

Neuroinflammation and Neurodegenerative Diseases: How Much Do We Still Not Know?



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
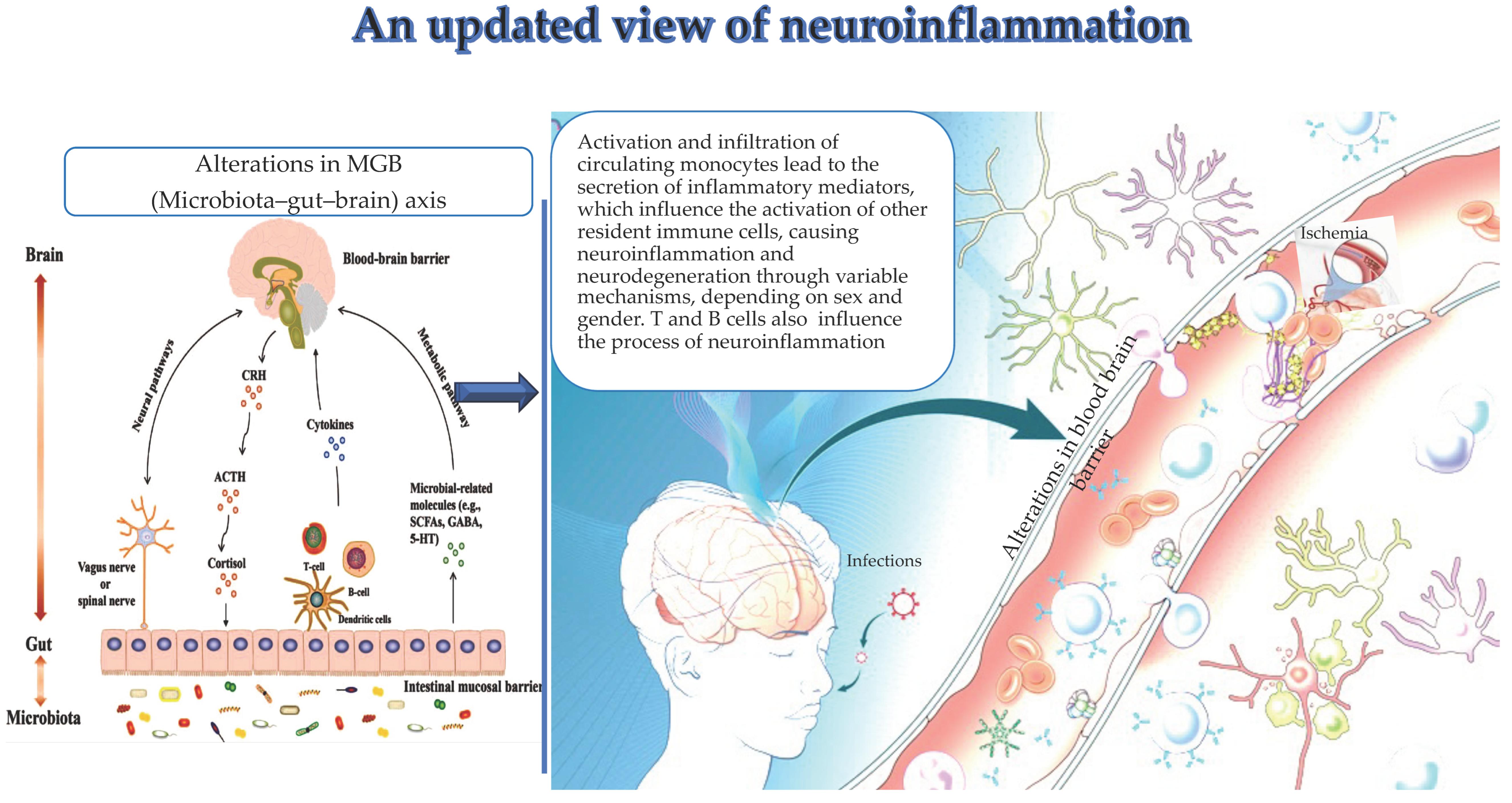
2. Recent Evidence on Peripheral and Infiltrating Monocytes and Clonotypic Immune Cells in Neuroinflammation and Their Sex- and Gender-Mediated Modulation
2.1. Monocytes in Neuroinflammation
2.2. Clonotypic Immune Cells in Neuroinflammation and ND
2.3. Considerations on Immune Cells Infiltrating the CNS and New Evidence on the Migration of Immune Cells outside the CNS
2.4. Sex/Gender Dimorphism: An Important Modifier of Immune System and Physiology of Brain, and a Crucial Differential Driver in Diseases
3. Changes in the MGB Axis and Neuroinflammation
4. Autophagy
5. Ferroptosis in Neuroinflammation
6. The Close Link of Endothelial Dysfunction with Neuroinflammation and ND
7. miRNAs and Epigenetic Factors in Neuroinflammation and ND
8. Transcriptional Factors and Related Pathways: Focus on NF-kB (Nuclear Factor Kappa-Light-Chain Enhancer of Activated B Cells) and Related Pathways
9. Circadian Cycle and Neuroinflammation
10. Chronic Low-Grade Inflammation and Neurodegenerative Diseases
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Disabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139 (Suppl. 2), 136–153. [Google Scholar] [CrossRef] [PubMed]
- Gilhus, N.E.; Deuschl, G. Neuroinflammation—A common thread in neurological disorders. Nat. Rev. Neurol. 2019, 15, 429–430. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in neurodegenerative diseases: Mechanism and potential therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 359. [Google Scholar] [CrossRef] [PubMed]
- Ní Chasaide, C.; Lynch, M.A. The role of the immune system in driving neuroinflammation. Brain Neurosci. Adv. 2020, 4, 2398212819901082. [Google Scholar] [CrossRef] [PubMed]
- Becher, B.; Spath, S.; Goverman, J. Cytokine networks in neuroinflammation. Nat. Rev. Immunol. 2017, 17, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Carata, E.; Muci, M.; Di Giulio, S.; Mariano, S.; Panzarini, E. Looking to the Future of the Role of Macrophages and Extracellular Vesicles in Neuroinflammation in ALS. Int. J. Mol. Sci. 2023, 24, 11251. [Google Scholar] [CrossRef] [PubMed]
- Schirò, G.; Iacono, S.; Balistreri, C.R. The Role of Human Microbiota in Myasthenia Gravis: A Narrative Review. Neurol. Int. 2023, 15, 392–404. [Google Scholar] [CrossRef]
- Celorrio, M.; Shumilov, K.; Friess, S.H. Gut microbial regulation of innate and adaptive immunity after traumatic brain injury. Neural Regen. Res. 2024, 19, 272–276. [Google Scholar] [CrossRef]
- Sanghai, N.; Tranmer, G.K. Biochemical and Molecular Pathways in Neurodegenerative Diseases: An Integrated View. Cells 2023, 12, 2318. [Google Scholar] [CrossRef]
- Li, L.; Guo, L.; Gao, R.; Yao, M.; Qu, X.; Sun, G.; Fu, Q.; Hu, C.; Han, G. Ferroptosis: A new regulatory mechanism in neuropathic pain. Front. Aging Neurosci. 2023, 15, 1206851. [Google Scholar] [CrossRef]
- Massaccesi, L.; Balistreri, C.R. Biomarkers of Oxidative Stress in Acute and Chronic Diseases. Antioxidants 2022, 11, 1766. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.B.; Jia, X.; Cao, Q.; Chen, Y.T.; Tong, J.; Lu, G.D.; Li, D.J.; Han, T.; Zhuang, C.L.; Wang, P. Ferroptosis-Regulated Cell Death as a Therapeutic Strategy for Neurodegenerative Diseases: Current Status and Future Prospects. ACS Chem. Neurosci. 2023, 14, 2995–3012. [Google Scholar] [CrossRef] [PubMed]
- Monastero, R.; Magro, D.; Venezia, M.; Pisano, C.; Balistreri, C.R. A promising therapeutic peptide and preventive/diagnostic biomarker for age-related diseases: The Elabela/Apela/Toddler peptide. Ageing Res. Rev. 2023, 91, 102076. [Google Scholar] [CrossRef] [PubMed]
- Schirò, G.; Iacono, S.; Ragonese, P.; Aridon, P.; Salemi, G.; Balistreri, C.R. A Brief Overview on BDNF-Trk Pathway in the Nervous System: A Potential Biomarker or Possible Target in Treatment of Multiple Sclerosis? Front. Neurol. 2022, 13, 917527. [Google Scholar] [CrossRef] [PubMed]
- Brites, D.; Fernandes, A. Neuroinflammation and Depression: Microglia Activation, Extracellular Microvesicles and microRNA Dysregulation. Front. Cell. Neurosci. 2015, 9, 476. [Google Scholar] [CrossRef] [PubMed]
- Tregub, P.P.; Ibrahimli, I.; Averchuk, A.S.; Salmina, A.B.; Litvitskiy, P.F.; Manasova, Z.S.; Popova, I.A. The Role of microRNAs in Epigenetic Regulation of Signaling Pathways in Neurological Pathologies. Int. J. Mol. Sci. 2023, 24, 12899. [Google Scholar] [CrossRef] [PubMed]
- Tassinari, V.; La Rosa, P.; Guida, E.; Colopi, A.; Caratelli, S.; De Paolis, F.; Gallo, A.; Cenciarelli, C.; Sconocchia, G.; Dolci, S.; et al. Contribution of A-to-I RNA editing, M6A RNA Methylation, and Alternative Splicing to physiological brain aging and neurodegenerative diseases. Mech. Ageing Dev. 2023, 212, 111807. [Google Scholar] [CrossRef]
- Xu, X.; Wang, J.; Chen, G. Circadian cycle and neuroinflammation. Open Life Sci. 2023, 18, 20220712. [Google Scholar] [CrossRef]
- Nair, A.L.; Groenendijk, L.; Overdevest, R.; Fowke, T.M.; Annida, R.; Mocellin, O.; de Vries, H.E.; Wevers, N.R. Human BBB-on-a-chip reveals barrier disruption, endothelial inflammation, and T cell migration under neuroinflammatory conditions. Front. Mol. Neurosci. 2023, 16, 1250123. [Google Scholar] [CrossRef]
- Bruno, M.; Bonomi, C.G.; Ricci, F.; Di Donna, M.G.; Mercuri, N.B.; Koch, G.; Martorana, A.; Motta, C. Blood-brain barrier permeability is associated with different neuroinflammatory profiles in Alzheimer’s disease. Eur. J. Neurol. 2023, 31, e16095, advance online publication. [Google Scholar] [CrossRef]
- Lauritsen, J.; Romero-Ramos, M. The systemic immune response in Parkinson’s disease: Focus on the peripheral immune component. Trends Neurosci. 2023, 46, 863–878. [Google Scholar] [CrossRef] [PubMed]
- Miró-Mur, F.; Pérez-de-Puig, I.; Ferrer-Ferrer, M.; Urra, X.; Justicia, C.; Chamorro, A.; Planas, A.M. Immature monocytes recruited to the ischemic mouse brain differentiate into macrophages with features of alternative activation. Brain Behav. Immun. 2016, 53, 18–33. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.A.; Maltby, S.; Plank, M.W.; Kluge, M.; Nilsson, M.; Foster, P.S.; Walker, F.R. Peripheral immune cells infiltrate into sites of secondary neurodegeneration after ischemic stroke. Brain Behav. Immun. 2018, 67, 299–307. [Google Scholar] [CrossRef]
- Kanazawa, M.; Ninomiya, I.; Hatakeyama, M.; Takahashi, T.; Shimohata, T. Microglia and Monocytes/Macrophages Polarization Reveal Novel Therapeutic Mechanism against Stroke. Int. J. Mol. Sci. 2017, 18, 2135. [Google Scholar] [CrossRef] [PubMed]
- Ritzel, R.M.; Li, Y.; Jiao, Y.; Doran, S.J.; Khan, N.; Henry, R.J.; Brunner, K.; Loane, D.J.; Faden, A.I.; Szeto, G.L.; et al. The brain-bone marrow axis and its implications for chronic traumatic brain injury. Res. Sq. 2023, rs.3.rs-3356007. [Google Scholar] [CrossRef]
- Côté, M.; Poirier, A.A.; Aubé, B.; Jobin, C.; Lacroix, S.; Soulet, D. Partial depletion of the proinflammatory monocyte population is neuroprotective in the myenteric plexus but not in the basal ganglia in a MPTP mouse model of Parkinson’s disease. Brain Behav. Immun. 2015, 46, 154–167. [Google Scholar] [CrossRef]
- Williams, D.W.; Veenstra, M.; Gaskill, P.J.; Morgello, S.; Calderon, T.M.; Berman, J.W. Monocytes mediate HIV neuropathogenesis: Mechanisms that contribute to HIV associated neurocognitive disorders. Curr. HIV Res. 2014, 12, 85–96. [Google Scholar] [CrossRef]
- Molnarfi, N.; Benkhoucha, M.; Bjarnadottir, K.; Juillard, C.; Lalive, P.H. Interferonbeta induces hepatocyte growth factor in monocytes of multiple sclerosis patients. PLoS ONE 2012, 7, e49882. [Google Scholar] [CrossRef]
- Man, S.; Tucky, B.; Cotleur, A.; Drazba, J.; Takeshita, Y.; Ransohoff, R.M. CXCL12- induced monocyte-endothelial interactions promote lymphocyte transmigration across an in vitro blood-brain barrier. Sci. Transl. Med. 2012, 4, 119ra114. [Google Scholar] [CrossRef]
- Schmitt, C.; Strazielle, N.; Ghersi-Egea, J.F. Brain leukocyte infiltration initiated by peripheral inflammation or experimental autoimmune encephalomyelitis occurs through pathways connected to the CSF-filled compartments of the forebrain and midbrain. J. Neuroinflammation 2012, 9, 187. [Google Scholar] [CrossRef]
- Vojdani, A.; Lambert, J. The role of Th17 in neuroimmune disorders: Target for CAM therapy. Part II. Evid. Based Complement. Altern. Med. 2011, 2011, 984965. [Google Scholar] [CrossRef] [PubMed]
- Kadiu, I.; Glanzer, J.G.; Kipnis, J.; Gendelman, H.E.; Thomas, M.P. Mononuclear phagocytes in the pathogenesis of neurodegenerative diseases. Neurotox. Res. 2005, 8, 25–50. [Google Scholar] [CrossRef] [PubMed]
- Cashman, J.R.; Gagliardi, S.; Lanier, M.; Ghirmai, S.; Abel, K.J.; Fiala, M. Curcumins promote monocytic gene expression related to beta-amyloid and superoxide dismutase clearance. Neurodegener. Dis. 2012, 10, 274–276. [Google Scholar] [CrossRef] [PubMed]
- Kohman, R.A. Aging microglia: Relevance to cognition and neural plasticity. Meth. Mol. Biol. 2012, 934, 193–218. [Google Scholar]
- Bottger, D.; Ullrich, C.; Humpel, C. Monocytes deliver bioactive nerve growth factor through a brain capillary endothelial cell-monolayer in vitro and counteract degeneration of cholinergic neurons. Brain Res. 2010, 1312, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Karlmark, K.R.; Tacke, F.; Dunay, I.R. Monocytes in health and disease—Minireview. Eur. J. Microbiol. Immunol. Bp 2012, 2, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Schirò, G.; Di Stefano, V.; Iacono, S.; Lupica, A.; Brighina, F.; Monastero, R.; Balistreri, C.R. Advances on Cellular Clonotypic Immunity in Amyotrophic Lateral Sclerosis. Brain Sci. 2022, 12, 1412. [Google Scholar] [CrossRef] [PubMed]
- Jorfi, M.; Park, J.; Hall, C.K.; Lin, C.J.; Chen, M.; von Maydell, D.; Kruskop, J.M.; Kang, B.; Choi, Y.; Prokopenko, D.; et al. Infiltrating CD8+ T cells exacerbate Alzheimer’s disease pathology in a 3D human neuroimmune axis model. Nat. Neurosci. 2023, 26, 1489–1504. [Google Scholar] [CrossRef]
- Kimura, K.; Nishigori, R.; Hamatani, M.; Sawamura, M.; Ashida, S.; Fujii, C.; Takata, M.; Lin, Y.; Sato, W.; Okamoto, T.; et al. Resident Memory-like CD8+ T Cells Are Involved in Chronic Inflammatory and Neurodegenerative Diseases in the CNS. Neurol. R Neuroimmunol. Neuroinflammation 2023, 11, e200172. [Google Scholar] [CrossRef]
- Harms, A.S.; Yang, Y.T.; Tansey, M.G. Central and peripheral innate and adaptive immunity in Parkinson’s disease. Sci. Transl. Med. 2023, 15, eadk3225. [Google Scholar] [CrossRef]
- Jia, Z.; Guo, M.; Ge, X.; Chen, F.; Lei, P. IL-33/ST2 Axis: A Potential Therapeutic Target in Neurodegenerative Diseases. Biomolecules 2023, 13, 1494. [Google Scholar] [CrossRef] [PubMed]
- Lutshumba, J.; Wilcock, D.M.; Monson, N.L.; Stowe, A.M. Sex-based differences in effector cells of the adaptive immune system during Alzheimer’s disease and related dementias. Neurobiol. Dis. 2023, 184, 106202. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Liu, J.; Qu, Q.; Zhao, X.; Zhang, J. JKAP, Th1 cells, and Th17 cells are dysregulated and inter-correlated, among them JKAP and Th17 cells relate to cognitive impairment progression in Alzheimer’s disease patients. Ir. J. Med. Sci. 2022, 191, 1855–1861. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wei, B.; Li, L.; Wang, B.; Sun, M. Th17 cells and inflammation in neurological disorders: Possible mechanisms of action. Front. Immunol. 2022, 13, 932152. [Google Scholar] [CrossRef] [PubMed]
- Oberstein, T.J.; Taha, L.; Spitzer, P.; Hellstern, J.; Herrmann, M.; Kornhuber, J.; Maler, J.M. Imbalance of Circulating Th17 and Regulatory T Cells in Alzheimer’s Disease: A Case Control Study. Front. Immunol. 2018, 9, 1213. [Google Scholar] [CrossRef]
- Jafarzadeh, A.; Sheikhi, A.; Jafarzadeh, Z.; Nemati, M. Differential roles of regulatory T cells in Alzheimer’s disease. Cell. Immunol. 2023, 393–394, 104778. [Google Scholar] [CrossRef]
- Fu, J.; Duan, J.; Mo, J.; Xiao, H.; Huang, Y.; Chen, W.; Xiang, S.; Yang, F.; Chen, Y.; Xu, S. Mild Cognitive Impairment Patients Have Higher Regulatory T-Cell Proportions Compared With Alzheimer’s Disease-Related Dementia Patients. Front. Aging Neurosci. 2021, 12, 624304. [Google Scholar] [CrossRef]
- Yeapuri, P.; Machhi, J.; Lu, Y.; Abdelmoaty, M.M.; Kadry, R.; Patel, M.; Bhattarai, S.; Lu, E.; Namminga, K.L.; Olson, K.E.; et al. Amyloid-β specific regulatory T cells attenuate Alzheimer’s disease pathobiology in APP/PS1 mice. Mol. Neurodegener. 2023, 18, 97. [Google Scholar] [CrossRef]
- Boillée, S.; Yamanaka, K.; Lobsiger, C.S.; Copeland, N.G.; Jenkins, N.A.; Kassiotis, G.; Kollias, G.; Cleveland, D.W. Onset and Progression in Inherited ALS Determined by Motor Neurons and Microglia. Science 2006, 312, 1389–1392. [Google Scholar] [CrossRef]
- Hooten, K.G.; Beers, D.R.; Zhao, W.; Appel, S.H. Protective and Toxic Neuroinflammation in Amyotrophic Lateral Sclerosis. Neurotherapeutics 2015, 12, 364–375. [Google Scholar] [CrossRef]
- Seksenyan, A.; Ron-Harel, N.; Azoulay, D.; Cahalon, L.; Cardon, M.; Rogeri, P.; Ko, M.K.; Weil, M.; Bulvik, S.; Rechavi, G.; et al. Thymic involution, a co-morbidity factor in amyotrophic lateral sclerosis. J. Cell. Mol. Med. 2009, 14, 2470–2482. [Google Scholar] [CrossRef] [PubMed]
- Ehrhart, J.; Smith, A.J.; Kuzmin-Nichols, N.; Zesiewicz, T.A.; Jahan, I.; Shytle, R.D.; Kim, S.-H.; Sanberg, C.D.; Vu, T.H.; Gooch, C.L.; et al. Humoral factors in ALS patients during disease progression. J. Neuroinflamm. 2015, 12, 127. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Beers, D.R.; Liao, B.; Henkel, J.S.; Appel, S.H. Regulatory T lymphocytes from ALS mice suppress microglia and effector T lymphocytes through different cytokine-mediated mechanisms. Neurobiol. Dis. 2012, 48, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; He, L.; Ren, M.; Lu, Y.; Meng, H.; Yin, D.; Chen, S.; Zhou, Q. Paraneoplastic Amyotrophic Lateral Sclerosis: Case Series and Literature Review. Brain Sci. 2022, 12, 1053. [Google Scholar] [CrossRef] [PubMed]
- Plantone, D.; Pardini, M.; Locci, S.; Nobili, F.; De Stefano, N. B Lymphocytes in Alzheimer’s Disease-A Comprehensive Review. J. Alzheimer’s Dis. JAD 2022, 88, 1241–1262. [Google Scholar] [CrossRef] [PubMed]
- Ashford, M.T.; Zhu, D.; Bride, J.; McLean, E.; Aaronson, A.; Conti, C.; Cypress, C.; Griffin, P.; Ross, R.; Duncan, T.; et al. Understanding Online Registry Facilitators and Barriers Experienced by Black Brain Health Registry Participants: The Community Engaged Digital Alzheimer’s Research (CEDAR) Study. J. Prev. Alzheimer’s Dis. 2023, 10, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Asher, S.; Priefer, R. Alzheimer’s disease failed clinical trials. Life Sci. 2022, 306, 120861. [Google Scholar] [CrossRef]
- Balistreri, C.R. New frontiers in ageing and longevity: Sex and gender medicine. Mech. Ageing Dev. 2023, 214, 111850. [Google Scholar] [CrossRef]
- Garofalo, S.; Cocozza, G.; Bernardini, G.; Savage, J.; Raspa, M.; Aronica, E.; Tremblay, M.E.; Ransohoff, R.M.; Santoni, A.; Limatola, C. Blocking immune cell infiltration of the central nervous system to tame Neuroinflammation in Amyotrophic lateral sclerosis. Brain Behav. Immun. 2022, 105, 1–14. [Google Scholar] [CrossRef]
- Rekik, A.; Aissi, M.; Rekik, I.; Mhiri, M.; Frih, M.A. Brain atrophy patterns in multiple sclerosis patients treated with natalizumab and its clinical correlates. Brain Behav. 2022, 12, e2573. [Google Scholar] [CrossRef]
- Laaker, C.; Baenen, C.; Kovács, K.G.; Sandor, M.; Fabry, Z. Immune cells as messengers from the CNS to the periphery: The role of the meningeal lymphatic system in immune cell migration from the CNS. Front. Immunol. 2023, 14, 1233908. [Google Scholar] [CrossRef] [PubMed]
- Schirò, G.; Balistreri, C.R. The close link between brain vascular pathological conditions and neurodegenerative diseases: Focus on some examples and potential treatments. Vasc. Pharmacol. 2022, 142, 106951. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, A.; Baschi, R.; Cicero, C.E.; Iacono, S.; Re, V.L.; Luca, A.; Schirò, G.; Monastero, R.; Gender Neurology Study Group of the Italian Society of Neurology. Sex and gender differences in Alzheimer’s disease, Parkinson’s disease, and Amyotrophic Lateral Sclerosis: A narrative review. Mech. Ageing Dev. 2023, 212, 111821. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Tellez, L.A.; Perkins, M.H.; Perez, I.O.; Qu, T.; Ferreira, J.; Ferreira, T.L.; Quinn, D.; Liu, Z.W.; Gao, X.B.; et al. A Neural Circuit for Gut-Induced Reward. Cell 2018, 175, 665–678.e23. [Google Scholar] [CrossRef] [PubMed]
- Agirman, G.; Yu, K.B.; Hsiao, E.Y. Signaling Inflammation across the Gut-Brain Axis. Science 2021, 374, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- McCarville, J.L.; Chen, G.Y.; Cuevas, V.D.; Troha, K.; Ayres, J.S. Microbiota Metabolites in Health and Disease. Annu. Rev. Immunol. 2020, 38, 147–170. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zeng, B.; Zeng, L.; Du, X.; Li, B.; Huo, R.; Liu, L.; Wang, H.; Dong, M.; Pan, J.; et al. Gut microbiota regulates mouse behaviors through glucocorticoid receptor pathway genes in the hippocampus. Transl. Psychiatry 2018, 8, 187. [Google Scholar] [CrossRef]
- O’Riordan, K.J.; Collins, M.K.; Moloney, G.M.; Knox, E.G.; Aburto, M.R.; Fülling, C.; Morley, S.J.; Clarke, G.; Schellekens, H.; Cryan, J.F. Short chain fatty acids: Microbial metabolites for gut-brain axis signalling. Mol. Cell. Endocrinol. 2022, 546, 111572. [Google Scholar] [CrossRef]
- Thibaut, M.M.; Bindels, L.B. Crosstalk between bile acid-activated receptors and microbiome in entero-hepatic inflammation. Trends Mol. Med. 2022, 28, 223–236. [Google Scholar] [CrossRef]
- Neumann, B.; Angstwurm, K.; Mergenthaler, P.; Kohler, S.; Schönenberger, S.; Bösel, J.; Neumann, U.; Vidal, A.; Huttner, H.B.; Gerner, S.T.; et al. Myasthenic crisis demanding mechanical ventilation: A multicenter analysis of 250 cases. Neurology 2020, 94, e299–e313. [Google Scholar] [CrossRef]
- Wang, H.; Li, X.; Zhang, Q.; Fu, C.; Jiang, W.; Xue, J.; Liu, S.; Meng, Q.; Ai, L.; Zhi, X.; et al. Autophagy in Disease Onset and Progression. Aging Dis. 2023. advance online publication. [Google Scholar] [CrossRef]
- Li, L.; Li, T.; Qu, X.; Sun, G.; Fu, Q.; Han, G. Stress/cell death pathways, neuroinflammation, and neuropathic pain. Immunol. Rev. 2023. advance online publication. [Google Scholar] [CrossRef] [PubMed]
- Qu, L.; Li, Y.; Liu, F.; Fang, Y.; He, J.; Ma, J.; Xu, T.; Wang, L.; Lei, P.; Dong, H.; et al. Microbiota-Gut-Brain Axis Dysregulation in Alzheimer’s Disease: Multi-Pathway Effects and Therapeutic Potential. Aging Dis. 2023. advance online publication. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, J.; Xing, Z.; Peng, C.; Li, D. Autophagy in Neuroinflammation: A Focus on Epigenetic Regulation. Aging Dis. 2023. advance online publication. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.W.; Zhu, X.X.; Tang, D.S.; Lu, J.H. Targeting autophagy in Alzheimer’s disease: Animal models and mechanisms. Zool. Res. 2023, 44, 1132–1145. [Google Scholar] [CrossRef] [PubMed]
- Nechushtai, L.; Frenkel, D.; Pinkas-Kramarski, R. Autophagy in Parkinson’s Disease. Biomolecules 2023, 13, 1435. [Google Scholar] [CrossRef] [PubMed]
- Azad, A.; Gökmen, Ü.R.; Uysal, H.; Köksoy, S.; Bilge, U.; Manguoğlu, A.E. Autophagy dysregulation plays a crucial role in regulatory T-cell loss and neuroinflammation in amyotrophic lateral sclerosis (ALS). Amyotroph. Lateral Scler. Front. Degener. 2023, 1–9, advance online publication. [Google Scholar] [CrossRef] [PubMed]
- Sil, S.; Niu, F.; Tom, E.; Liao, K.; Periyasamy, P.; Buch, S. Cocaine Mediated Neuroinflammation: Role of Dysregulated Autophagy in Pericytes. Mol. Neurobiol. 2019, 56, 3576–3590. [Google Scholar] [CrossRef]
- Singh, S.; Thangaraj, A.; Chivero, E.T.; Guo, M.L.; Periyasamy, P.; Buch, S. Role of Dysregulated Autophagy in HIV Tat, Cocaine, and cART Mediated NLRP3 Activation in Microglia. J. Neuroimmune Pharmacol. 2023, 18, 327–347. [Google Scholar] [CrossRef]
- Elkhoely, A.; Estfanous, R.S.; Alrobaian, M.; Borg, H.M.; Kabel, A.M. Repositioning itraconazole for amelioration of bleomycin-induced pulmonary fibrosis: Targeting HMGB1/TLR4 Axis, NLRP3 inflammasome/NF-κB signaling, and autophagy. Life Sci. 2023, 313, 121288. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, X.; Zhang, H.; Lin, X.; Chen, X.; Zhang, Y.; Lin, X.; Huang, L.; Zhuge, Q. Dimethyl itaconate inhibits LPS-induced microglia inflammation and inflammasome-mediated pyroptosis via inducing autophagy and regulating the Nrf-2/HO-1 signaling pathway. Mol. Med. Rep. 2021, 24, 672. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Huang, P.; Lai, F.; Zhang, T.; Guan, J.; Wan, H.; He, Y. Mechanisms of Ferritinophagy and Ferroptosis in Diseases. Mol. Neurobiol. 2023. advance online publication. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, H.; He, Q.; Zou, R.; Cai, J.; Zhang, L. Ferroptosis: Underlying mechanisms and involvement in neurodegenerative diseases. Apoptosis Int. J. Program. Cell Death 2023. advance online publication. [Google Scholar] [CrossRef] [PubMed]
- Chavoshinezhad, S.; Beirami, E.; Izadpanah, E.; Feligioni, M.; Hassanzadeh, K. Molecular mechanism and potential therapeutic targets of necroptosis and ferroptosis in Alzheimer’s disease. Biomed. Pharmacother. Biomed. Pharmacother. 2023, 168, 115656. [Google Scholar] [CrossRef] [PubMed]
- Dar, N.J.; John, U.; Bano, N.; Khan, S.; Bhat, S.A. Oxytosis/Ferroptosis in Neurodegeneration: The Underlying Role of Master Regulator Glutathione Peroxidase 4 (GPX4). Mol. Neurobiol. 2023. advance online publication. [Google Scholar] [CrossRef]
- Balistreri, C.R. Vascular ageing and the related complications in the brain: New insights on related mechanisms and their translational applications. Mech. Ageing Dev. 2021, 196, 111469. [Google Scholar] [CrossRef]
- Allegra, A.; Giarratana, R.M.; Scola, L.; Balistreri, C.R. The close link between the fetal programming imprinting and neurodegeneration in adulthood: The key role of “hemogenic endothelium” programming. Mech. Ageing Dev. 2021, 195, 111461. [Google Scholar] [CrossRef]
- Balistreri, C.R. Promising Strategies for Preserving Adult Endothelium Health and Reversing Its Dysfunction: From Liquid Biopsy to New Omics Technologies and Noninvasive Circulating Biomarkers. Int. J. Mol. Sci. 2022, 23, 7548. [Google Scholar] [CrossRef]
- Olivieri, F.; Pompilio, G.; Balistreri, C.R. Endothelial progenitor cells in ageing. Mech. Ageing Dev. 2016, 159, 1–3. [Google Scholar] [CrossRef]
- Ho, P.T.B.; Clark, I.M.; Le, L.T.T. MicroRNA-Based Diagnosis and Therapy. Int. J. Mol. Sci. 2022, 23, 7167. [Google Scholar] [CrossRef]
- Ferrante, M.; Conti, G.O. Environment and Neurodegenerative Diseases: An Update on miRNA Role. MicroRNA 2017, 6, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, A.; Gu, R.; Tong, Y.; Cheng, J. Role and regulatory mechanism of microRNA mediated neuroinflammation in neuronal system diseases. Front. Immunol. 2023, 14, 1238930. [Google Scholar] [CrossRef]
- Rastegar-Moghaddam, S.H.; Ebrahimzadeh-Bideskan, A.; Shahba, S.; Malvandi, A.M.; Mohammadipour, A. Roles of the miR-155 in Neuroinflammation and Neurological Disorders: A Potent Biological and Therapeutic Target. Cell. Mol. Neurobiol. 2023, 43, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Li, H.; Xie, W.; Wei, N.; Liu, M. MicroRNA-195 triggers neuroinflammation in Parkinson’s disease in a Rho-associated kinase 1-dependent manner. Mol. Med. Rep. 2019, 19, 5153–5161. [Google Scholar] [CrossRef]
- Zhang, J.; Hu, M.; Teng, Z.; Tang, Y.P.; Chen, C. Synaptic and cognitive improvements by inhibition of 2-AG metabolism are through upregulation of microRNA-188-3p in a mouse model of Alzheimer’s disease. J. Neurosci. 2014, 34, 14919–14933. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Tan, J.; Zhang, J. miR-137 attenuates Aβ-induced neurotoxicity through inactivation of NF-κB pathway by targeting TNFAIP1 in Neuro2a cells. Biochem. Biophys. Res. Commun. 2017, 490, 941–947. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Gong, Z.; Dong, J.; Bi, H.; Wang, B.; Du, K.; Zhang, C.; Chen, L. lncRNA XIST inhibition promotes M2 polarization of microglial and aggravates the spinal cord injury via regulating miR-124-3p/IRF1 axis. Heliyon 2023, 9, e17852. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Yang, H.; Ma, J.; Luo, S.; Chen, S.; Gu, Q. MicroRNA-30e regulates neuroinflammation in MPTP model of Parkinson’s disease by targeting Nlrp3. Hum. Cell 2018, 31, 106–115. [Google Scholar] [CrossRef]
- Singh, V.; Kushwaha, S.; Ansari, J.A.; Gangopadhyay, S.; Mishra, S.K.; Dey, R.K.; Giri, A.K.; Patnaik, S.; Ghosh, D. MicroRNA-129-5p-regulated microglial expression of the surface receptor CD200R1 controls neuroinflammation. J. Biol. Chem. 2022, 298, 101521. [Google Scholar] [CrossRef]
- Nasirishargh, A.; Kumar, P.; Ramasubramanian, L.; Clark, K.; Hao, D.; Lazar, S.V.; Wang, A. Exosomal microRNAs from mesenchymal stem/stromal cells: Biology and applications in neuroprotection. World J. Stem Cells 2021, 13, 776–794. [Google Scholar] [CrossRef]
- Tang, Y.; Bao, J.S.; Su, J.H.; Huang, W. MicroRNA-139 modulates Alzheimer’s-associated pathogenesis in SAMP8 mice by targeting cannabinoid receptor type 2. Genet. Mol. Res. GMR 2017, 16, gmr16019166. [Google Scholar] [CrossRef] [PubMed]
- Balistreri, C.R.; Candore, G.; Accardi, G.; Colonna-Romano, G.; Lio, D. NF-κB pathway activators as potential ageing biomarkers: Targets for new therapeutic strategies. Immun. Ageing 2013, 10, 24. [Google Scholar] [CrossRef] [PubMed]
- Balistreri, C.R.; Colonna-Romano, G.; Lio, D.; Candore, G.; Caruso, C. TLR4 polymorphisms and ageing: Implications for the pathophysiology of age-related diseases. J. Clin. Immunol. 2009, 29, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wise, L.; Fukuchi, K.I. TLR4 Cross-Talk With NLRP3 Inflammasome and Complement Signaling Pathways in Alzheimer’s Disease. Front. Immunol. 2020, 11, 724. [Google Scholar] [CrossRef] [PubMed]
- Gurram, P.C.; Manandhar, S.; Satarker, S.; Mudgal, J.; Arora, D.; Nampoothiri, M. Dopaminergic Signaling as a Plausible Modulator of Astrocytic Toll-Like Receptor 4: A Crosstalk between Neuroinflammation and Cognition. CNS Neurol. Disord. Drug Targets 2023, 22, 539–557. [Google Scholar] [CrossRef]
- Sivamaruthi, B.S.; Raghani, N.; Chorawala, M.; Bhattacharya, S.; Prajapati, B.G.; Elossaily, G.M.; Chaiyasut, C. NF-κB Pathway and Its Inhibitors: A Promising Frontier in the Management of Alzheimer’s Disease. Biomedicines 2023, 11, 2587. [Google Scholar] [CrossRef]
- Cui, D.; Chen, Y.; Ye, B.; Guo, W.; Wang, D.; He, J. Natural products for the treatment of neurodegenerative diseases. Phytomedicine Int. J. Phytother. Phytopharm. 2023, 121, 155101. [Google Scholar] [CrossRef]
- Decandia, D.; Gelfo, F.; Landolfo, E.; Balsamo, F.; Petrosini, L.; Cutuli, D. Dietary Protection against Cognitive Impairment, Neuroinflammation and Oxidative Stress in Alzheimer’s Disease Animal Models of Lipopolysaccharide-Induced Inflammation. Int. J. Mol. Sci. 2023, 24, 5921. [Google Scholar] [CrossRef]
- Balistreri, C.R. Anti-Inflamm-Ageing and/or Anti-Age-Related Disease Emerging Treatments: A Historical Alchemy or Revolutionary Effective Procedures? Mediat. Inflamm. 2018, 2018, 3705389. [Google Scholar] [CrossRef]
- Dias-Carvalho, A.; Sá, S.I.; Carvalho, F.; Fernandes, E.; Costa, V.M. Inflammation as common link to progressive neurological diseases. Arch. Toxicol. 2023. advance online publication. [Google Scholar] [CrossRef]
- Koole, L.; Martinez-Martinez, P.; Amelsvoort, T.V.; Evelo, C.T.; Ehrhart, F. Interactive neuroinflammation pathways and transcriptomics-based identification of drugs and chemical compounds for schizophrenia. World J. Biol. Psychiatry 2023, 1–14, advance online publication. [Google Scholar] [CrossRef] [PubMed]
- Lashgari, N.A.; Roudsari, N.M.; Shamsnia, H.S.; Shayan, M.; Momtaz, S.; Abdolghaffari, A.H. TLR/mTOR inflammatory signaling pathway: Novel insight for the treatment of schizophrenia. Can. J. Physiol. Pharmacol. 2023. advance online publication. [Google Scholar] [CrossRef]
- Sarapultsev, A.; Gusev, E.; Komelkova, M.; Utepova, I.; Luo, S.; Hu, D. JAK-STAT signaling in inflammation and stress-related diseases: Implications for therapeutic interventions. Mol. Biomed. 2023, 4, 40. [Google Scholar] [CrossRef] [PubMed]
- Islam, F.; Roy, S.; Zehravi, M.; Paul, S.; Sutradhar, H.; Yaidikar, L.; Kumar, B.R.; Dogiparthi, L.K.; Prema, S.; Nainu, F.; et al. Polyphenols Targeting MAP Kinase Signaling Pathway in Neurological Diseases: Understanding Molecular Mechanisms and Therapeutic Targets. Mol. Neurobiol. 2023. advance online publication. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Zhang, X.; Zhang, R.; Wang, Z.; Gan, J.; Gao, Q.; Yang, L.; Xu, P.; Jiang, X. Inflammatory signaling pathways in the treatment of Alzheimer’s disease with inhibitors, natural products and metabolites (Review). Int. J. Mol. Med. 2023, 52, 111. [Google Scholar] [CrossRef] [PubMed]
- Singh, L.; Bhatti, R. Signaling Pathways Involved in the Neuroprotective Effect of Osthole: Evidence and Mechanisms. Mol. Neurobiol. 2023. advance online publication. [Google Scholar] [CrossRef]
- Mohawk, J.A.; Green, C.B.; Takahashi, J.S. Central and peripheral circadian clocks in mammals. Annu. Rev. Neurosci. 2012, 35, 445–462. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Routh, B.N.; Gaudet, A.D.; Fonken, L.K. Circadian Regulation of the Neuroimmune Environment Across the Lifespan: From Brain Development to Aging. J. Biol. Rhythm. 2023, 38, 419–446. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Sachdeva, P.; Sarkar, J.; Izhaar, R. Circadian dysfunction and Alzheimer’s disease—An updated review. Aging Med. 2022, 6, 71–81. [Google Scholar] [CrossRef]
- Meng, D.; Yang, M.; Zhang, H.; Zhang, L.; Song, H.; Liu, Y.; Zeng, Y.; Yang, B.; Wang, X.; Chen, Y.; et al. Microglia activation mediates circadian rhythm disruption-induced cognitive impairment in mice. J. Neuroimmunol. 2023, 379, 578102. [Google Scholar] [CrossRef]
- Srinivasan, M.; Walker, C. Circadian Clock, Glucocorticoids and NF-κB Signaling in Neuroinflammation- Implicating Glucocorticoid Induced Leucine Zipper as a Molecular Link. ASN Neuro 2022, 14, 17590914221120190. [Google Scholar] [CrossRef]
- Aho, V.; Ollila, H.M.; Rantanen, V.; Kronholm, E.; Surakka, I.; van Leeuwen, W.M.; Lehto, M.; Matikainen, S.; Ripatti, S.; Härmä, M.; et al. Partial sleep restriction activates immune response-related gene expression pathways: Experimental and epidemiological studies in humans. PLoS ONE 2013, 8, e77184. [Google Scholar] [CrossRef] [PubMed]
- Taishi, P.; Chen, Z.; Obál, F., Jr.; Hansen, M.K.; Zhang, J.; Fang, J.; Krueger, J.M. Sleep-associated changes in interleukin-1beta mRNA in the brain. J. Interferon Cytokine Res. 1998, 18, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Szentirmai, É.; Kapás, L. Sleep and body temperature in TNFα knockout mice: The effects of sleep deprivation, β3-AR stimulation and exogenous TNFα. Brain Behav. Immun. 2019, 81, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.; Gibbons, C.M.; Dykstra-Aiello, C.; Ellingsen, R.; Koh, K.M.S.; Taishi, P.; Krueger, J.M. Interleukin-1 receptor accessory proteins are required for normal homeostatic responses to sleep deprivation. J. Appl. Physiol. 2019, 127, 770–780. [Google Scholar] [CrossRef]
- Choudhury, M.E.; Miyanishi, K.; Takeda, H.; Islam, A.; Matsuoka, N.; Kubo, M.; Matsumoto, S.; Kunieda, T.; Nomoto, M.; Yano, H.; et al. Phagocytic elimination of synapses by microglia during sleep. Glia 2020, 68, 44–59. [Google Scholar] [CrossRef] [PubMed]
- Klarica, M.; Radoš, M.; Orešković, D. The Movement of Cerebrospinal Fluid and Its Relationship with Substances Behavior in Cerebrospinal and Interstitial Fluid. Neuroscience 2019, 414, 28–48. [Google Scholar] [CrossRef] [PubMed]
- Hand, L.E.; Gray, K.J.; Dickson, S.H.; Simpkins, D.A.; Ray, D.W.; Konkel, J.E.; Hepworth, M.R.; Gibbs, J.E. Regulatory T cells confer a circadian signature on inflammatory arthritis. Nat. Commun. 2020, 11, 1658. [Google Scholar] [CrossRef]
- García-García, A.; Korn, C.; García-Fernández, M.; Domingues, O.; Villadiego, J.; Martín-Pérez, D.; Isern, J.; Bejarano-García, J.A.; Zimmer, J.; Pérez-Simón, J.A.; et al. Dual cholinergic signals regulate daily migration of hematopoietic stem cells and leukocytes. Blood 2019, 133, 224–236. [Google Scholar] [CrossRef]
- Adrover, J.M.; Del Fresno, C.; Crainiciuc, G.; Cuartero, M.I.; Casanova-Acebes, M.; Weiss, L.A.; Huerga-Encabo, H.; Silvestre-Roig, C.; Rossaint, J.; Cossío, I.; et al. A Neutrophil Timer Coordinates Immune Defense and Vascular Protection. Immunity 2019, 50, 390–402.e10. [Google Scholar] [CrossRef]
- O’Siorain, J.R.; Curtis, A.M. Circadian Control of Redox Reactions in the Macrophage Inflammatory Response. Antioxid. Redox Signal. 2022, 37, 664–678. [Google Scholar] [CrossRef]
- Blacher, E.; Tsai, C.; Litichevskiy, L.; Shipony, Z.; Iweka, C.A.; Schneider, K.M.; Chuluun, B.; Heller, H.C.; Menon, V.; Thaiss, C.A.; et al. Aging disrupts circadian gene regulation and function in macrophages. Nat. Immunol. 2022, 23, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Jin, X.; Xu, F.; Wang, S.; Liu, L.; Li, X.; Lin, H.; Du, M. Circadian rhythm-associated Rev-erbα modulates polarization of decidual macrophage via the PI3K/Akt signaling pathway. Am. J. Reprod. Immunol. 2021, 86, e13436. [Google Scholar] [CrossRef] [PubMed]
- Nobis, C.C.; Dubeau Laramée, G.; Kervezee, L.; De Sousa, D.M.; Labrecque, N.; Cermakian, N. The circadian clock of CD8 T cells modulates their early response to vaccination and the rhythmicity of related signaling pathways. Proc. Natl. Acad. Sci. USA 2019, 116, 20077–20086. [Google Scholar] [CrossRef] [PubMed]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Monastero, R.; Caruso, C.; Vasto, S. Alzheimer’s disease and infections, where we stand and where we go. Immun. Ageing 2014, 11, 26. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Proctor, M.J.; McMillan, D.C.; Horgan, P.G.; Fletcher, C.D.; Talwar, D.; Morrison, D.S. Systemic inflammation predicts all-cause mortality: A glasgow inflammation outcome study. PLoS ONE 2015, 10, e0116206. [Google Scholar] [CrossRef]
- Maggio, M.; Guralnik, J.M.; Longo, D.L.; Ferrucci, L. Interleukin-6 in aging and chronic disease: A magnificent pathway. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2006, 61, 575–584. [Google Scholar] [CrossRef]
- Feghali, C.A.; Wright, T.M. Cytokines in acute and chronic inflammation. Front. Biosci. A J. Virtual Libr. 1997, 2, d12–d26. [Google Scholar] [CrossRef]
- Parra-Torres, V.; Melgar-Rodríguez, S.; Muñoz-Manríquez, C.; Sanhueza, B.; Cafferata, E.A.; Paula-Lima, A.C.; Díaz-Zúñiga, J. Periodontal bacteria in the brain-Implication for Alzheimer’s disease: A systematic review. Oral Dis. 2023, 29, 21–28. [Google Scholar] [CrossRef]
- Liu, S.; Dashper, S.G.; Zhao, R. Association Between Oral Bacteria and Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Alzheimer’s Dis. JAD 2023, 91, 129–150. [Google Scholar] [CrossRef] [PubMed]
- Beydoun, M.A.; Beydoun, H.A.; Hossain, S.; El-Hajj, Z.W.; Weiss, J.; Zonderman, A.B. Clinical and Bacterial Markers of Periodontitis and Their Association with Incident All-Cause and Alzheimer’s Disease Dementia in a Large National Survey. J. Alzheimer’s Dis. JAD 2020, 75, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Larvin, H.; Gao, C.; Kang, J.; Aggarwal, V.R.; Pavitt, S.; Wu, J. The impact of study factors in the association of periodontal disease and cognitive disorders: Systematic review and meta-analysis. Age Ageing 2023, 52, afad015. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jin, Y.; Li, K.; Qiu, H.; Jiang, Z.; Zhu, J.; Chen, S.; Xie, W.; Chen, G.; Yang, D. Is There an Association Between Parkinson’s Disease and Periodontitis? A Systematic Review and Meta-Analysis. J. Park. Dis. 2023, 13, 1107–1125. [Google Scholar] [CrossRef]
- Yazdanpanahi, N.; Etemadifar, M.; Kardi, M.T.; Shams, E.; Shahbazi, S. Investigation of ERG Gene Expression in Iranian Patients with Multiple Sclerosis. Immunol. Investig. 2018, 47, 351–359. [Google Scholar] [CrossRef]
- Pisano, C.; Terriaca, S.; Scioli, M.G.; Nardi, P.; Altieri, C.; Orlandi, A.; Ruvolo, G.; Balistreri, C.R. The Endothelial Transcription Factor ERG Mediates a Differential Role in the Aneurysmatic Ascending Aorta with Bicuspid or Tricuspid Aorta Valve: A Preliminary Study. Int. J. Mol. Sci. 2022, 23, 10848. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balistreri, C.R.; Monastero, R. Neuroinflammation and Neurodegenerative Diseases: How Much Do We Still Not Know? Brain Sci. 2024, 14, 19. https://doi.org/10.3390/brainsci14010019
Balistreri CR, Monastero R. Neuroinflammation and Neurodegenerative Diseases: How Much Do We Still Not Know? Brain Sciences. 2024; 14(1):19. https://doi.org/10.3390/brainsci14010019
Chicago/Turabian StyleBalistreri, Carmela Rita, and Roberto Monastero. 2024. "Neuroinflammation and Neurodegenerative Diseases: How Much Do We Still Not Know?" Brain Sciences 14, no. 1: 19. https://doi.org/10.3390/brainsci14010019
APA StyleBalistreri, C. R., & Monastero, R. (2024). Neuroinflammation and Neurodegenerative Diseases: How Much Do We Still Not Know? Brain Sciences, 14(1), 19. https://doi.org/10.3390/brainsci14010019