




Epigallocatechin-3-Gallate and Genistein for Decreasing Gut Dysbiosis, Inhibiting Inflammasomes, and Aiding Autophagy in Alzheimer’s Disease



Abstract
1. Introduction
2. Prescription Therapeutic Options for AD
3. Bioflavonoids as Novel Therapeutic Option for AD
4. An Overview of the Gut Microbiota
4.1. Activity of Gut Microbiota in the Human Body
4.2. Onset Factors in the Microbiota for Dysbiosis
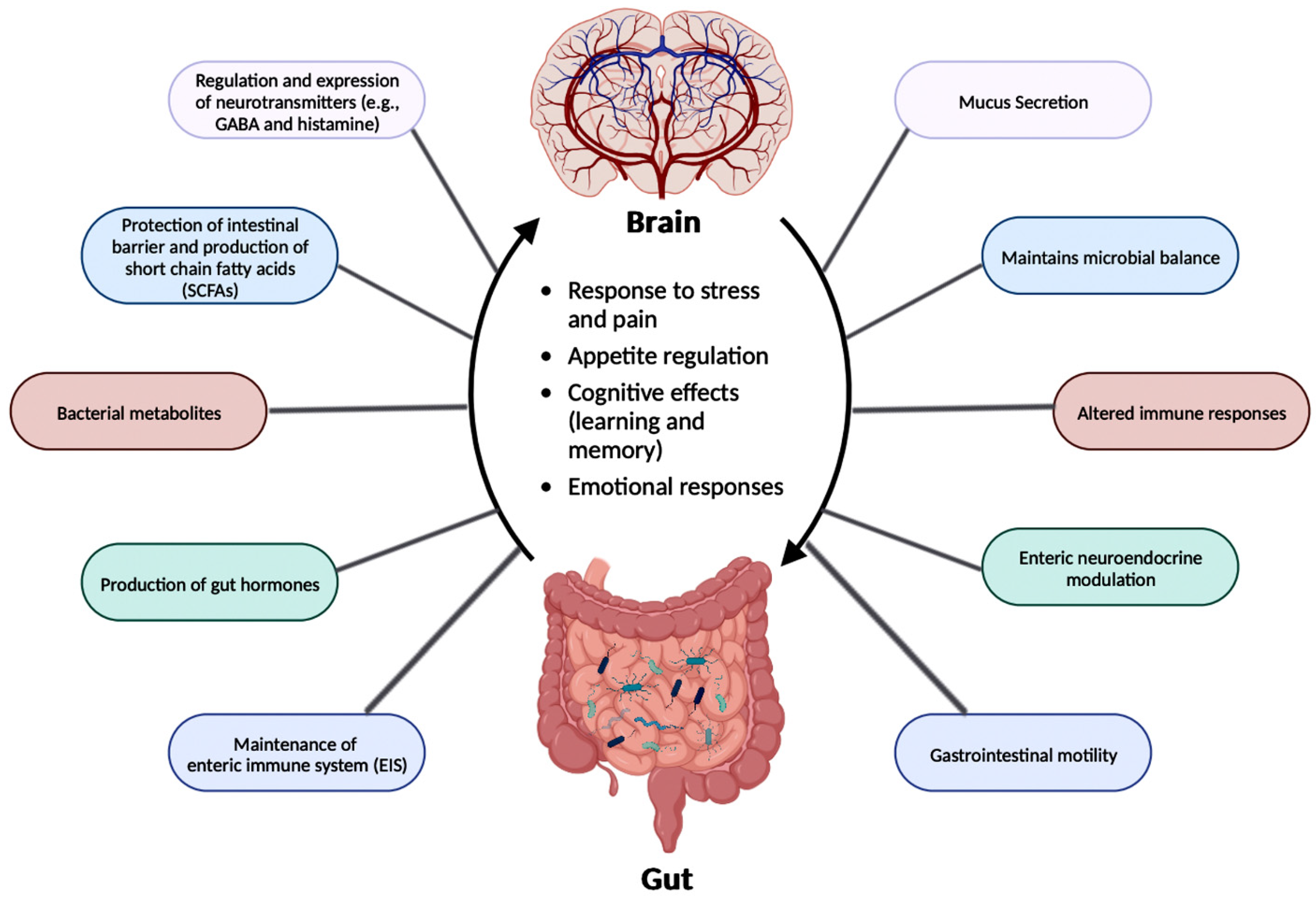
4.3. Gut-Brain Axis (GBA) and Dysbiosis
4.4. GBA and AD
5. Neuroinflammation in AD
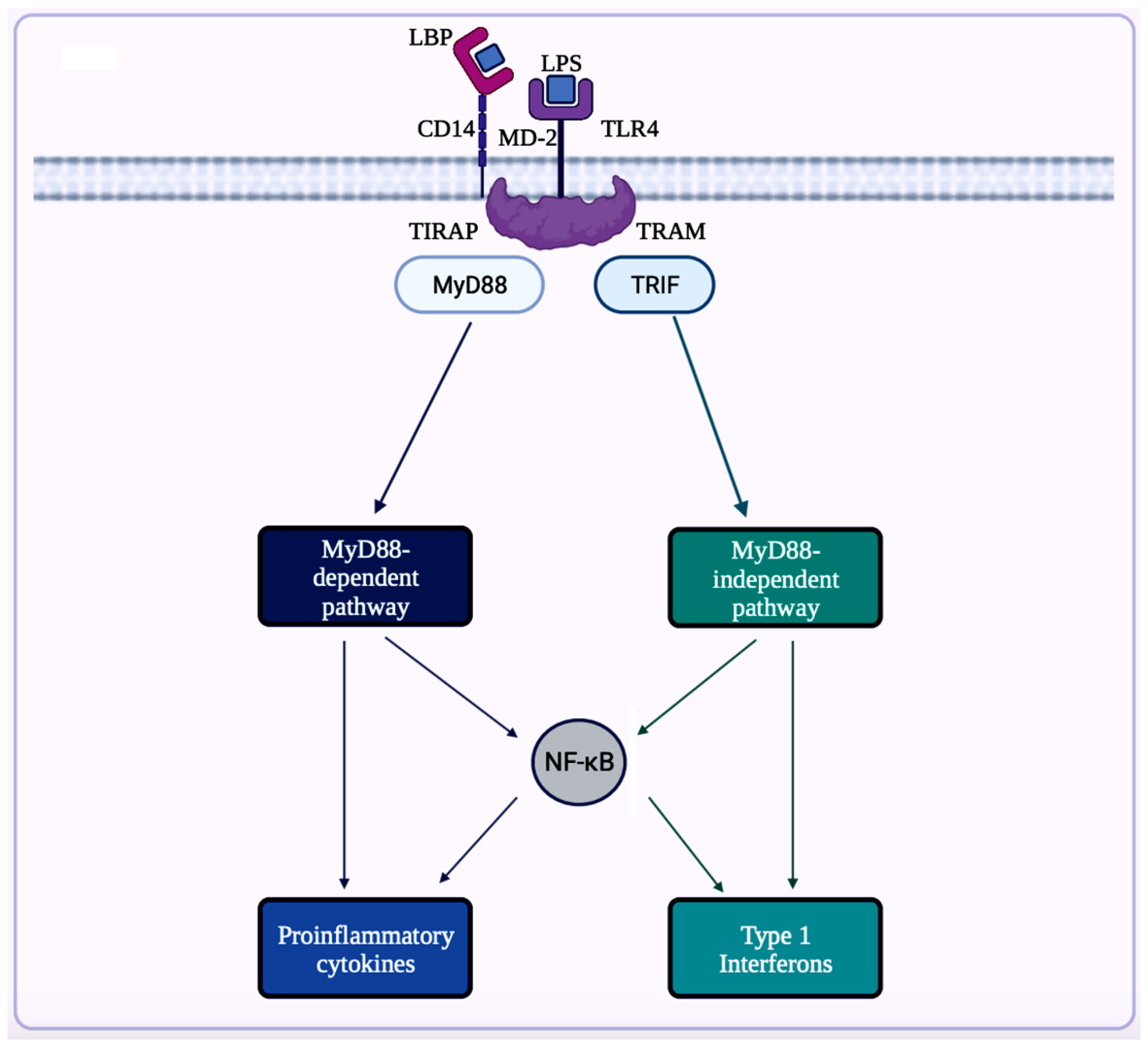
5.1. Implications of Gut Microbiota in Neuroinflammation in AD
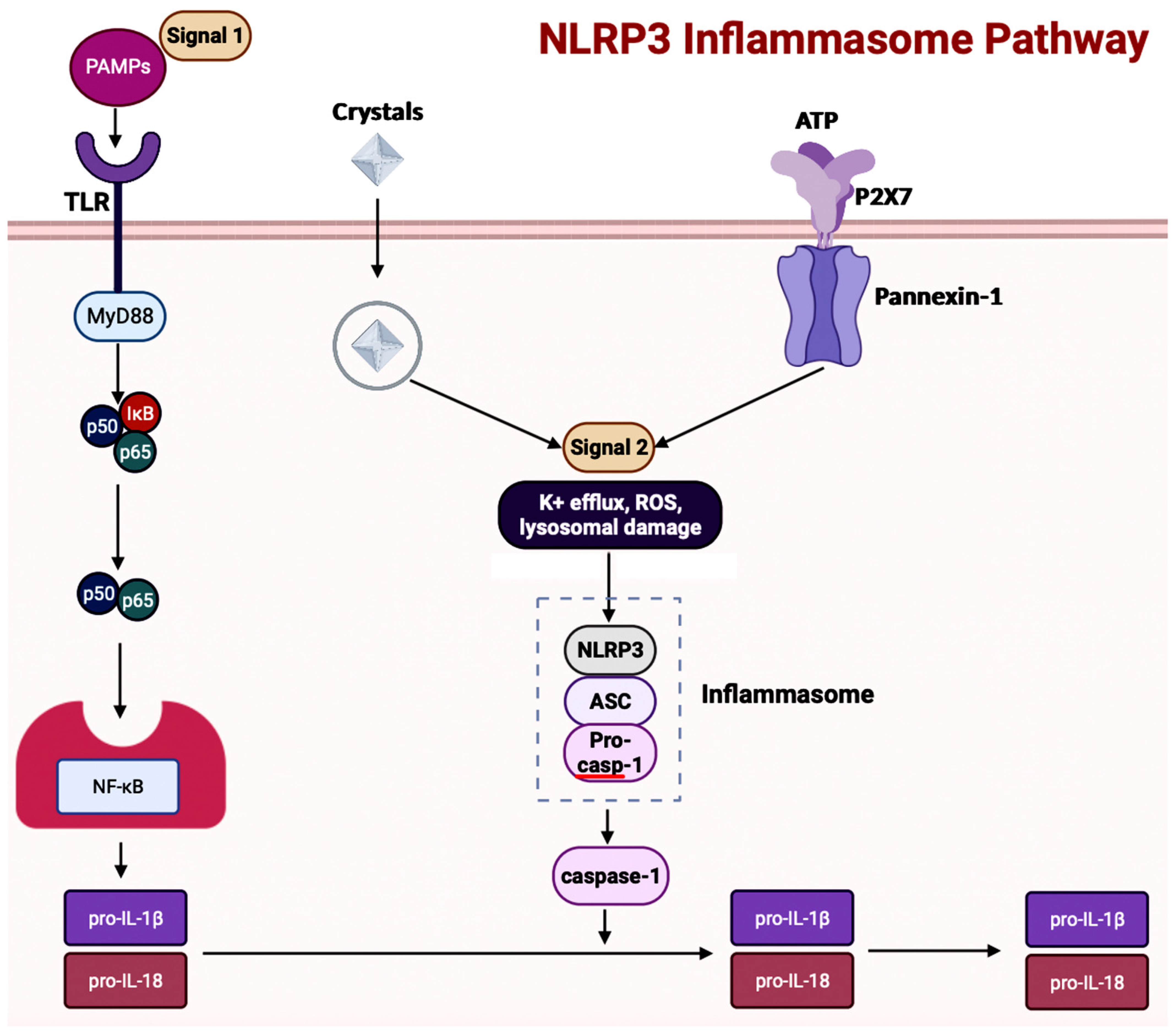
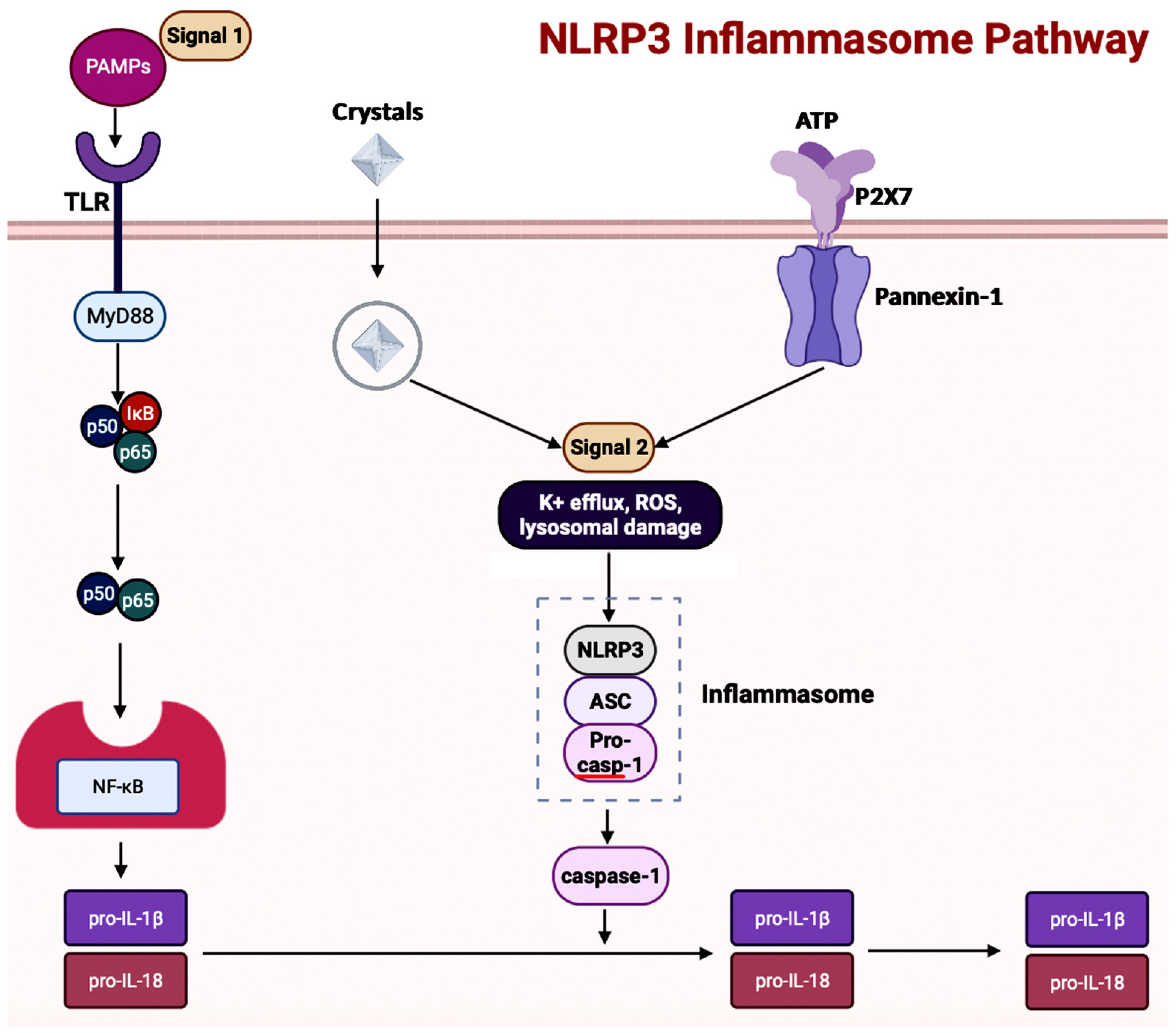
5.2. Inflammasomes in AD
5.3. Inflammasomes and GBA in AD
5.4. NETosis
5.5. NETosis and Gut Microbiota
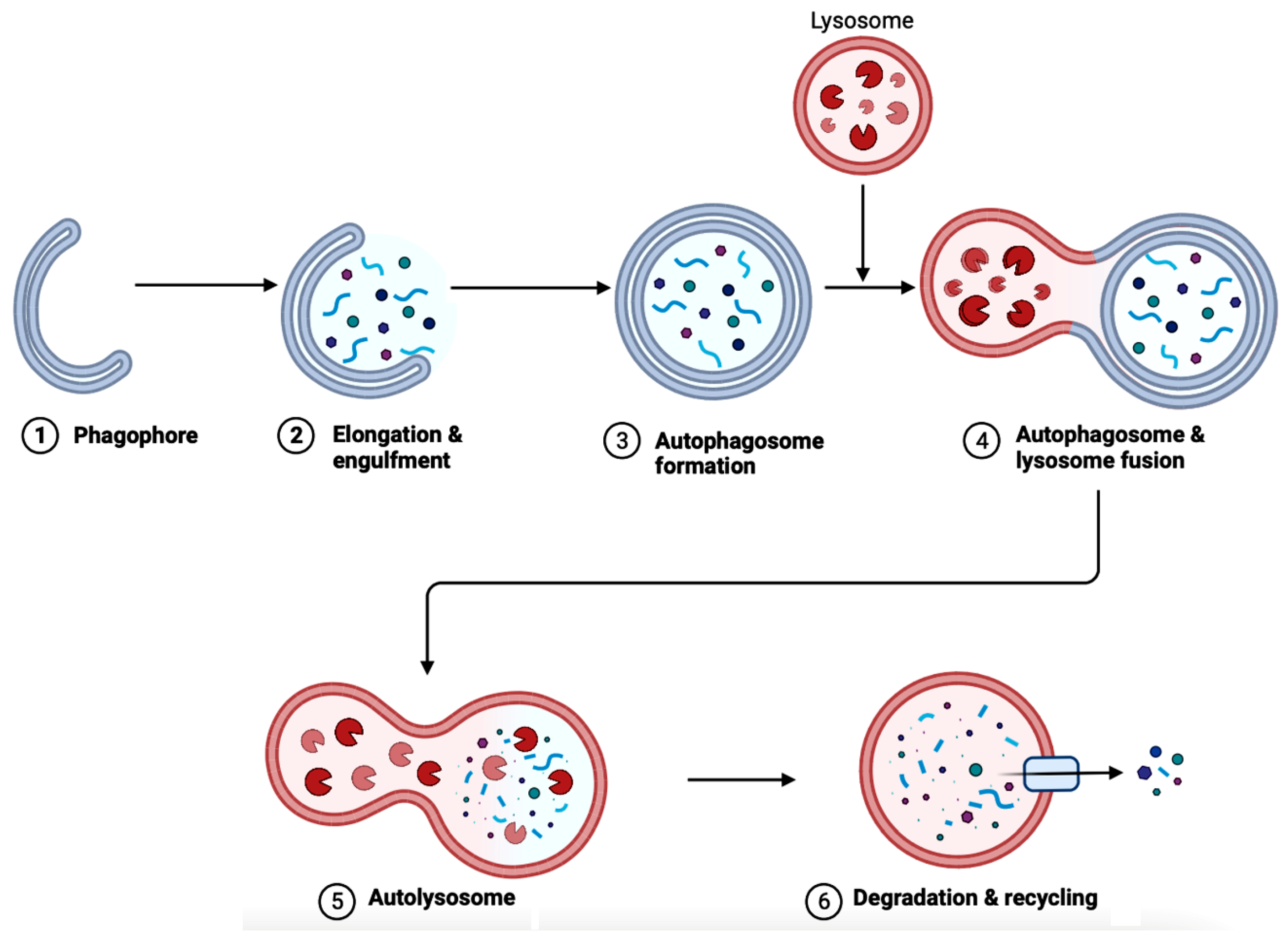
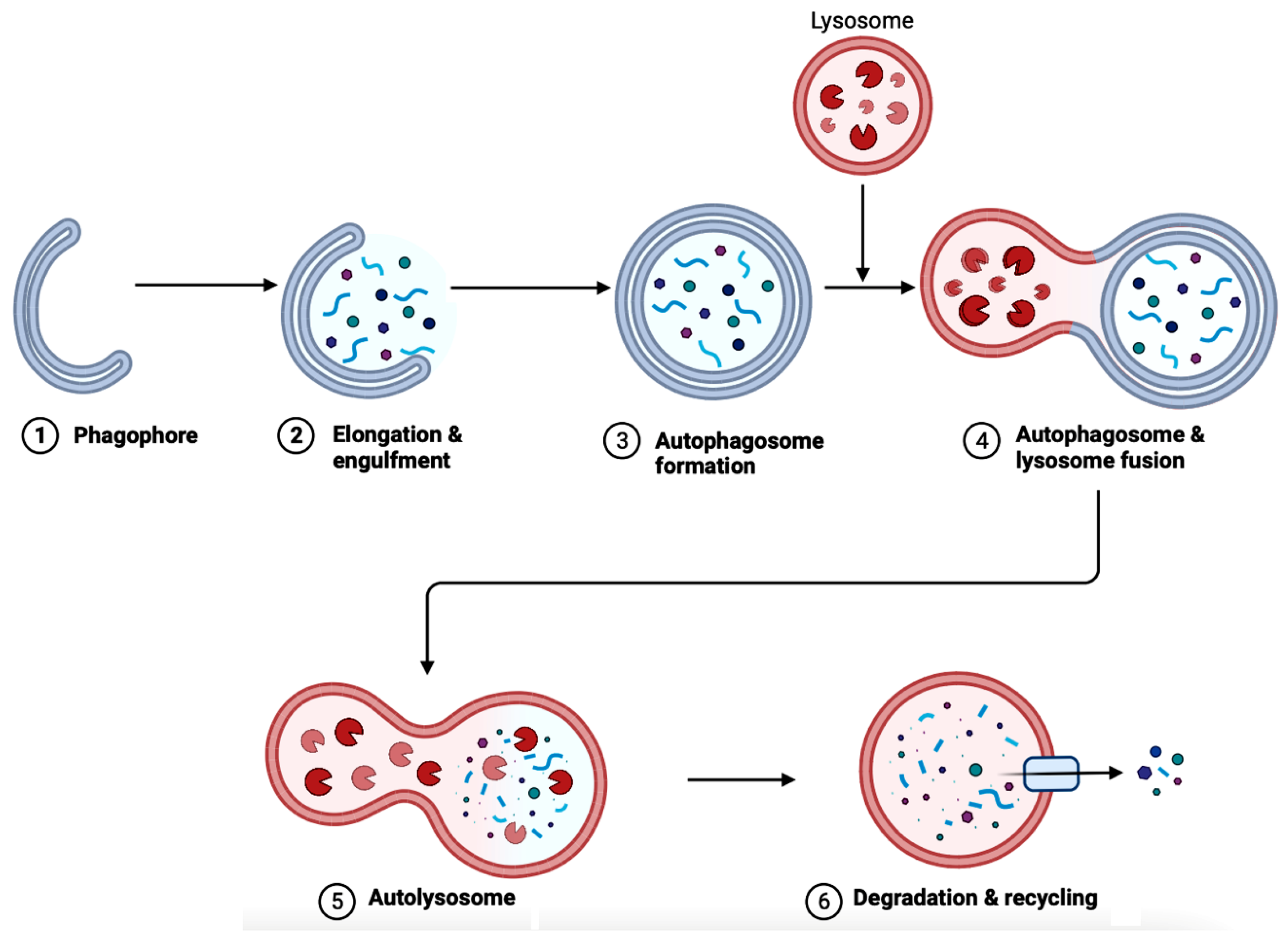
6. Autophagy in AD
Implications of Gut Microbiota in Autophagy in AD
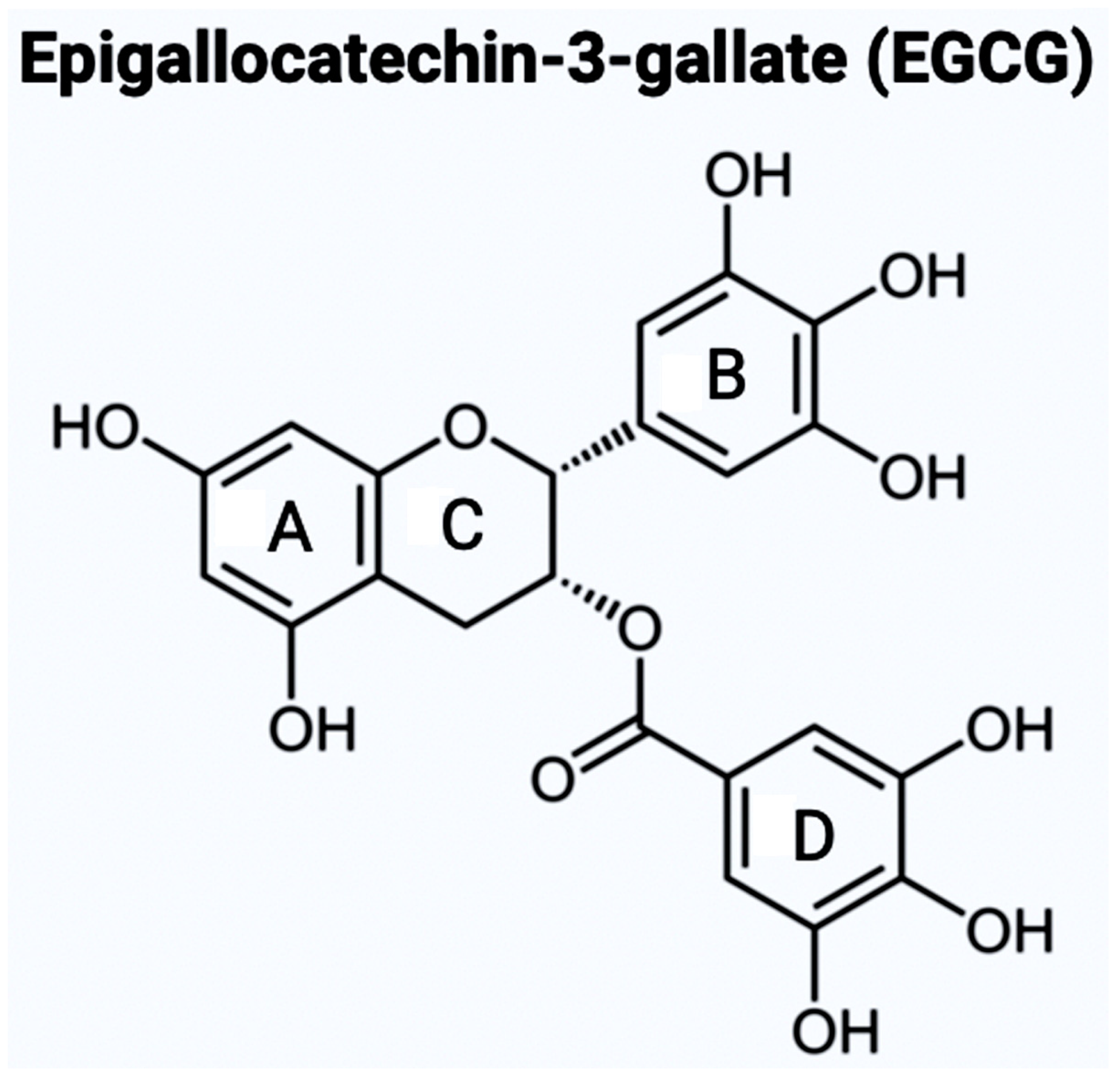
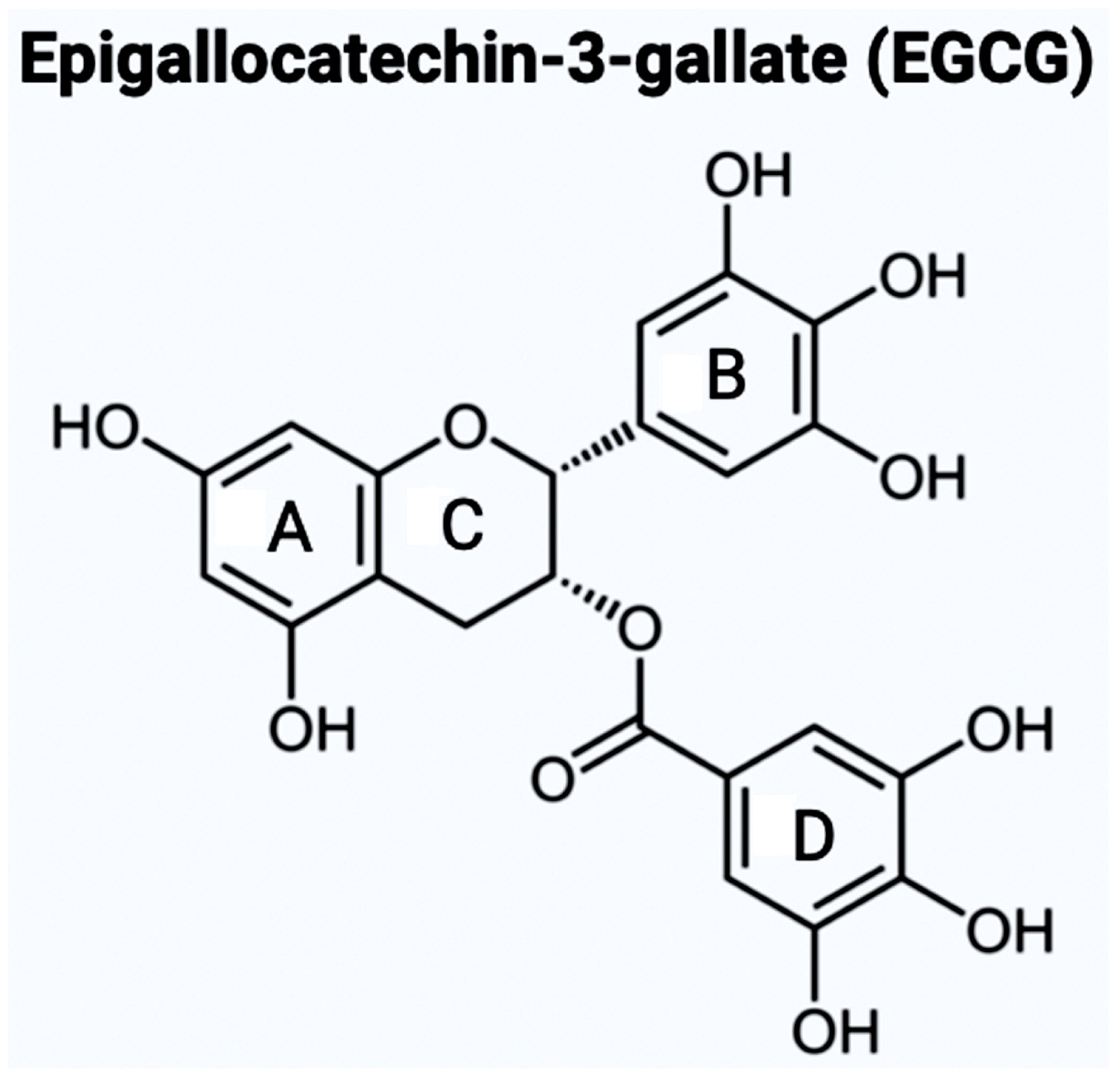
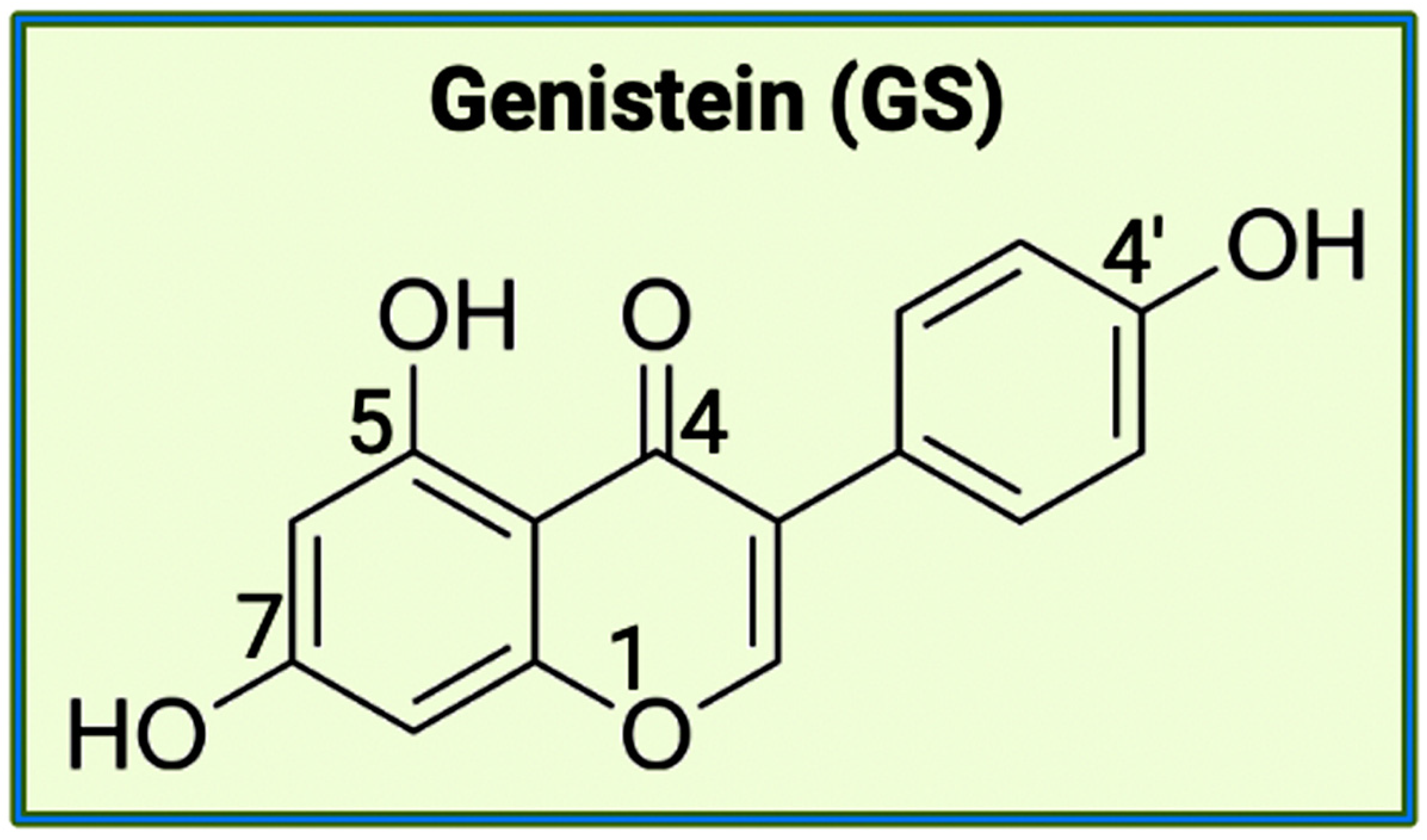
7. The Bioflavonoids EGCG and GS as Therapeutic Agents for AD
7.1. Overview of Regulating Cell Signaling by EGCG and GS
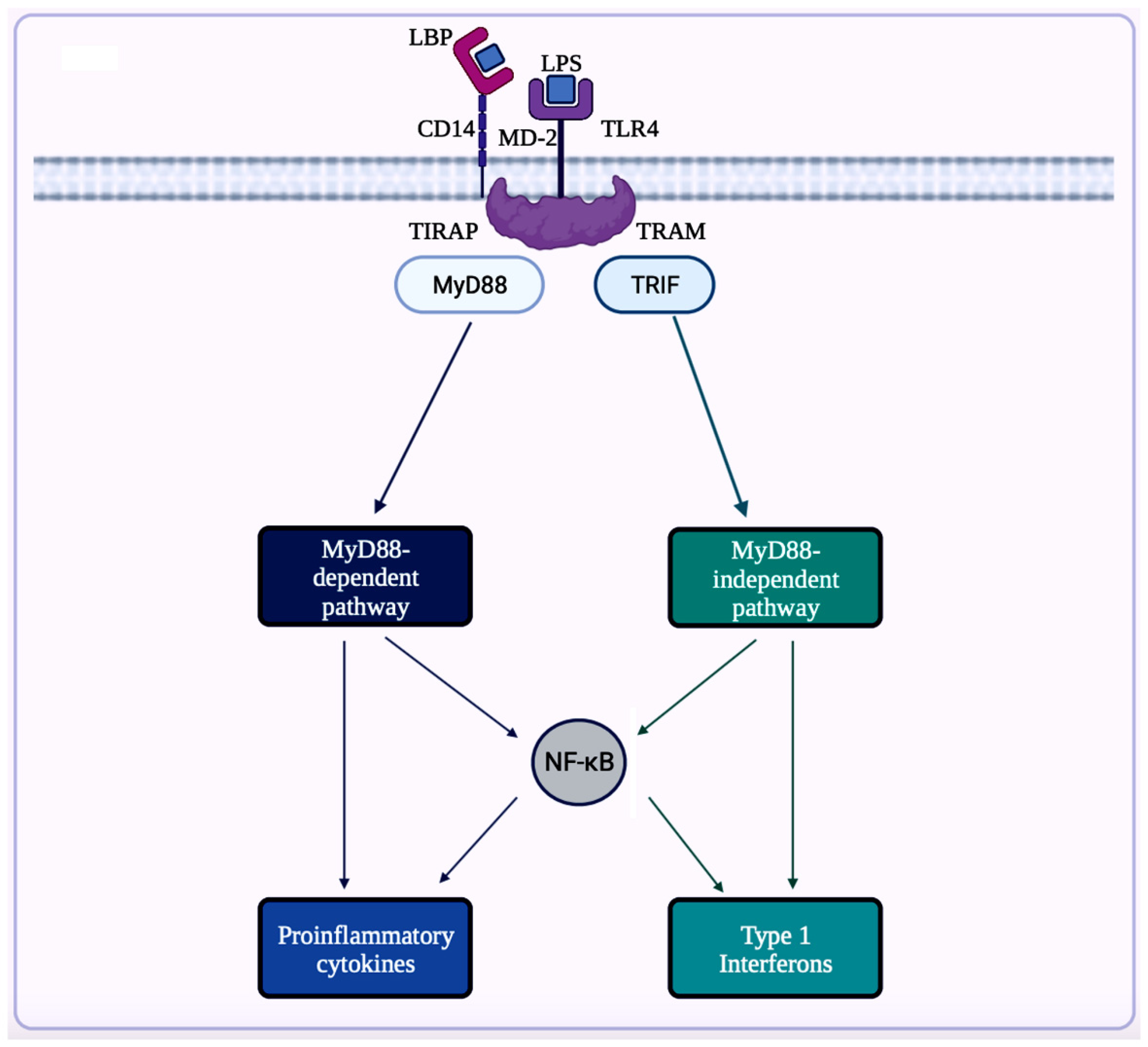
7.1.1. NF-κB Signaling Pathway
7.1.2. MAPK Signaling Pathway
7.1.3. EGFR Signaling Pathway
7.1.4. IGF Signal Transduction Pathway
7.1.5. mTOR Pathway
7.1.6. 5-Hydroxytryptamine Signaling
7.2. Exploration of Bioflavonoids as Therapeutic Options in AD
7.2.1. AChE Inhibitor
7.2.2. Diet
7.2.3. Fecal Microbiota Transplantation (FMT)
7.2.4. Neural Stem Cell Therapy
7.2.5. Nanomaterials
8. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Breijyeh, Z.; Karaman, R. Comprehensive review on Alzheimer’s disease: Causes and treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef] [PubMed]
- Graff-Radford, J.; Yong, K.X.; Apostolova, L.G.; Bouwman, F.H.; Carrillo, M.; Dickerson, B.C.; Rabinovici, G.D.; Schott, J.M.; Jones, D.T.; Murray, M.E. New insights into atypical Alzheimer’s disease in the era of biomarkers. Lancet Neurol. 2021, 20, 222–234. [Google Scholar] [CrossRef] [PubMed]
- U.S. Department of Health and Human Services. Alzheimer’s Disease Fact Sheet. National Institute on Aging. Available online: https://www.nia.nih.gov/health/alzheimers-disease-fact-sheet#:~:text=Alzheimer%27s%20disease%20is%20a%20brain,first%20appear%20later%20in%20life (accessed on 20 May 2023).
- Soria Lopez, J.A.; González, H.M.; Léger, G.C. Alzheimer’s disease. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2019; Volume 167, pp. 231–255. [Google Scholar] [CrossRef]
- Landau, S.M.; Lu, M.; Joshi, A.D.; Pontecorvo, M.; Mintun, M.A.; Trojanowski, J.Q.; Shaw, L.M.; Jagust, W.J. Comparing positron emission tomography imaging and cerebrospinal fluid measurements of β-amyloid. Ann. Neurol. 2013, 74, 826–836. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blennow, K.; et al. Preclinical Alzheimer’s disease: Definition, natural history, and Diagnostic Criteria. Alzheimers Dement. 2016, 12, 292–323. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. Medications for Memory, Cognition and Dementia-Related Behaviors. Alzheimer’s Disease and Dementia. Available online: https://www.alz.org/alzheimers-dementia/treatments/medications-for-memory (accessed on 20 May 2023).
- Rutsch, A.; Kantsjö, J.B.; Ronchi, F. The gut-brain axis: How microbiota and host Inflammasome Influence Brain Physiology and pathology. Front. Immunol. 2020, 11, 604179. [Google Scholar] [CrossRef]
- Sharma, K. Cholinesterase inhibitors as Alzheimer’s therapeutics (review). Mol. Med. Rep. 2019, 20, 1479–1487. [Google Scholar] [CrossRef]
- Weinstock, M. Selectivity of cholinesterase inhibition. CNS Drugs 1999, 12, 307–323. [Google Scholar] [CrossRef]
- Rogers, S.L.; Farlow, M.R.; Doody, R.S.; Mohs, R.; Friedhoff, L.T. A 24-week, double-blind, placebo-controlled trial of donepezil in patients with Alzheimer’s disease. Neurology 1998, 50, 136–145. [Google Scholar] [CrossRef]
- López-Arrieta, J.; Schneider, L. Metrifonate for Alzheimer’s disease. Cochrane Database Syst. Rev. 2006, 2, CD003155. [Google Scholar] [CrossRef]
- Russo, A.; Acquaviva, R.; Campisi, A.; Sorrenti, V.; Di Giacomo, C.; Virgata, G.; Barcellona, M.L.; Vanella, A. Bioflavonoids as antiradicals, antioxidants and DNA cleavage protectors. Cell Biol. Toxicol. 2000, 16, 91–98. [Google Scholar] [CrossRef]
- Del Rio, D.; Rodriguez-Mateos, A.; Spencer, J.P.E.; Tognolini, M.; Borges, G.; Crozier, A. Dietary (poly)phenolics in human health: Structures, bioavailability, and evidence of protective effects against chronic diseases. Antioxid. Redox Signal. 2013, 18, 1818–1892. [Google Scholar] [CrossRef] [PubMed]
- Godos, J.; Currenti, W.; Angelino, D.; Mena, P.; Castellano, S.; Caraci, F.; Galvano, F.; Del Rio, D.; Ferri, R.; Grosso, G. Diet and mental health: Review of the recent updates on molecular mechanisms. Antioxidants 2020, 9, 346. [Google Scholar] [CrossRef] [PubMed]
- De Bruyne, T.; Steenput, B.; Roth, L.; De Meyer, G.; Santos, C.; Valentová, K.; Dambrova, M.; Hermans, N. Dietary polyphenols targeting arterial stiffness: Interplay of contributing mechanisms and gut microbiome-related metabolism. Nutrients 2019, 11, 578. [Google Scholar] [CrossRef]
- WebMD. 10 Foods High in Flavonoids and Why You Need Them. 2023. Available online: https://www.webmd.com/diet/foods-high-in-flavonoids (accessed on 20 May 2023).
- Flanagan, E.; Müller, M.; Hornberger, M.; Vauzour, D. Impact of flavonoids on cellular and molecular mechanisms underlying age-related cognitive decline and neurodegeneration. Curr. Nutr. Rep. 2018, 7, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.J.; Spencer, J.P.E. Flavonoids, cognition, and dementia: Actions, mechanisms, and potential therapeutic utility for alzheimer disease. Free. Radic. Biol. Med. 2012, 52, 35–45. [Google Scholar] [CrossRef]
- Narayana, R.K.; Reddy, S.M.; Chaluvadi, M.R.; Krishna, D.R. Bioflavonoids classification, pharmacological, biochemical effects and therapeutic potential. Indian J. Pharmacol. 2001, 33, 2–16. [Google Scholar]
- Zhang, Z.; Zhang, Y.; Li, J.; Fu, C.; Zhang, X. The neuroprotective effect of tea polyphenols on the regulation of intestinal flora. Molecules 2021, 26, 3692. [Google Scholar] [CrossRef]
- Hole, K.L.; Williams, R.J. Flavonoids as an intervention for Alzheimer’s disease: Progress and hurdles towards defining a mechanism of Action1. Brain Plast. 2021, 6, 167–192. [Google Scholar] [CrossRef]
- Roseiro, L.B.; Rauter, A.P.; Serralheiro, M.L. Polyphenols as acetylcholinesterase inhibitors: Structural specificity and impact on human disease. Nutr. Aging 2012, 1, 99–111. [Google Scholar] [CrossRef]
- Potenza, M.A.; Iacobazzi, D.; Sgarra, L.; Montagnani, M. The intrinsic virtues of EGCG, an extremely good cell guardian, on prevention and treatment of diabesity complications. Molecules 2020, 25, 3061. [Google Scholar] [CrossRef]
- Goh, Y.X.; Jalil, J.; Lam, K.W.; Husain, K.; Premakumar, C.M. Genistein: A review on its anti-inflammatory properties. Front. Pharmacol. 2022, 13, 820969. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Zhang, R.; Liu, Y.; Wu, Z.; Weng, P. The interaction effect between tea polyphenols and intestinal microbiota: Role in ameliorating neurological diseases. J. Food Biochem. 2021, 46, e13870. [Google Scholar] [CrossRef] [PubMed]
- Sharifi-Rad, J.; Quispe, C.; Imran, M.; Rauf, A.; Nadeem, M.; Gondal, T.A.; Ahmad, B.; Atif, M.; Mubarak, M.S.; Sytar, O.; et al. Genistein: An integrative overview of its mode of action, pharmacological properties, and Health Benefits. Oxidative Med. Cell. Longev. 2021, 2021, 3268136. [Google Scholar] [CrossRef] [PubMed]
- Berg, G.; Rybakova, D.; Fischer, D.; Cernava, T.; Vergès, M.-C.C.; Charles, T.; Chen, X.; Cocolin, L.; Eversole, K.; Corral, G.H.; et al. Microbiome definition re-visited: Old concepts and new challenges. Microbiome 2020, 8, 103. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; Cassader, M. Obesity, diabetes, and gut microbiota. Diabetes Care 2010, 33, 2277–2284. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.S.; Ikram, M.; Park, J.S.; Park, T.J.; Kim, M.O. Gut Microbiota, its role in induction of Alzheimer’s disease pathology, and possible therapeutic interventions: Special focus on anthocyanins. Cells 2020, 9, 853. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, S.; Macfarlane, G.T. Regulation of short-chain fatty acid production. Proc. Nutr. Soc. 2003, 62, 67–72. [Google Scholar] [CrossRef]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef]
- Tan, C.; Wu, Q.; Wang, H.; Gao, X.; Xu, R.; Cui, Z.; Zhu, J.; Zeng, X.; Zhou, H.; He, Y.; et al. Dysbiosis of gut microbiota and short-chain fatty acids in acute ischemic stroke and the subsequent risk for poor functional outcomes. J. Parenter. Enter. Nutr. 2020, 45, 518–529. [Google Scholar] [CrossRef]
- Chidambaram, S.B.; Essa, M.M.; Rathipriya, A.G.; Bishir, M.; Ray, B.; Mahalakshmi, A.M.; Tousif, A.H.; Sakharkar, M.K.; Kashyap, R.S.; Friedland, R.P.; et al. Gut dysbiosis, defective autophagy and altered immune responses in neurodegenerative diseases: Tales of a vicious cycle. Pharmacol. Ther. 2021, 231, 107988. [Google Scholar] [CrossRef]
- Martínez-Cerdeño, V.; Noctor, S.C. Neural progenitor cell terminology. Front. Neuroanat. 2018, 12, 104. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.L.; Millischer, V.; Rodin, S.; MacFabe, D.F.; Villaescusa, J.C.; Lavebratt, C. Enteric short-chain fatty acids promote proliferation of human neural progenitor cells. J. Neurochem. 2019, 154, 635–646. [Google Scholar] [CrossRef]
- Lakhdari, O.; Tap, J.; Béguet-Crespel, F.; Le Roux, K.; de Wouters, T.; Cultrone, A.; Nepelska, M.; Lefèvre, F.; Doré, J.; Blottière, H.M. Identification of NF-ΚB modulation capabilities within human intestinal commensal bacteria. J. Biomed. Biotechnol. 2011, 2011, 282356. [Google Scholar] [CrossRef] [PubMed]
- Yan, F.; Polk, D.B. Disruption of NF-B signaling by ancient microbial molecules: Novel therapies of the future? Gut 2010, 59, 421–426. [Google Scholar] [CrossRef]
- WebMD. Dysbiosis: Gut Imbalance, IBD, and More. WebMD, 2022. Available online: https://www.webmd.com/digestive-disorders/what-is-dysbiosis (accessed on 22 May 2023).
- DeGruttola, A.K.; Low, D.; Mizoguchi, A.; Mizoguchi, E. Current understanding of dysbiosis in disease in human and animal models. Inflamm. Bowel Dis. 2016, 22, 1137–1150. [Google Scholar] [CrossRef]
- Liu, S.; Gao, J.; Zhu, M.; Liu, K.; Zhang, H.-L. Gut Microbiota and dysbiosis in Alzheimer’s disease: Implications for pathogenesis and treatment. Mol. Neurobiol. 2020, 57, 5026–5043. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, P.J.; Dowden, R.A.; Campbell, S.C. Role of dietary lipids in modulating inflammation through the gut microbiota. Nutrients 2019, 11, 117. [Google Scholar] [CrossRef]
- Nyangale, E.P.; Mottram, D.S.; Gibson, G.R. Gut microbial activity, implications for health and disease: The potential role of metabolite analysis. J. Proteome Res. 2012, 11, 5573–5585. [Google Scholar] [CrossRef]
- Wen, L.; Duffy, A. Factors influencing the gut microbiota, inflammation, and type 2 diabetes. J. Nutr. 2017, 147, 1468S–1475S. [Google Scholar] [CrossRef]
- Kulas, T.; Bursac, D.; Zegarac, Z.; PlaninicRados, G.; Hrgovic, Z. New views on cesarean section, its possible complications and long-term consequences for children’s health. Med. Arch. 2013, 67, 460–463. [Google Scholar] [CrossRef]
- Carabotti, M.; Scirocco, A.; Maselli, M.A.; Severi, C. The gut-brain axis: Interactions between enteric microbiota, central and enteric nervous systems. Ann. Gastroenterol. 2015, 28, 203–209. [Google Scholar] [PubMed]
- Tsigos, C.; Chrousos, G.P. Hypothalamic–pituitary–adrenal axis, neuroendocrine factors and stress. J. Psychosom. Res. 2002, 53, 865–871. [Google Scholar] [CrossRef]
- Mayer, E.A.; Savidge, T.; Shulman, R.J. Brain–gut microbiome interactions and functional bowel disorders. Gastroenterology 2014, 146, 1500–1512. [Google Scholar] [CrossRef] [PubMed]
- Shabbir, U.; Arshad, M.S.; Sameen, A.; Oh, D.-H. Crosstalk between gut and brain in Alzheimer’s disease: The role of Gut Microbiota Modulation Strategies. Nutrients 2021, 13, 690. [Google Scholar] [CrossRef]
- Chakrabarti, A.; Geurts, L.; Hoyles, L.; Iozzo, P.; Kraneveld, A.D.; La Fata, G.; Miani, M.; Patterson, E.; Pot, B.; Shortt, C.; et al. The microbiota–gut–brain axis: Pathways to Better Brain Health. perspectives on what we know, what we need to investigate and how to put knowledge into practice. Cell. Mol. Life Sci. 2022, 79, 80. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Y.; Xiayu, X.; Shi, C.; Chen, W.; Song, N.; Fu, X.; Zhou, R.; Xu, Y.-F.; Huang, L.; et al. Altered Gut Microbiota in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2017, 60, 1241–1257. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Park, Y.J.; Gonzales-Portillo, B.; Saft, M.; Cozene, B.; Sadanandan, N.; Borlongan, C.V. Gut dysbiosis in stroke and its implications on Alzheimer’s disease-like cognitive dysfunction. CNS Neurosci. Ther. 2021, 27, 505–514. [Google Scholar] [CrossRef]
- Fakharian, F.; Asgari, B.; Nabavi-Rad, A.; Sadeghi, A.; Soleimani, N.; Yadegar, A.; Zali, M.R. The interplay between helicobacter pylori and the gut microbiota: An emerging driver influencing the immune system homeostasis and gastric carcinogenesis. Front. Cell. Infect. Microbiol. 2022, 12, 953718. [Google Scholar] [CrossRef]
- Huynh, V.A.; Takala, T.M.; Murros, K.E.; Diwedi, B.; Saris, P.E. Desulfovibrio bacteria enhance alpha-synuclein aggregation in a Caenorhabditis elegans model of parkinson’s disease. Front. Cell. Infect. Microbiol. 2023, 13, 502. [Google Scholar] [CrossRef]
- Minter, M.R.; Zhang, C.; Leone, V.; Ringus, D.L.; Zhang, X.; Oyler-Castrillo, P.; Musch, M.W.; Liao, F.; Ward, J.F.; Holtzman, D.M.; et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Sci. Rep. 2016, 6, 30028. [Google Scholar] [CrossRef]
- Bäuerl, C.; Collado, M.C.; Diaz Cuevas, A.; Viña, J.; Pérez Martínez, G. Shifts in gut microbiota composition in an app/pss1 transgenic mouse model of Alzheimer’s disease during lifespan. Lett. Appl. Microbiol. 2018, 66, 464–471. [Google Scholar] [CrossRef]
- Poole, S.; Singhrao, S.K.; Chukkapalli, S.; Rivera, M.; Velsko, I.; Kesavalu, L.; Crean, S. Active invasion of porphyromonas gingivalis and infection-induced complement activation in apoe-/- mice brains. J. Alzheimers Dis. 2014, 43, 67–80. [Google Scholar] [CrossRef]
- Cattaneo, A.; Cattane, N.; Galluzzi, S.; Provasi, S.; Lopizzo, N.; Festari, C.; Ferrari, C.; Guerra, U.P.; Paghera, B.; Muscio, C.; et al. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol. Aging 2017, 49, 60–68. [Google Scholar] [CrossRef]
- Sun, Y.; Sommerville, N.R.; Liu, J.Y.; Ngan, M.P.; Poon, D.; Ponomarev, E.D.; Lu, Z.; Kung, J.S.; Rudd, J.A. Intra-gastrointestinal amyloid-β1–42 oligomers perturb enteric function and induce Alzheimer’s disease pathology. J. Physiol. 2020, 598, 4209–4223. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; Khoury, J.E.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Das, A.; Ray, S.K.; Banik, N.L. Role of pro-inflammatory cytokines released from microglia in Neurodegenerative Diseases. Brain Res. Bull. 2012, 87, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.-Q.; Ma, C.-G.; Ding, Z.-B.; Song, L.-J.; Wang, Q.; Kumar, G. Astrocytes: A double-edged sword in neurodegenerative diseases. Neural Regen. Res. 2021, 16, 1702–1710. [Google Scholar] [CrossRef] [PubMed]
- Mohr, A.E.; Crawford, M.; Jasbi, P.; Fessler, S.; Sweazea, K.L. Lipopolysaccharide and the gut microbiota: Considering structural variation. FEBS Lett. 2022, 596, 849–875. [Google Scholar] [CrossRef]
- Waldstein, S.R.; Wendell, C.R.; Seliger, S.L.; Ferrucci, L.; Metter, E.J.; Zonderman, A.B. Nonsteroidal anti-inflammatory drugs, aspirin, and cognitive function in the Baltimore Longitudinal Study of Aging. J. Am. Geriatr. Soc. 2010, 58, 38–43. [Google Scholar] [CrossRef]
- Mira-Pascual, L.; Cabrera-Rubio, R.; Ocon, S.; Costales, P.; Parra, A.; Suarez, A.; Moris, F.; Rodrigo, L.; Mira, A.; Collado, M.C. Microbial mucosal colonic shifts associated with the development of colorectal cancer reveal the presence of different bacterial and archaeal biomarkers. J. Gastroenterol. 2014, 50, 167–179. [Google Scholar] [CrossRef]
- Al Bander, Z.; Nitert, M.D.; Mousa, A.; Naderpoor, N. The gut microbiota and inflammation: An overview. Int. J. Environ. Res. Public Health 2020, 17, 7618. [Google Scholar] [CrossRef]
- Di Sabatino, A.; Cazzola, P.; Ciccocioppo, R.; Morera, R.; Biancheri, P.; Rovedatti, L.; Cantoro, L.; Vanoli, A.; Tinozzi, F.P.; Tinozzi, S.; et al. Efficacy of butyrate in the treatment of mild to moderate Crohn’s disease. Dig. Liver Dis. Suppl. 2007, 1, 31–35. [Google Scholar] [CrossRef]
- Long, S.L.; Gahan, C.G.M.; Joyce, S.A. Interactions between gut bacteria and bile in health and disease. Mol. Asp. Med. 2017, 56, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Just, S.; Mondot, S.; Ecker, J.; Wegner, K.; Rath, E.; Gau, L.; Streidl, T.; Hery-Arnaud, G.; Schmidt, S.; Lesker, T.R.; et al. The gut microbiota drives the impact of bile acids and fat source in diet on mouse metabolism. Microbiome 2018, 6, 134. [Google Scholar] [CrossRef]
- Tedelind, S.; Westberg, F.; Kjerrulf, M.; Vidal, A. Anti-inflammatory properties of the short-chain fatty acids acetate and propionate: A study with relevance to inflammatory bowel disease. World J. Gastroenterol. 2007, 13, 2826–2832. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Zheng, L.J.; Zhang, L.J. Neuroinflammation, Gut Microbiome, and Alzheimer’s disease. Mol. Neurobiol. 2018, 55, 8243–8250. [Google Scholar] [CrossRef]
- Watanabe, D.; Guo, Y.; Kamada, N. Interaction between the inflammasome and commensal microorganisms in gastrointestinal health and disease. EMBO Mol. Med. 2021, 13, e13452. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Callaway, J.B.; Ting, J.P.-Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef]
- Man, S.M. Inflammasomes in the gastrointestinal tract: Infection, cancer and gut microbiota homeostasis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 721–737. [Google Scholar] [CrossRef]
- Liang, T.; Zhang, Y.; Wu, S.; Chen, Q.; Wang, L. The role of NLRP3 inflammasome in Alzheimer’s disease and potential therapeutic targets. Front. Pharmacol. 2022, 13, 845185. [Google Scholar] [CrossRef]
- Blevins, H.M.; Xu, Y.; Biby, S.; Zhang, S. The NLRP3 inflammasome pathway: A review of mechanisms and inhibitors for the treatment of inflammatory diseases. Front. Aging Neurosci. 2022, 14, 879021. [Google Scholar] [CrossRef] [PubMed]
- Barczuk, J.; Siwecka, N.; Lusa, W.; Rozpędek-Kamińska, W.; Kucharska, E.; Majsterek, I. Targeting NLRP3-mediated neuroinflammation in Alzheimer’s disease treatment. Int. J. Mol. Sci. 2022, 23, 8979. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Dempsey, C.; Rubio Araiz, A.; Bryson, K.J.; Finucane, O.; Larkin, C.; Mills, E.L.; Robertson, A.A.B.; Cooper, M.A.; O’Neill, L.A.J.; Lynch, M.A. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-β and cognitive function in app/PS1 mice. Brain Behav. Immun. 2017, 61, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Schieber, A.M.; O’Connor, C.P.; Leblanc, M.; Michel, D.; Ayres, J.S. Pathogen-mediated inhibition of anorexia promotes host survival and transmission. Cell 2017, 168, 503–516.e12. [Google Scholar] [CrossRef] [PubMed]
- Shukla, P.K.; Delotterie, D.F.; Xiao, J.; Pierre, J.F.; Rao, R.; McDonald, M.P.; Khan, M.M. Alterations in the gut-microbial-inflammasome-brain axis in a mouse model of Alzheimer’s disease. Cells 2021, 10, 779. [Google Scholar] [CrossRef]
- Shen, H.; Guan, Q.; Zhang, X.; Yuan, C.; Tan, Z.; Zhai, L.; Hao, Y.; Gu, Y.; Han, C. New mechanism of neuroinflammation in Alzheimer’s disease: The activation of NLRP3 inflammasome mediated by gut microbiota. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2020, 100, 109884. [Google Scholar] [CrossRef]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2017, 18, 134–147. [Google Scholar] [CrossRef]
- Vorobjeva, N.V.; Chernyak, B.V. Netosis: Molecular mechanisms, role in physiology and pathology. Biochemistry 2020, 85, 1178–1190. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Liu, K.; Hua, T.; Zhang, C.; Sun, B.; Guan, Y. PET imaging of neutrophils infiltration in Alzheimer’s disease transgenic mice. Front. Neurol. 2020, 11, 523798. [Google Scholar] [CrossRef]
- Zenaro, E.; Pietronigro, E.; Bianca, V.D.; Piacentino, G.; Marongiu, L.; Budui, S.; Turano, E.; Rossi, B.; Angiari, S.; Dusi, S.; et al. Neutrophils promote Alzheimer’s disease–like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 2015, 21, 880–886. [Google Scholar] [CrossRef]
- Ascher, S.; Wilms, E.; Pontarollo, G.; Formes, H.; Bayer, F.; Müller, M.; Malinarich, F.; Grill, A.; Bosmann, M.; Saffarzadeh, M.; et al. Gut microbiota restrict netosis in acute mesenteric ischemia-reperfusion injury. Arter. Thromb. Vasc. Biol. 2020, 40, 2279–2292. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Shao, L.-H.; Wang, F.; Shen, X.-F.; Xia, X.-F.; Kang, X.; Song, P.; Wang, M.; Lu, X.-F.; Wang, C.; et al. Netting gut disease: Neutrophil extracellular trap in intestinal pathology. Oxidative Med. Cell. Longev. 2021, 2021, 5541222. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yang, X.; Song, Y.-Q.; Tu, J. Autophagy in Alzheimer’s disease pathogenesis: Therapeutic potential and future perspectives. Ageing Res. Rev. 2021, 72, 101464. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Stachowiak, A.; Mamun, A.A.; Tzvetkov, N.T.; Takeda, S.; Atanasov, A.G.; Bergantin, L.B.; Abdel-Daim, M.M.; Stankiewicz, A.M. Autophagy and Alzheimer’s disease: From molecular mechanisms to therapeutic implications. Front. Aging Neurosci. 2018, 10, 4. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and Molecular Mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Manea, A.J.; Ray, S.K. Regulation of autophagy as a therapeutic option in glioblastoma. Apoptosis 2021, 26, 574–599. [Google Scholar] [CrossRef]
- Zare-shahabadi, A.; Masliah, E.; Johnson, G.V.W.; Rezaei, N. Autophagy in Alzheimer’s disease. Rev. Neurosci. 2015, 26, 385–395. [Google Scholar] [CrossRef]
- Wang, L.; Klionsky, D.J.; Shen, H.-M. The emerging mechanisms and functions of microautophagy. Nat. Rev. Mol. Cell Biol. 2022, 24, 186–203. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.S.L.; Cheung, Z.H.; Ip, N.Y. Molecular machinery of macroautophagy and its deregulation in diseases. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2011, 1812, 1490–1497. [Google Scholar] [CrossRef] [PubMed]
- Lapaquette, P.; Bizeau, J.-B.; Acar, N.; Bringer, M.-A. Reciprocal interactions between gut microbiota and autophagy. World J. Gastroenterol. 2021, 27, 8283–8301. [Google Scholar] [CrossRef] [PubMed]
- Shoubridge, A.P.; Fourrier, C.; Choo, J.M.; Proud, C.G.; Sargeant, T.J.; Rogers, G.B. Gut microbiome regulation of autophagic flux and Neurodegenerative Disease Risks. Front. Microbiol. 2021, 12, 817433. [Google Scholar] [CrossRef]
- Nixon, R.A. The role of autophagy in Neurodegenerative Disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Afaq, F.; Saleem, M.; Ahmad, N.; Mukhtar, H. Targeting multiple signaling pathways by green tea polyphenol (−)-epigallocatechin-3-gallate. Cancer Res. 2006, 66, 2500–2505. [Google Scholar] [CrossRef]
- Joo, S.-Y.; Song, Y.-A.; Park, Y.-L.; Myung, E.; Chung, C.-Y.; Park, K.-J.; Cho, S.-B.; Lee, W.-S.; Kim, H.-S.; Rew, J.-S.; et al. Epigallocatechin-3-gallate inhibits LPS-induced NF-ΚB and MAPK signaling pathways in bone marrow-derived macrophages. Gut Liver 2012, 6, 188–196. [Google Scholar] [CrossRef]
- Jiang, J.; Mo, Z.-C.; Yin, K.; Zhao, G.-J.; Lv, Y.-C.; Ouyang, X.-P.; Jiang, Z.-S.; Fu, Y.; Tang, C.-K. Epigallocatechin-3-gallate prevents TNF-α-induced NF-ΚB activation thereby upregulating ABCA1 via the nrf2/KEAP1 pathway in macrophage foam cells. Int. J. Mol. Med. 2012, 29, 946–956. [Google Scholar] [CrossRef]
- Duan, X.; Li, Y.; Xu, F.; Ding, H. Study on the neuroprotective effects of genistein on Alzheimer’s disease. Brain Behav. 2021, 11, e02100. [Google Scholar] [CrossRef]
- Morrison, D.K. MAP Kinase Pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a011254. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Guo, Y.; Pan, W.; Liu, S.; Shen, Z.; Xu, Y.; Hu, L. Erk/MAPK signalling pathway and tumorigenesis (review). Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Mokra, D.; Joskova, M.; Mokry, J. Therapeutic effects of green tea polyphenol (−)-epigallocatechin-3-gallate (EGCG) in relation to molecular pathways controlling inflammation, oxidative stress, and apoptosis. Int. J. Mol. Sci. 2022, 24, 340. [Google Scholar] [CrossRef] [PubMed]
- Payne, A.; Nahashon, S.; Taka, E.; Adinew, G.M.; Soliman, K.F. Epigallocatechin-3-gallate (EGCG): New therapeutic perspectives for neuroprotection, aging, and neuroinflammation for the modern age. Biomolecules 2022, 12, 371. [Google Scholar] [CrossRef]
- Xu, D.; Peng, S.; Guo, R.; Yao, L.; Mo, H.; Li, H.; Song, H.; Hu, L. EGCG alleviates oxidative stress and inhibits aflatoxin B1 biosynthesis via MAPK signaling pathway. Toxins 2021, 13, 693. [Google Scholar] [CrossRef]
- Uddin, M.S.; Kabir, M.T. Emerging signal regulating potential of genistein against Alzheimer’s disease: A promising molecule of interest. Front. Cell Dev. Biol. 2019, 7, 197. [Google Scholar] [CrossRef]
- Wee, P.; Wang, Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef]
- Creative Diagnostics. EGF/EGFR Signaling Pathway. Available online: https://www.creative-diagnostics.com/egf-egfr-signaling-pathway.htm#:~:text=EGFR%20signaling%20pathway%20is%20one,regulate%20intercellular%20communication%20during%20development (accessed on 15 June 2023).
- Yarden, Y.; Shilo, B.Z. SnapShot: EGFR signaling pathway. Cell 2007, 131, 1018. [Google Scholar] [CrossRef]
- Mansour, H.M.; Fawzy, H.M.; El-Khatib, A.S.; Khattab, M.M. Repurposed anti-cancer epidermal growth factor receptor inhibitors: Mechanisms of neuroprotective effects in Alzheimer’s disease. Neural Regen. Res. 2022, 17, 1913–1918. [Google Scholar] [CrossRef]
- Ettcheto, M.; Cano, A.; Sanchez-López, E.; Verdaguer, E.; Folch, J.; Auladell, C.; Camins, A. Masitinib for the treatment of Alzheimer’s disease. Neurodegener. Dis. Manag. 2021, 11, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Minnelli, C.; Cianfruglia, L.; Laudadio, E.; Mobbili, G.; Galeazzi, R.; Armeni, T. Effect of epigallocatechin-3-gallate on EGFR signaling and migration in non-small cell lung cancer. Int. J. Mol. Sci. 2021, 22, 11833. [Google Scholar] [CrossRef]
- Farabegoli, F.; Govoni, M.; Spisni, E.; Papi, A. EGFR inhibition by (-)-epigallocatechin-3-gallate and IIF treatments reduces breast cancer cell invasion. Biosci. Rep. 2017, 37, BSR20170168. [Google Scholar] [CrossRef] [PubMed]
- Mas-Bargues, C.; Borrás, C.; Viña, J. The multimodal action of genistein in Alzheimer’s and other age-related diseases. Free. Radic. Biol. Med. 2022, 183, 127–137. [Google Scholar] [CrossRef]
- Hakuno, F.; Takahashi, S.-I. 40 years of IGF1: IGF1 receptor signaling pathways. Mol. Endocrinol. 2018, 61, T69–T86. [Google Scholar] [CrossRef]
- Hua, H.; Kong, Q.; Yin, J.; Zhang, J.; Jiang, Y. Insulin-like growth factor receptor signaling in tumorigenesis and drug resistance: A challenge for cancer therapy. J. Hematol. Oncol. 2020, 13, 64. [Google Scholar] [CrossRef]
- Westwood, A.J.; Beiser, A.; DeCarli, C.; Harris, T.B.; Chen, T.C.; He, X.-M.; Roubenoff, R.; Pikula, A.; Au, R.; Braverman, L.E.; et al. Insulin-like growth factor-1 and risk of alzheimer dementia and brain atrophy. Neurology 2014, 82, 1613–1619. [Google Scholar] [CrossRef]
- Shimizu, M.; Shirakami, Y.; Sakai, H.; Tatebe, H.; Nakagawa, T.; Hara, Y.; Weinstein, I.B.; Moriwaki, H. EGCG inhibits activation of the insulin-like growth factor (IGF)/IGF-1 receptor axis in human hepatocellular carcinoma cells. Cancer Lett. 2008, 262, 10–18. [Google Scholar] [CrossRef]
- Sakai, H.; Shimizu, M.; Shirakami, Y.; Weinstein, I.; Moriwaki, H. Effects of EGCG on activation of the IGF/IGF-1R system in human hepatoma cells. Cancer Res. 2007, 67 (Suppl. S9), 1573. Available online: https://aacrjournals.org/cancerres/article/67/9_Supplement/1573/535894/Effects-of-EGCG-on-activation-of-the-IGF-IGF-1R (accessed on 20 May 2023).
- Chen, J.; Duan, Y.; Zhang, X.; Ye, Y.; Ge, B.; Chen, J. Genistein induces apoptosis by the inactivation of the IGF-1R/P-akt signaling pathway in MCF-7 human breast cancer cells. Food Funct. 2015, 6, 995–1000. [Google Scholar] [CrossRef]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. MTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef]
- Sohrabi, M.; Floden, A.M.; Manocha, G.D.; Klug, M.G.; Combs, C.K. IGF-1R inhibitor ameliorates neuroinflammation in an Alzheimer’s disease transgenic mouse model. Front. Cell. Neurosci. 2020, 14, 200. [Google Scholar] [CrossRef]
- Wimmer, R.J.; Russell, S.J.; Schneider, M.F. Green tea component EGCG, insulin and IGF-1 promote nuclear efflux of atrophy-associated transcription factor FOXO1 in skeletal muscle fibers. J. Nutr. Biochem. 2015, 26, 1559–1567. [Google Scholar] [CrossRef] [PubMed]
- Mueed, Z.; Tandon, P.; Maurya, S.K.; Deval, R.; Kamal, M.A.; Poddar, N.K. Tau and mtor: The hotspots for multifarious diseases in Alzheimer’s development. Front. Neurosci. 2019, 12, 1017. [Google Scholar] [CrossRef]
- Oddo, S. The role of mtor signaling in Alzheimer disease. Front. Biosci. 2012, S4, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Van Aller, G.S.; Carson, J.D.; Tang, W.; Peng, H.; Zhao, L.; Copeland, R.A.; Tummino, P.J.; Luo, L. Epigallocatechin gallate (EGCG), a major component of green tea, is a dual phosphoinositide-3-kinase/mtor inhibitor. Biochem. Biophys. Res. Commun. 2011, 406, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Holczer, M.; Besze, B.; Zámbó, V.; Csala, M.; Bánhegyi, G.; Kapuy, O. Epigallocatechin-3-gallate (EGCG) promotes autophagy-dependent survival via influencing the balance of mTOR-AMPK pathways upon endoplasmic reticulum stress. Oxidative Med. Cell. Longev. 2018, 2018, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Javed, Z.; Khan, K.; Herrera-Bravo, J.; Naeem, S.; Iqbal, M.J.; Sadia, H.; Qadri, Q.R.; Raza, S.; Irshad, A.; Akbar, A.; et al. Genistein as a regulator of signaling pathways and microRNAs in different types of cancers. Cancer Cell Int. 2021, 21, 388. [Google Scholar] [CrossRef] [PubMed]
- Sahu, A.; Gopalakrishnan, L.; Gaur, N.; Chatterjee, O.; Mol, P.; Modi, P.K.; Dagamajalu, S.; Advani, J.; Jain, S.; Keshava Prasad, T.S. The 5-hydroxytryptamine signaling map: An overview of serotonin-serotonin receptor mediated signaling network. J. Cell Commun. Signal. 2018, 12, 731–735. [Google Scholar] [CrossRef]
- Uceda, S.; Echeverry-Alzate, V.; Reiriz-Rojas, M.; Martínez-Miguel, E.; Pérez-Curiel, A.; Gómez-Senent, S.; Beltrán-Velasco, A.I. Gut microbial metabolome and dysbiosis in neurodegenerative diseases: Psychobiotics and fecal microbiota transplantation as a therapeutic approach—A comprehensive narrative review. Int. J. Mol. Sci. 2023, 24, 13294. [Google Scholar] [CrossRef]
- Dunham, S.J.; McNair, K.A.; Adams, E.D.; Avelar-Barragan, J.; Forner, S.; Mapstone, M.; Whiteson, K.L. Longitudinal analysis of the microbiome and metabolome in the 5xfad mouse model of Alzheimer’s disease. mBio 2022, 13, e0179422. [Google Scholar] [CrossRef]
- Li, G.; Yang, J.; Wang, X.; Zhou, C.; Zheng, X.; Lin, W. Effects of EGCG on depression-related behavior and serotonin concentration in a rat model of chronic unpredictable mild stress. Food Funct. 2020, 11, 8780–8787. [Google Scholar] [CrossRef] [PubMed]
- Thangavel, P.; Puga-Olguín, A.; Rodríguez-Landa, J.F.; Zepeda, R.C. Genistein as potential therapeutic candidate for menopausal symptoms and other related diseases. Molecules 2019, 24, 3892. [Google Scholar] [CrossRef] [PubMed]
- Tayeb, H.O.; Yang, H.D.; Price, B.H.; Tarazi, F.I. Pharmacotherapies for Alzheimer’s disease: Beyond cholinesterase inhibitors. Pharmacol. Ther. 2012, 134, 8–25. [Google Scholar] [CrossRef]
- Kim, J.-W.; Im, S.; Jeong, H.-R.; Jung, Y.-S.; Lee, I.; Kim, K.J.; Park, S.K.; Kim, D.-O. Neuroprotective effects of Korean red pine (pinus densiflora) bark extract and its phenolics. J. Microbiol. Biotechnol. 2018, 28, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Okello, E.J.; Mather, J. Comparative kinetics of acetyl- and butyryl-cholinesterase inhibition by green tea catechins|relevance to the symptomatic treatment of Alzheimer’s disease. Nutrients 2020, 12, 1090. [Google Scholar] [CrossRef] [PubMed]
- Okello, E.J.; Leylabi, R.; McDougall, G.J. Inhibition of acetylcholinesterase by green and white tea and their simulated intestinal metabolites. Food Funct. 2012, 3, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Gao, M.; Kang, G.; Huang, H. The potential role of phytonutrients flavonoids influencing gut microbiota in the prophylaxis and treatment of inflammatory bowel disease. Front. Nutr. 2021, 8, 798038. [Google Scholar] [CrossRef] [PubMed]
- Andreu Fernández, V.; Almeida Toledano, L.; Pizarro Lozano, N.; Navarro Tapia, E.; Gómez Roig, M.D.; De la Torre Fornell, R.; García Algar, Ó. Bioavailability of epigallocatechin gallate administered with different nutritional strategies in healthy volunteers. Antioxidants 2020, 9, 440. [Google Scholar] [CrossRef]
- Li, R.; Robinson, M.; Ding, X.; Geetha, T.; Al-Nakkash, L.; Broderick, T.L.; Babu, J.R. Genistein: A focus on several neurodegenerative diseases. J. Food Biochem. 2022, 46, e14155. [Google Scholar] [CrossRef]
- Yang, Z.; Kulkarni, K.; Zhu, W.; Hu, M. Bioavailability and pharmacokinetics of Genistein: Mechanistic studies on its ADME. Anti-Cancer Agents Med. Chem. 2012, 12, 1264–1280. [Google Scholar] [CrossRef] [PubMed]
- Pamer, E.G. Fecal microbiota transplantation: Effectiveness, complexities, and lingering concerns. Mucosal Immunol. 2014, 7, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Ma, X.; Geng, S.; Jiang, X.; Li, Y.; Hu, L.; Li, J.; Wang, Y.; Han, X. Fecal microbiota transplantation beneficially regulates intestinal mucosal autophagy and alleviates gut barrier injury. mSystems 2018, 3, e00137-18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ishikawa, D.; Ohkusa, T.; Fukuda, S.; Nagahara, A. Hot topics on fecal microbiota transplantation for the treatment of inflammatory bowel disease. Front. Med. 2022, 9, 106856. [Google Scholar] [CrossRef]
- Elangovan, S.; Borody, T.J.; Holsinger, R.M. Fecal microbiota transplantation reduces pathology and improves cognition in a mouse model of Alzheimer’s disease. Cells 2022, 12, 119. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Huang, S.; Li, T.; Li, N.; Han, D.; Zhang, B.; Xu, Z.Z.; Zhang, S.; Pang, J.; Wang, S.; et al. Gut microbiota from green tea polyphenol-dosed mice improves intestinal epithelial homeostasis and ameliorates experimental colitis. Microbiome 2021, 9, 184. [Google Scholar] [CrossRef] [PubMed]
- Hou, Q.; Huang, J.; Zhao, L.; Pan, X.; Liao, C.; Jiang, Q.; Lei, J.; Guo, F.; Cui, J.; Guo, Y.; et al. Dietary genistein increases microbiota-derived short chain fatty acid levels, modulates homeostasis of the aging gut, and extends healthspan and lifespan. Pharmacol. Res. 2023, 188, 106676. [Google Scholar] [CrossRef]
- Zhao, L.; Liu, J.-W.; Shi, H.-Y.; Ma, Y.-M. Neural stem cell therapy for brain disease. World J. Stem Cells 2021, 13, 1278–1292. [Google Scholar] [CrossRef]
- Kim, H.-J. Regulation of neural stem cell fate by natural products. Biomol. Ther. 2019, 27, 15–24. [Google Scholar] [CrossRef]
- Ager, R.R.; Davis, J.L.; Agazaryan, A.; Benavente, F.; Poon, W.W.; LaFerla, F.M.; Blurton-Jones, M. Human neural stem cells improve cognition and promote synaptic growth in two complementary transgenic models of Alzheimer’s disease and neuronal loss. Hippocampus 2015, 25, 813–826. [Google Scholar] [CrossRef]
- Li, X.; Zhu, H.; Sun, X.; Zuo, F.; Lei, J.; Wang, Z.; Bao, X.; Wang, R. Human neural stem cell transplantation rescues cognitive defects in App/PS1 model of Alzheimer’s disease by enhancing neuronal connectivity and metabolic activity. Front. Aging Neurosci. 2016, 8, 282. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.K.; Cho, T.; Wang, Y.T.; McLarnon, J.G. Neural progenitor cells attenuate inflammatory reactivity and neuronal loss in an animal model of inflamed ad brain. J. Neuroinflamm. 2009, 6, 39. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, M.M.; Goodkey, K.; Voronova, A. Regulation of microglia function by neural stem cells. Front. Cell. Neurosci. 2023, 17, 1130205. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Sun, J.; Zhao, H.; Guo, H.; Li, J. Functional mechanism on stem cells by tea (Camellia sinensis) bioactive compounds. Food Sci. Hum. Wellness 2022, 11, 579–586. [Google Scholar] [CrossRef]
- Zhang, Y.; He, Q.; Dong, J.; Jia, Z.; Hao, F.; Shan, C. Effects of epigallocatechin-3-gallate on proliferation and differentiation of mouse cochlear neural stem cells: Involvement of PI3K/akt signaling pathway. Eur. J. Pharm. Sci. 2016, 88, 267–273. [Google Scholar] [CrossRef]
- Zhang, J.-C.; Xu, H.; Yuan, Y.; Chen, J.-Y.; Zhang, Y.-J.; Lin, Y.; Yuan, S.-Y. Delayed treatment with green tea polyphenol EGCG promotes neurogenesis after ischemic stroke in adult mice. Mol. Neurobiol. 2016, 54, 3652–3664. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.; Han, H.; Zhong, C.; Geng, Q. Effects of genistein and daidzein on hippocampus neuronal cell proliferation and BDNF expression in H19-7 neural cell line. J. Nutr. Health Aging 2011, 16, 389–394. [Google Scholar] [CrossRef]
- Squillaro, T.; Cimini, A.; Peluso, G.; Giordano, A.; Melone, M.A.B. Nano-delivery systems for encapsulation of dietary polyphenols: An experimental approach for neurodegenerative diseases and brain tumors. Biochem. Pharmacol. 2018, 154, 303–317. [Google Scholar] [CrossRef]
- TWI. What are Nanoparticles? Definition, Size, Uses and Properties. Available online: https://www.twi-global.com/technical-knowledge/faqs/what-are-nanoparticles (accessed on 4 August 2023).
- Li, B.; Du, W.; Jin, J.; Du, Q. Preservation of (−)-epigallocatechin-3-gallate antioxidant properties loaded in heat treated β-lactoglobulin nanoparticles. J. Agric. Food Chem. 2012, 60, 3477–3484. [Google Scholar] [CrossRef]
- Dai, W.; Ruan, C.; Zhang, Y.; Wang, J.; Han, J.; Shao, Z.; Sun, Y.; Liang, J. Bioavailability enhancement of EGCG by structural modification and nano-delivery: A Review. J. Funct. Foods 2020, 65, 103732. [Google Scholar] [CrossRef]
- Rassu, G.; Porcu, E.; Fancello, S.; Obinu, A.; Senes, N.; Galleri, G.; Migheli, R.; Gavini, E.; Giunchedi, P. Intranasal delivery of genistein-loaded nanoparticles as a potential preventive system against neurodegenerative disorders. Pharmaceutics 2018, 11, 8. [Google Scholar] [CrossRef]
- Chakrabarti, M.; McDonald, A.J.; Will Reed, J.; Moss, M.A.; Das, B.C.; Ray, S.K. Molecular signaling mechanisms of natural and synthetic retinoids for inhibition of pathogenesis in Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 50, 335–352. [Google Scholar] [CrossRef]
- Das, B.; Dasgupta, S.; Ray, S.K. Potential therapeutic roles of retinoids for prevention of Neuroinflammation and neurodegeneration in Alzheimer’s disease. Neural Regen. Res. 2019, 14, 1880–1892. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AChE Inhibitor | Dosage (mg/d) | Outcomes and Side Effects | Availability for Clinical Use | Reference |
|---|---|---|---|---|
| Tacrine | 80–160 | Nausea and abnormal liver functionality | No | [10] |
| Physostigmine | 36 | Nausea, diarrhea, and dizziness | No | [11] |
| Rivastigmine | 6–12 | Nausea, vomiting, and diarrhea | Yes | [11] |
| Galantamine * | 20–50 | Nausea, vomiting, and diarrhea | Yes | [11] |
| Donepezil | 10 | Higher cognitive improvements and reduced inflammatory cytokines as well as oxidative stress | Yes | [12] |
| 5 | Reduced inflammatory cytokines and oxidative stress | |||
| Metrifonate | NA | Bradycardia, rhinitis, abdominal pain, neuromuscular dysfunction, and respiratory failure | No | [13] |
| AD Animal Model | Change in Gut Microbiota in AD Mice | Observed Pathological Symptoms | Reference |
|---|---|---|---|
| AD model mice (with varying ages) | Decreased microbial diversity and reduced SCFA levels | Amyloid deposition and ultrastructural abnormalities in the intestine, cognitive dysfunction, and signaling pathway alterations | [51] |
| APP/PSEN1 mice | Decreased microbial diversity | Cognitive dysfunction | [52] |
| APPSWE/PSIΔE9 mice | Varied gut microbial composition | Increased cerebral Aβ pathology | [55] |
| APP/PS1 mice | Increased pro-inflammatory bacteria during aging | Autism and inflammatory-related disorders | [56] |
| ApoE-/- mice | Porphyromonas gingivalis infection | Neuronal injury | [57] |
| Signaling Pathway in AD | Associated Functions | EGCG and GS | References |
|---|---|---|---|
| NF-κB pathway | Regulates pro-inflammatory genes | Both can inhibit the pathway | [33,92,93,94,95] |
| MAPK pathway | Regulates apoptosis, differentiation, etc. | Both can inhibit the pathway | [96,99,100,101,102] |
| EGFR pathway | Regulates gene expression and cell proliferation | Both can inhibit the pathway | [103,104,105,106,107,108,109,110] |
| IGF signal transduction pathway | Regulates cell differentiation, cell survival, and cell maintenance | Both can inhibit the pathway | [110,111,112,113,114,115,116,117,118] |
| mTOR pathway | Regulates cell proliferation, apoptosis, and autophagy | Both can inhibit the pathway | [119,120,121,122,123] |
| 5-Hydroxytryptamine signaling pathway | Regulates serotonin production | Both can facilitate the pathway | [124,125,126,127] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muraleedharan, A.; Ray, S.K. Epigallocatechin-3-Gallate and Genistein for Decreasing Gut Dysbiosis, Inhibiting Inflammasomes, and Aiding Autophagy in Alzheimer’s Disease. Brain Sci. 2024, 14, 96. https://doi.org/10.3390/brainsci14010096
Muraleedharan A, Ray SK. Epigallocatechin-3-Gallate and Genistein for Decreasing Gut Dysbiosis, Inhibiting Inflammasomes, and Aiding Autophagy in Alzheimer’s Disease. Brain Sciences. 2024; 14(1):96. https://doi.org/10.3390/brainsci14010096
Chicago/Turabian StyleMuraleedharan, Ahalya, and Swapan K. Ray. 2024. "Epigallocatechin-3-Gallate and Genistein for Decreasing Gut Dysbiosis, Inhibiting Inflammasomes, and Aiding Autophagy in Alzheimer’s Disease" Brain Sciences 14, no. 1: 96. https://doi.org/10.3390/brainsci14010096
APA StyleMuraleedharan, A., & Ray, S. K. (2024). Epigallocatechin-3-Gallate and Genistein for Decreasing Gut Dysbiosis, Inhibiting Inflammasomes, and Aiding Autophagy in Alzheimer’s Disease. Brain Sciences, 14(1), 96. https://doi.org/10.3390/brainsci14010096