biol. Res. 2015, 48, 354–362. [Google Scholar] [CrossRef]
- Virmani, M.A.; Cirulli, M. The Role of l-Carnitine in Mitochondria, Prevention of Metabolic Inflexibility and Disease Initiation. Int. J. Mol. Sci. 2022, 23, 2717. [Google Scholar] [CrossRef]
- Shanti, B.; Joy, Y.-L. Riboflavin metabolism: Role in mitochondrial function. J. Transl. Genet. Genom. 2020, 4, 285–306. [Google Scholar]
- Marcé-Grau, A.; Martí-Sánchez, L.; Baide-Mairena, H.; Ortigoza-Escobar, J.D.; Pérez-Dueñas, B. Genetic defects of thiamine transport and metabolism: A review of clinical phenotypes, genetics, and functional studies. J. Inherit. Metab. Dis. 2019, 42, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.; Rao, S.N.; Kumar, M.K. Genetic heterogeneity of mitochondrial genome in thiamine deficient Leigh syndrome patients. J. Neurol. Sci. 2019, 404, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Liu, J. The Effects and Mechanisms of Mitochondrial Nutrient α-Lipoic Acid on Improving Age-Associated Mitochondrial and Cognitive Dysfunction: An Overview. Neurochem. Res. 2008, 33, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Fava, A.; Pirritano, D.; Plastino, M.; Cristiano, D.; Puccio, G.; Colica, C.; Ermio, C.; De Bartolo, M.; Mauro, G.; Bosco, D. The Effect of Lipoic Acid Therapy on Cognitive Functioning in Patients with Alzheimer’s Disease. J. Neurodegener. Dis. 2013, 2013, 454253. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zhao, X.; Liu, L.; Zhang, H.; Xuan, M.; Guo, Z.; Wang, H.; Liu, C. Neurochemical effects of the R form of α-lipoic acid and its neuroprotective mechanism in cellular models of Parkinson’s disease. Int. J. Biochem. Cell Biol. 2017, 87, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Pineda, M.; Ormazabal, A.; López-Gallardo, E.; Nascimento, A.; Solano, A.; Herrero, M.D.; Vilaseca, M.A.; Briones, P.; Ibáñez, L.; Montoya, J.; et al. Cerebral folate deficiency and leukoencephalopathy caused by a mitochondrial DNA deletion. Ann. Neurol. 2006, 59, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Quijada-Fraile, P.; O’Callaghan, M.; Martin-Hernandez, E.; Montero, R.; García-Cazorla, A.; de Aragón, A.M.; Muchart, J.; Malaga, I.; Pardo, R.; García-Gonzalez, P.; et al. Follow-up of folinic acid supplementation for patients with cerebral folate deficiency and Kearns-Sayre syndrome. Orphanet J. Rare Dis. 2014, 9, 217. [Google Scholar] [CrossRef] [PubMed]
- Ramaekers, V.T.; Weis, J.; Sequeira, J.M.; Quadros, E.V.; Blau, N. Mitochondrial complex I encephalomyopathy and cerebral 5-methyltetrahydrofolate deficiency. Neuropediatrics 2007, 38, 184–187. [Google Scholar] [CrossRef]
- Jauslin, M.L.; Meier, T.; Smith, R.A.J.; Murphy, P.M. Mitochondria-targeted antioxidants protect Friedreich Ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. FASEB J. 2003, 17, 1972–1974. [Google Scholar] [CrossRef] [PubMed]
- Sulaimon, L.A.; Afolabi, L.O.; Adisa, R.A.; Ayankojo, A.G.; Afolabi, M.O.; Adewolu, A.M.; Wan, X. Pharmacological significance of MitoQ in ameliorating mitochondria-related diseases. Adv. Redox Res. 2022, 5, 100037. [Google Scholar] [CrossRef]
- Lapatto, H.A.K.; Kuusela, M.; Heikkinen, A.; Muniandy, M.; van der Kolk, B.W.; Gopalakrishnan, S.; Pöllänen, N.; Sandvik, M.; Schmidt, M.S.; Heinonen, S.; et al. Nicotinamide riboside improves muscle mitochondrial biogenesis, satellite cell differentiation, and gut microbiota in a twin study. Sci. Adv. 2023, 9, eadd5163. [Google Scholar] [CrossRef]
- Airhart, S.E.; Shireman, L.M.; Risler, L.J.; Anderson, G.D.; Gowda, G.A.N.; Raftery, D.; Tian, R.; Shen, D.D.; O’Brien, K.D. An open-label, non-randomized study of the pharmacokinetics of the nutritional supplement nicotinamide riboside (NR) and its effects on blood NAD+ levels in healthy volunteers. PLoS ONE 2017, 12, e0186459. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Emrick, L.T.; Hsu, J.W.; Chanprasert, S.; Almannai, M.; Craigen, W.J.; Jahoor, F.; Scaglia, F. Impaired nitric oxide production in children with MELAS syndrome and the effect of arginine and citrulline supplementation. Mol. Genet. Metab. 2016, 117, 407–412. [Google Scholar] [CrossRef]
- Chatfield, K.C.; Sparagna, G.C.; Chau, S.; Phillips, E.K.; Ambardekar, A.V.; Aftab, M.; Mitchell, M.B.; Sucharov, C.C.; Miyamoto, S.D.; Stauffer, B.L. Elamipretide Improves Mitochondrial Function in the Failing Human Heart. JACC Basic Transl. Sci. 2019, 4, 147–157. [Google Scholar] [CrossRef]
- Sabbah, H.N. Elamipretide for Barth syndrome cardiomyopathy: Gradual rebuilding of a failed power grid. Heart Fail. Rev. 2022, 27, 1911–1923. [Google Scholar] [CrossRef]
- Hornby, B.; Thompson, W.R.; Almuqbil, M.; Manuel, R.; Abbruscato, A.; Carr, J.; Vernon, H.J. Natural history comparison study to assess the efficacy of elamipretide in patients with Barth syndrome. Orphanet J. Rare Dis. 2022, 17, 336. [Google Scholar] [CrossRef] [PubMed]
- de Haas, R.; Das, D.; Garanto, A.; Renkema, H.G.; Greupink, R.; van den Broek, P.; Pertijs, J.; Collin, R.W.J.; Willems, P.; Beyrath, J.; et al. Therapeutic effects of the mitochondrial ROS-redox modulator KH176 in a mammalian model of Leigh Disease. Sci. Rep. 2017, 7, 11733. [Google Scholar] [CrossRef] [PubMed]
- Seo, K.-S.; Kim, J.-H.; Min, K.-N.; Moon, J.-A.; Roh, T.-C.; Lee, M.-J.; Lee, K.-W.; Min, J.-E.; Lee, Y.-M. KL1333, a Novel NAD(+) Modulator, Improves Energy Metabolism and Mitochondrial Dysfunction in MELAS Fibroblasts. Front. Neurol. 2018, 9, 552. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, H.; Zeng, W.; Wei, J. Edaravone protects against hyperosmolarity-induced oxidative stress and apoptosis in primary human corneal epithelial cells. PLoS ONE 2017, 12, e0174437. [Google Scholar] [CrossRef]
- Jacoby, E.; Blumkin, M.; Anikster, Y.; Varda-Bloom, N.; Pansheen, J.; Bar Yoseph, O.; Gruber, N.; Lahav, E.; Besser, M.J.; Schachter, J.; et al. First-in-Human Mitochondrial Augmentation of Hematopoietic Stem Cells in Pearson Syndrome. Blood 2018, 132 (Suppl. S1), 1024. [Google Scholar] [CrossRef]
- Zelissen, R.; Ahmadian, S.; Montilla-Rojo, J.; Timmer, E.; Ummelen, M.; Hopman, A.; Smeets, H.; van Tienen, F. Fusion of Wild-Type Mesoangioblasts with Myotubes of mtDNA Mutation Carriers Leads to a Proportional Reduction in mtDNA Mutation Load. Int. J. Mol. Sci. 2023, 24, 2679. [Google Scholar] [CrossRef] [PubMed]
- van Tienen, F.; Zelissen, R.; Timmer, E.; van Gisbergen, M.; Lindsey, P.; Quattrocelli, M.; Sampaolesi, M.; Mulder-den Hartog, E.; de Coo, I.; Smeets, H. Healthy, mtDNA-mutation free mesoangioblasts from mtDNA patients qualify for autologous therapy. Stem Cell Res. Ther. 2019, 10, 405. [Google Scholar] [CrossRef]
- Zaidman, I.; Elhasid, R.; Gefen, A.; Dovrat, A.Y.; Mutaz, S.; Shaoul, R.; Adiv, O.E.; Mandel, H.; Tal, G. Hematopoietic stem cell transplantation for mitochondrial neurogastrointestinal encephalopathy: A single-center experience underscoring the multiple factors involved in the prognosis. Pediatr. Blood Cancer 2021, 68, e28926. [Google Scholar] [CrossRef]
- Halter, J.P.; Michael, W.; Schüpbach, M.; Mandel, H.; Casali, C.; Orchard, K.; Collin, M.; Valcarcel, D.; Rovelli, A.; Filosto, M.; et al. Allogeneic haematopoietic stem cell transplantation for mitochondrial neurogastrointestinal encephalomyopathy. Brain 2015, 138, 2847–2858. [Google Scholar] [CrossRef]





| Differential Diagnosis of Chronic Progressive External Ophthalmoplegia | |||
|---|---|---|---|
| Myopathic | Neuropathic | Neuromuscular Junction | Other |
| Orbital myositis | Multiple sclerosis | Myasthenia gravis | Botulism |
| Graves’ disease | Miller Fisher syndrome | Congenital myasthenic syndrome | Medications: Statins |
| Myotonic dystrophy types 1 and 2 | A-beta lipoproteinemia | Lambert–Eaton myasthenic syndrome (LEMS) | |
| Tolosa-Hunt syndrome | |||
| WEBINO syndrome | |||
| CAPOS syndrome | |||
| CANOMAD syndrome | |||
| Congenital myopathies | Supranuclear ophthalmoplegia: Hereditary ataxias HSP SCA 1, 2, 3, 7, 9, 11, 28 Congenital cranial dysinnervation disorders: CFEOM, Moebius syndrome, Duane syndrome | ||
| OPMD | |||
| OPDM | |||
| LGMD with ophthalmoplegia | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, A.; Esmaeil, A.; Behbehani, R. Mitochondrial Chronic Progressive External Ophthalmoplegia. Brain Sci. 2024, 14, 135. https://doi.org/10.3390/brainsci14020135
Ali A, Esmaeil A, Behbehani R. Mitochondrial Chronic Progressive External Ophthalmoplegia. Brain Sciences. 2024; 14(2):135. https://doi.org/10.3390/brainsci14020135
Chicago/Turabian StyleAli, Ali, Ali Esmaeil, and Raed Behbehani. 2024. "Mitochondrial Chronic Progressive External Ophthalmoplegia" Brain Sciences 14, no. 2: 135. https://doi.org/10.3390/brainsci14020135
APA StyleAli, A., Esmaeil, A., & Behbehani, R. (2024). Mitochondrial Chronic Progressive External Ophthalmoplegia. Brain Sciences, 14(2), 135. https://doi.org/10.3390/brainsci14020135