
The Role of Brain-Derived Neurotrophic Factor (BDNF) in Diagnosis and Treatment of Epilepsy, Depression, Schizophrenia, Anorexia Nervosa and Alzheimer’s Disease as Highly Drug-Resistant Diseases: A Narrative Review

 ,
,  , ,
, , {kind=link}
{kind=link}
Abstract
1. Introduction
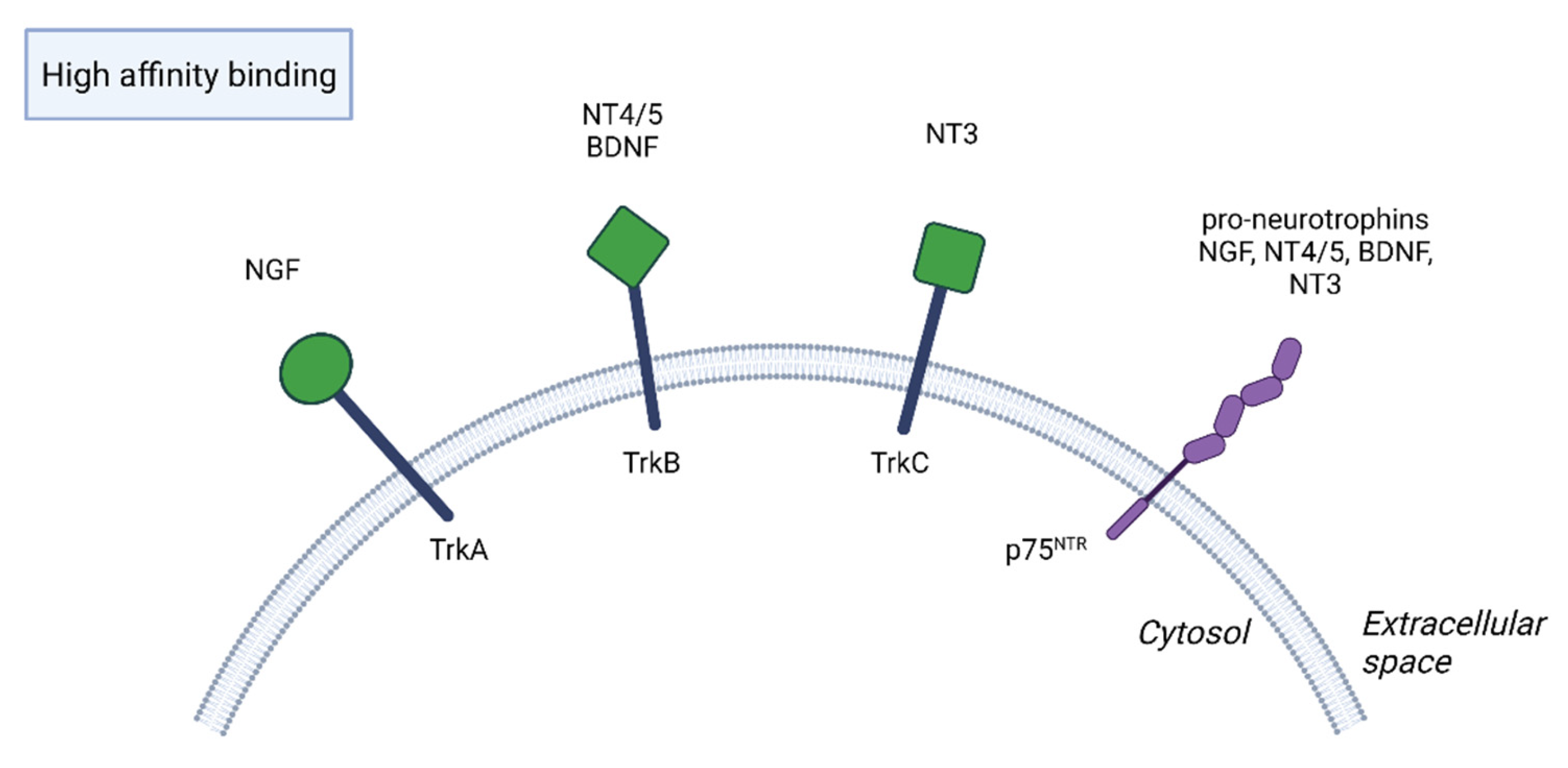
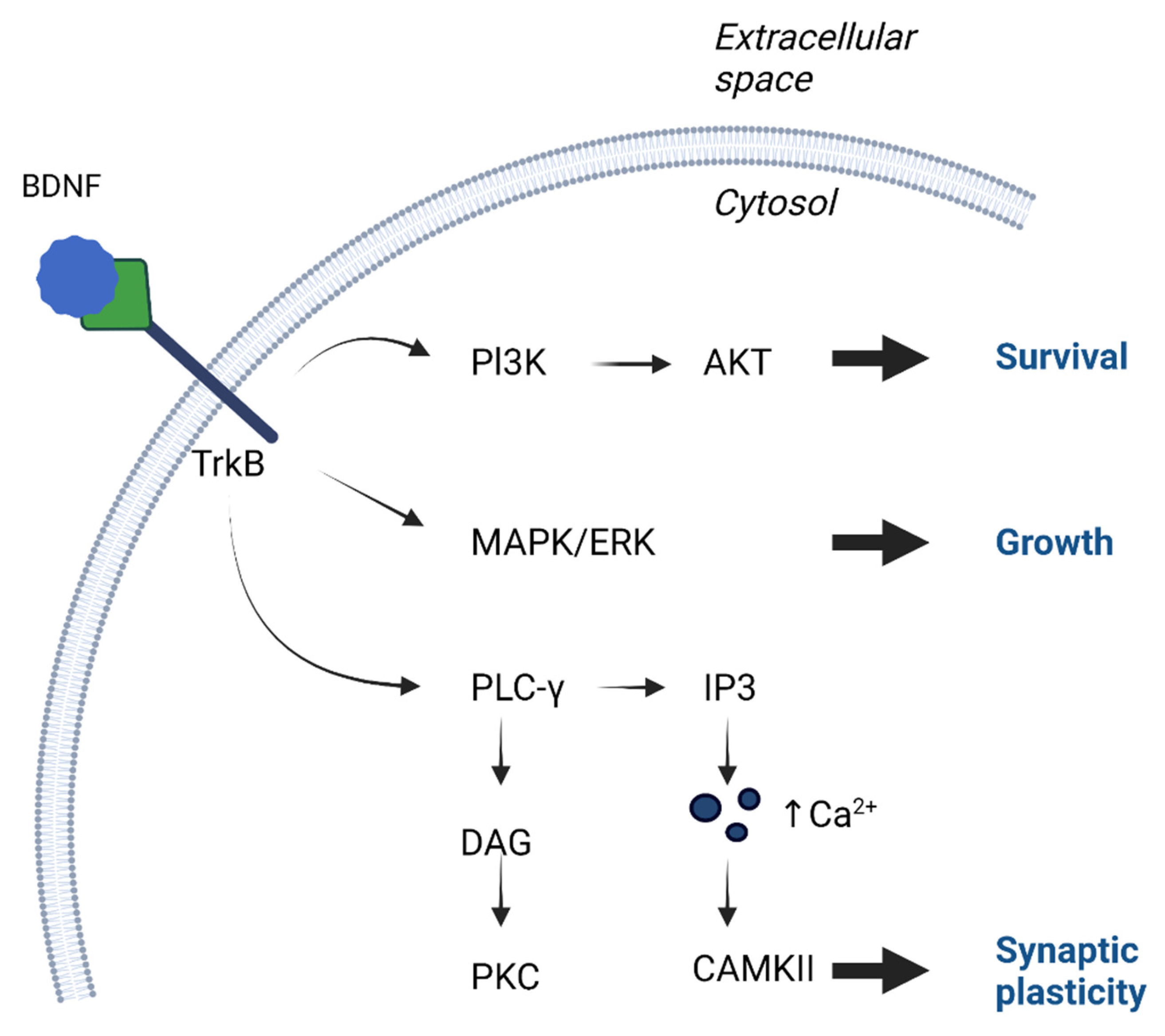
2. Function of BDNF in the Nervous System
3. Polymorphisms of BDNF Gene and Receptors
4. Clinical Implications
4.1. Epilepsy
4.2. Depression
4.3. Hunger Control Disorders
4.4. Schizophrenia
4.5. Alzheimer’s Disease
4.6. Therapeutic Properties of BDNF
5. Conclusions
6. Methodology
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Binder, D.K.; Scharfman, H.E. Mini Review. Growth Factors 2004, 22, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Iughetti, L.; Lucaccioni, L.; Fugetto, F.; Predieri, B.; Berardi, A.; Ferrari, F. Brain-derived neurotrophic factor and epilepsy: A systematic review. Neuropeptides 2018, 72, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Giacobbo, B.L.; Doorduin, J.; Klein, H.C.; Dierckx, R.A.J.O.; Bromberg, E.; de Vries, E.F.J. Brain-Derived Neurotrophic Factor in Brain Disorders: Focus on Neuroinflammation. Mol. Neurobiol. 2019, 56, 3295–3312. [Google Scholar] [CrossRef] [PubMed]
- Dechant, G.; Neumann, H. Neurotrophins. Adv. Exp. Med. Biol. 2002, 513, 303–334. [Google Scholar] [CrossRef]
- Miranda-Lourenço, C.; Ribeiro-Rodrigues, L.; Fonseca-Gomes, J.; Tanqueiro, S.R.; Belo, R.F.; Ferreira, C.B.; Rei, N.; Ferreira-Manso, M.; de Almeida-Borlido, C.; Costa-Coelho, T.; et al. Challenges of BDNF-based therapies: From common to rare diseases. Pharmacol. Res. 2020, 162, 105281. [Google Scholar] [CrossRef] [PubMed]
- Bathina, S.; Das, U.N. Brain-derived neurotrophic factor and its clinical implications. Arch. Med. Sci. 2015, 11, 1164–1178. [Google Scholar] [CrossRef]
- Yang, T.; Nie, Z.; Shu, H.; Kuang, Y.; Chen, X.; Cheng, J.; Yu, S.; Liu, H. The Role of BDNF on Neural Plasticity in Depression. Front. Cell. Neurosci. 2020, 14, 82. [Google Scholar] [CrossRef]
- Ghassabian, A.; Sundaram, R.; Chahal, N.; McLain, A.C.; Bell, E.; Lawrence, D.A.; Yeung, E.H. Determinants of neonatal brain-derived neurotrophic factor and association with child development. Dev. Psychopathol. 2017, 29, 1499–1511. [Google Scholar] [CrossRef]
- Favalli, G.; Li, J.; Belmonte-De-Abreu, P.; Wong, A.H.; Daskalakis, Z.J. The role of BDNF in the pathophysiology and treatment of schizophrenia. J. Psychiatr. Res. 2012, 46, 1–11. [Google Scholar] [CrossRef]
- Xia, M.; Zhao, T.; Wang, X.; Li, Y.; Li, Y.; Zheng, T.; Li, J.; Feng, Y.; Wei, Y.; Sun, P. Brain-Derived Neurotrophic Factor and Its Applications through Nanosystem Delivery. Iran. J. Pharm. Res. 2021, 20, 137–151. [Google Scholar] [CrossRef]
- Endlich, N.; Lange, T.; Kuhn, J.; Klemm, P.; Kotb, A.M.; Siegerist, F.; Kindt, F.; Lindenmeyer, M.T.; Cohen, C.D.; Kuss, A.W.; et al. BDNF: mRNA expression in urine cells of patients with chronic kidney disease and its role in kidney function. J. Cell. Mol. Med. 2018, 22, 5265–5277. [Google Scholar] [CrossRef]
- Prakash, Y.; Martin, R.J. Brain-derived neurotrophic factor in the airways. Pharmacol. Ther. 2014, 143, 74–86. [Google Scholar] [CrossRef]
- Urbina-Varela, R.; Soto-Espinoza, M.I.; Vargas, R.; Quiñones, L.; del Campo, A. Influence of BDNF Genetic Polymorphisms in the Pathophysiology of Aging-Related Diseases. Aging Dis. 2020, 11, 1513. [Google Scholar] [CrossRef]
- Miranda, M.; Morici, J.F.; Zanoni, M.B.; Bekinschtein, P. Brain-Derived Neurotrophic Factor: A Key Molecule for Memory in the Healthy and the Pathological Brain. Front. Cell. Neurosci. 2019, 13, 363. [Google Scholar] [CrossRef]
- Autry, A.E.; Monteggia, L.M. Brain-Derived Neurotrophic Factor and Neuropsychiatric Disorders. Pharmacol. Rev. 2012, 64, 238–258. [Google Scholar] [CrossRef]
- Numakawa, T.; Odaka, H.; Adachi, N. Actions of Brain-Derived Neurotrophin Factor in the Neurogenesis and Neuronal Function, and Its Involvement in the Pathophysiology of Brain Diseases. Int. J. Mol. Sci. 2018, 19, 3650. [Google Scholar] [CrossRef]
- Tao, Y.S.; Piao, S.G.; Jin, Y.S.; Jin, J.Z.; Zheng, H.L.; Zhao, H.Y.; Lim, S.W.; Yang, C.W.; Li, C. Expression of brain-derived neurotrophic factor in kidneys from normal and cyclosporine-treated rats. BMC Nephrol. 2018, 19, 1–12. [Google Scholar] [CrossRef]
- Citri, A.; Malenka, R.C. Synaptic Plasticity: Multiple Forms, Functions, and Mechanisms. Neuropsychopharmacology 2008, 33, 18–41. [Google Scholar] [CrossRef]
- Serra-Millàs, M. Are the changes in the peripheral brain-derived neurotrophic factor levels due to platelet activation? World J. Psychiatry 2016, 6, 84–101. [Google Scholar] [CrossRef]
- Castrén, E.; Monteggia, L.M. Brain-Derived Neurotrophic Factor Signaling in Depression and Antidepressant Action. Biol. Psychiatry 2021, 90, 128–136. [Google Scholar] [CrossRef]
- Brandys, M.K.; Kas, M.J.H.; van Elburg, A.A.; Campbell, I.C.; Adan, R.A.H. A meta-analysis of circulating BDNF concentrations in anorexia nervosa. World J. Biol. Psychiatry 2011, 12, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Bahr, R.; Lopez, A.; Rey, J.A. Intranasal Esketamine (SpravatoTM) for Use in Treatment-Resistant Depression In Conjunction With an Oral Antidepressant. Pharm. Ther. 2019, 44, 340–375. [Google Scholar]
- Autry, A.E.; Adachi, M.; Nosyreva, E.; Na, E.S.; Los, M.F.; Cheng, P.-F.; Kavalali, E.T.; Monteggia, L.M. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 2011, 475, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Ignácio, Z.M.; Réus, G.Z.; Arent, C.O.; Abelaira, H.M.; Pitcher, M.R.; Quevedo, J. New perspectives on the involvement of mTOR in depression as well as in the action of antidepressant drugs. Br. J. Clin. Pharmacol. 2016, 82, 1280–1290. [Google Scholar] [CrossRef] [PubMed]
- Joanna, O.; Agata, M.-B.; Wojciech, R.; Elżbieta, Ś.; Małgorzata, S.; Katarzyna, Z. Serum Brain-Derived Neutrophic Factor in Girls with Anorexia Nervosa and Obesity. Pediatr. Endocrinol. 2018, 62, 27–38. [Google Scholar]
- Glud, M.; Christiansen, T.; Larsen, L.H.; Richelsen, B.; Bruun, J.M. Changes in Circulating BDNF in relation to Sex, Diet, and Exercise: A 12-Week Randomized Controlled Study in Overweight and Obese Participants. J. Obes. 2019, 2019, 4537274. [Google Scholar] [CrossRef]
- Altman, J.; DAS, G.D. Post-Natal Origin of Microneurones in the Rat Brain. Nature 1965, 207, 953–956. [Google Scholar] [CrossRef]
- Respondek, M.; Buszman, E. Regulation of neurogenesis: Factors affecting of new neurons formation in adult mammals brain. Postep. Hig. I Med. Dosw. 2015, 69, 1451–1461. [Google Scholar]
- Martínez-Levy, G.A.; Rocha, L.; Rodríguez-Pineda, F.; Alonso-Vanegas, M.A.; Nani, A.; Buentello-García, R.M.; Briones-Velasco, M.; San-Juan, D.; Cienfuegos, J.; Cruz-Fuentes, C.S. Increased Expression of Brain-Derived Neurotrophic Factor Transcripts I and VI, cAMP Response Element Binding, and Glucocorticoid Receptor in the Cortex of Patients with Temporal Lobe Epilepsy. Mol. Neurobiol. 2017, 55, 1–11. [Google Scholar] [CrossRef]
- Pencea, V.; Bingaman, K.D.; Wiegand, S.J.; Luskin, M.B. Infusion of Brain-Derived Neurotrophic Factor into the Lateral Ventricle of the Adult Rat Leads to New Neurons in the Parenchyma of the Striatum, Septum, Thalamus, and Hypothalamus. J. Neurosci. 2001, 21, 6706–6717. [Google Scholar] [CrossRef]
- Lin, T.W.; Harward, S.C.; Huang, Y.Z.; McNamara, J.O. Targeting BDNF/TrkB pathways for preventing or suppressing epilepsy. Neuropharmacology 2020, 167, 107734. [Google Scholar] [CrossRef]
- Porcher, C.; Medina, I.; Gaiarsa, J.-L. Mechanism of BDNF Modulation in GABAergic Synaptic Transmission in Healthy and Disease Brains. Front. Cell. Neurosci. 2018, 12, 273. [Google Scholar] [CrossRef]
- Terracciano, A.; Piras, M.G.; Lobina, M.; Mulas, A.; Meirelles, O.; Sutin, A.R.; Chan, W.; Sanna, S.; Uda, M.; Crisponi, L.; et al. Genetics of serum BDNF: Meta-analysis of the Val66Met and genome-wide association study. World J. Biol. Psychiatry 2013, 14, 583–589. [Google Scholar] [CrossRef]
- Verhagen, M.; van der Meij, A.; Deurzen, P.A.M.V.; Janzing, J.G.E.; Arias-Vásquez, A.; Buitelaar, J.K.; Franke, B. Meta-analysis of the BDNF Val66Met polymorphism in major depressive disorder: Effects of gender and ethnicity. Mol. Psychiatry 2010, 15, 260–271. [Google Scholar] [CrossRef]
- Zhao, M.; Chen, L.; Yang, J.; Han, D.; Fang, D.; Qiu, X.; Yang, X.; Qiao, Z.; Ma, J.; Wang, L.; et al. BDNF Val66Met polymorphism, life stress and depression: A meta-analysis of gene-environment interaction. J. Affect. Disord. 2018, 227, 226–235. [Google Scholar] [CrossRef]
- Brandys, M.K.; Kas, M.; Van Elburg, A.A.; Ophoff, R.; Landt, M.C.T.S.-O.; Middeldorp, C.M.; Boomsma, R.I.; Van Furth, E.F.; Slagboom, P.E.; Adan, R.A.H. The Val66Met polymorphism of the BDNF gene in anorexia nervosa: New data and a meta-analysis. World J. Biol. Psychiatry 2013, 14, 441–451. [Google Scholar] [CrossRef]
- Lim, Y.Y.; Maruff, P.; Barthélemy, N.R.; Goate, A.; Hassenstab, J.; Sato, C.; Fagan, A.M.; Benzinger, T.L.S.; Xiong, C.; Cruchaga, C.; et al. Association of BDNF Val66Met With Tau Hyperphosphorylation and Cognition in Dominantly Inherited Alzheimer Disease. JAMA Neurol. 2022, 79, 261. [Google Scholar] [CrossRef]
- Li, M.; Chang, H.; Xiao, X. BDNF Val66Met polymorphism and bipolar disorder in European populations: A risk association in case-control, family-based and GWAS studies. Neurosci. Biobehav. Rev. 2016, 68, 218–233. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, J.; Guo, Y.; Dong, G.; Zou, W.; Chen, Z. Association between BDNF G196A (Val66Met) polymorphism and cognitive impairment in patients with Parkinson's disease: A meta-analysis. Braz. J. Med. Biol. Res. 2019, 52, e8443. [Google Scholar] [CrossRef]
- Kishi, T.; Yoshimura, R.; Ikuta, T.; Iwata, N. Brain-Derived Neurotrophic Factor and Major Depressive Disorder: Evidence from Meta-Analyses. Front. Psychiatry 2017, 8, 308. [Google Scholar] [CrossRef]
- Nowroozi, A.; Salehi, M.A.; Mohammadi, S. Brain-derived neurotrophic factor in patients with epilepsy: A systematic review and meta-analysis. Epilepsy Res. 2021, 178, 106794. [Google Scholar] [CrossRef] [PubMed]
- Ho, B.-C.; Andreasen, N.C.; Dawson, J.D.; Wassink, T.H. Association Between Brain-Derived Neurotrophic Factor Val66Met Gene Polymorphism and Progressive Brain Volume Changes in Schizophrenia. Am. J. Psychiatry 2007, 164, 1890–1899. [Google Scholar] [CrossRef] [PubMed]
- Beghi, E. The Epidemiology of Epilepsy. Neuroepidemiology 2020, 54, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Dziema, H.; Lee, K.H.; Choi, Y.-S.; Obrietan, K. CRE-mediated transcription and COX-2 expression in the pilocarpine model of status epilepticus. Neurobiol. Dis. 2007, 25, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qi, J.-S.; Kong, S.; Sun, Y.; Fan, J.; Jiang, M.; Chen, G. BDNF-TrkB signaling pathway mediates the induction of epileptiform activity induced by a convulsant drug cyclothiazide. Neuropharmacology 2009, 57, 49–59. [Google Scholar] [CrossRef]
- Li, L.; Deng, J.; Liu, C.; Luo, H.; Guan, Y.; Zhou, J.; Qi, X.; Li, T.; Xu, Z.D.; Luan, G.-M. Altered expression of neuropeptide Y receptors caused by focal cortical dysplasia in human intractable epilepsy. Oncotarget 2016, 7, 15329–15338. [Google Scholar] [CrossRef]
- Gall, C.M.; Isackson, P.J. Limbic Seizures Increase Neuronal Production of Messenger RNA for Nerve Growth Factor. Science 1989, 245, 758–761. [Google Scholar] [CrossRef]
- LaFrance, W.C.; Leaver, K.; Stopa, E.G.; Papandonatos, G.D.; Blum, A.S. Decreased serum BDNF levels in patients with epileptic and psychogenic nonepileptic seizures. Neurology 2010, 75, 1285–1291. [Google Scholar] [CrossRef]
- Bittigau, P.; Sifringer, M.; Genz, K.; Reith, E.; Pospischil, D.; Govindarajalu, S.; Dzietko, M.; Pesditschek, S.; Mai, I.; Dikranian, K.; et al. Antiepileptic drugs and apoptotic neurodegeneration in the developing brain. Proc. Natl. Acad. Sci. USA 2002, 99, 15089–15094. [Google Scholar] [CrossRef]
- Aungst, S.; England, P.M.; Thompson, S.M. Critical role of trkB receptors in reactive axonal sprouting and hyperexcitability after axonal injury. J. Neurophysiol. 2013, 109, 813–824. [Google Scholar] [CrossRef]
- Gu, F.; Parada, I.; Yang, T.; Longo, F.M.; Prince, D.A. Partial TrkB receptor activation suppresses cortical epileptogenesis through actions on parvalbumin interneurons. Neurobiol. Dis. 2018, 113, 45–58. [Google Scholar] [CrossRef]
- Falcicchia, C.; Paolone, G.; Emerich, D.F.; Lovisari, F.; Bell, W.J.; Fradet, T.; Wahlberg, L.U.; Simonato, M. Seizure-Suppressant and Neuroprotective Effects of Encapsulated BDNF-Producing Cells in a Rat Model of Temporal Lobe Epilepsy. Mol. Ther. Methods Clin. Dev. 2018, 9, 211–224. [Google Scholar] [CrossRef]
- Wang, X.; Hu, Z.; Zhong, K. The Role of Brain-Derived Neurotrophic Factor in Epileptogenesis: An Update. Front. Pharmacol. 2021, 12, 3405. [Google Scholar] [CrossRef]
- Gałecki, P.; Bliźniewska-Kowalska, K. Treatment-resistant depression—recommendations of the National Consultant in the field of psychiatry. Psychiatr. Polska 2021, 55, 7–21. [Google Scholar] [CrossRef]
- Zhao, X.; Li, Y.; Tian, Q.; Zhu, B.; Zhao, Z. Repetitive transcranial magnetic stimulation increases serum brain-derived neurotrophic factor and decreases interleukin-1β and tumor necrosis factor-α in elderly patients with refractory depression. J. Int. Med. Res. 2019, 47, 1848–1855. [Google Scholar] [CrossRef]
- Leiknes, K.A.; Cooke, M.J.; Schweder, L.J.-V.; Harboe, I.; Høie, B. Electroconvulsive therapy during pregnancy: A systematic review of case studies. Arch. Women's Ment. Health 2015, 18, 1–39. [Google Scholar] [CrossRef]
- Zanos, P.; Gould, T.D. Mechanisms of ketamine action as an antidepressant. Mol. Psychiatry 2018, 23, 801–811. [Google Scholar] [CrossRef]
- Gkesoglou, T.; Bargiota, S.I.; Iordanidou, E.; Vasiliadis, M.; Bozikas, V.-P.; Agorastos, A. Prognostic Significance of Blood-Based Baseline Biomarkers in Treatment-Resistant Depression: A Literature Review of Available Studies on Treatment Response. Brain Sci. 2022, 12, 940. [Google Scholar] [CrossRef]
- Daly, E.J.; Trivedi, M.H.; Janik, A.; Li, H.; Zhang, Y.; Li, X.; Lane, R.; Lim, P.; Duca, A.R.; Hough, D.; et al. Efficacy of Esketamine Nasal Spray Plus Oral Antidepressant Treatment for Relapse Prevention in Patients With Treatment-Resistant Depression: A Randomized Clinical Trial. JAMA Psychiatry 2019, 76, 893–903. [Google Scholar] [CrossRef]
- Chan, S.W.; Harmer, C.J.; Norbury, R.; O’Sullivan, U.; Goodwin, G.M.; Portella, M.J. Hippocampal volume in vulnerability and resilience to depression. J. Affect. Disord. 2016, 189, 199–202. [Google Scholar] [CrossRef]
- Sheline, Y.I. Depression and the Hippocampus: Cause or Effect? Biol. Psychiatry 2011, 70, 308–309. [Google Scholar] [CrossRef] [PubMed]
- Molendijk, M.; Spinhoven, P.; Polak, M.; Bus, B.A.A.; Penninx, B.W.J.H.; Elzinga, B.M. Serum BDNF concentrations as peripheral manifestations of depression: Evidence from a systematic review and meta-analyses on 179 associations (N=9484). Mol. Psychiatry 2014, 19, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Arosio, B.; Guerini, F.R.; Voshaar, R.C.O.; Aprahamian, I. Blood Brain-Derived Neurotrophic Factor (BDNF) and Major Depression: Do We Have a Translational Perspective? Front. Behav. Neurosci. 2021, 15, 626906. [Google Scholar] [CrossRef] [PubMed]
- Duman, R.S.; Heninger, G.R.; Nestler, E.J. A Molecular and Cellular Theory of Depression. Arch. Gen. Psychiatry 1997, 54, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Groves, J.O. Is it time to reassess the BDNF hypothesis of depression? Mol. Psychiatry 2007, 12, 1079–1088. [Google Scholar] [CrossRef]
- Chen, B.; Dowlatshahi, D.; MacQueen, G.M.; Wang, J.-F.; Young, L. Increased hippocampal bdnf immunoreactivity in subjects treated with antidepressant medication. Biol. Psychiatry 2001, 50, 260–265. [Google Scholar] [CrossRef]
- Sen, S.; Duman, R.; Sanacora, G. Serum Brain-Derived Neurotrophic Factor, Depression, and Antidepressant Medications: Meta-Analyses and Implications. Biol. Psychiatry 2008, 64, 527–532. [Google Scholar] [CrossRef]
- Hieronymus, F.; Nilsson, S.; Eriksson, E. A mega-analysis of fixed-dose trials reveals dose-dependency and a rapid onset of action for the antidepressant effect of three selective serotonin reuptake inhibitors. Transl. Psychiatry 2016, 6, e834. [Google Scholar] [CrossRef]
- Brunoni, A.R.; Lopes, M.; Fregni, F. A systematic review and meta-analysis of clinical studies on major depression and BDNF levels: Implications for the role of neuroplasticity in depression. Int. J. Neuropsychopharmacol. 2008, 11, 1169–1180. [Google Scholar] [CrossRef]
- Bocchio-Chiavetto, L.; Bagnardi, V.; Zanardini, R.; Molteni, R.; Nielsen, M.G.; Placentino, A.; Giovannini, C.; Rillosi, L.; Ventriglia, M.; Riva, M.A.; et al. Serum and plasma BDNF levels in major depression: A replication study and meta-analyses. World J. Biol. Psychiatry 2010, 11, 763–773. [Google Scholar] [CrossRef]
- Niitsu, T.; Fabbri, C.; Bentini, F.; Serretti, A. Pharmacogenetics in major depression: A comprehensive meta-analysis. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2013, 45, 183–194. [Google Scholar] [CrossRef]
- Jelen, L.A.; Stone, J.M. Ketamine for depression. Int. Rev. Psychiatry 2021, 33, 207–228. [Google Scholar] [CrossRef]
- Pan, W.; Banks, W.A.; Fasold, M.B.; Bluth, J.; Kastin, A.J. Transport of brain-derived neurotrophic factor across the blood–brain barrier. Neuropharmacology 1998, 37, 1553–1561. [Google Scholar] [CrossRef]
- Porter, G.A.; O’Connor, J.C. Brain-derived neurotrophic factor and inflammation in depression: Pathogenic partners in crime? World J. Psychiatry 2022, 12, 77–97. [Google Scholar] [CrossRef]
- Zhang, J.-C.; Wu, J.; Fujita, Y.; Yao, W.; Ren, Q.; Yang, C.; Li, S.-X.; Shirayama, Y.; Hashimoto, K. Antidepressant Effects of TrkB Ligands on Depression-Like Behavior and Dendritic Changes in Mice After Inflammation. Int. J. Neuropsychopharmacol. 2015, 18, 1–12. [Google Scholar] [CrossRef]
- Sorri, A.; Järventausta, K.; Kampman, O.; Lehtimäki, K.; Björkqvist, M.; Tuohimaa, K.; Hämäläinen, M.; Moilanen, E.; Leinonen, E. Effect of electroconvulsive therapy on brain-derived neurotrophic factor levels in patients with major depressive disorder. Brain Behav. 2018, 8, e01101. [Google Scholar] [CrossRef]
- Pelosof, R.; dos Santos, L.A.; Farhat, L.C.; Gattaz, W.F.; Talib, L.; Brunoni, A.R. BDNF blood levels after electroconvulsive therapy in patients with mood disorders: An updated systematic review and meta-analysis. World J. Biol. Psychiatry 2022, 15, 411–418. [Google Scholar] [CrossRef]
- Brunoni, A.R.; Baeken, C.; Machado-Vieira, R.; Gattaz, W.F.; Vanderhasselt, M.-A. BDNF blood levels after electroconvulsive therapy in patients with mood disorders: A systematic review and meta-analysis. World J. Biol. Psychiatry 2014, 15, 411–418. [Google Scholar] [CrossRef]
- van Eeden, A.E.; van Hoeken, D.; Hoek, H.W. Incidence, prevalence and mortality of anorexia nervosa and bulimia nervosa. Curr. Opin. Psychiatry 2021, 34, 515–524. [Google Scholar] [CrossRef]
- Berends, T.; Van Meijel, B.; Nugteren, W.; Deen, M.; Danner, U.N.; Hoek, H.W.; Van Elburg, A.A. Rate, timing and predictors of relapse in patients with anorexia nervosa following a relapse prevention program: A cohort study. BMC Psychiatry 2016, 16, 316. [Google Scholar] [CrossRef]
- Borsdorf, B.; Dahmen, B.; Buehren, K.; Dempfle, A.; Egberts, K.; Ehrlich, S.; Fleischhaker, C.; Konrad, K.; Schwarte, R.; Timmesfeld, N.; et al. BDNF levels in adolescent patients with anorexia nervosa increase continuously to supranormal levels 2.5 years after first hospitalization. J. Psychiatry Neurosci. 2021, 46, E568–E578. [Google Scholar] [CrossRef] [PubMed]
- Chooi, Y.C.; Ding, C.; Magkos, F. The epidemiology of obesity. Metabolism 2019, 92, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Kansra, A.R.; Lakkunarajah, S.; Jay, M.S. Childhood and Adolescent Obesity: A Review. Front. Pediatr. 2021, 8, 581461. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, C.; Izquierdo, A.; Slattery, M.; Becker, K.R.; Plessow, F.; Thomas, J.J.; Eddy, K.T.; Lawson, E.A.; Misra, M. Changes in appetite-regulating hormones following food intake are associated with changes in reported appetite and a measure of hedonic eating in girls and young women with anorexia nervosa. Psychoneuroendocrinology 2020, 113, 104556. [Google Scholar] [CrossRef] [PubMed]
- Tyszkiewicz-Nwafor, M.; Rybakowski, F.; Dmitrzak-Weglarz, M.; Skibinska, M.; Paszyńska, E.; Dutkiewicz, A.; Słopien, A. Brain-Derived Neurotrophic Factor and Oxytocin Signaling in Association With Clinical Symptoms in Adolescent Inpatients With Anorexia Nervosa—A Longitudinal Study. Front. Psychiatry 2020, 10, 1032. [Google Scholar] [CrossRef]
- Cordeira, J.; Rios, M. Weighing in the Role of BDNF in the Central Control of Eating Behavior. Mol. Neurobiol. 2011, 44, 441–448. [Google Scholar] [CrossRef]
- Piotrowicz, Z.; Chalimoniuk, M.; Czuba, M.; Langfort, J. Rola neurotroficznego czynnika pochodzenia mózgowego w kontroli łaknienia. Postep. Biochem. 2020, 66, 205–212. [Google Scholar] [CrossRef]
- Camerino, C.; Zayzafoon, M.; Rymaszewski, M.; Heiny, J.; Rios, M.; Hauschka, P.V. Central depletion of brain-derived neurotrophic factor in mice results in high bone mass and metabolic phenotype. Endocrinology 2012, 153, 5394–5405. [Google Scholar] [CrossRef]
- Pelleymounter, M.A.; Cullen, M.J.; Wellman, C.L. Characteristics of BDNF-induced weight loss. Exp. Neurol. 1995, 131, 229–238. [Google Scholar] [CrossRef]
- Kernie, S.; Liebl, D.J.; Parada, L.F. BDNF regulates eating behavior and locomotor activity in mice. EMBO J. 2000, 19, 1290–1300. [Google Scholar] [CrossRef]
- Liao, G.-Y.; An, J.J.; Gharami, K.; Waterhouse, E.G.; Vanevski, F.; Jones, K.; Xu, B. Dendritically targeted Bdnf mRNA is essential for energy balance and response to leptin. Nat. Med. 2012, 18, 564–571. [Google Scholar] [CrossRef]
- Yeo, G.S.H.; Hung, C.-C.C.; Rochford, J.; Keogh, J.; Gray, J.; Sivaramakrishnan, S.; O'Rahilly, S.; Farooqi, S. A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat. Neurosci. 2004, 7, 1187–1189. [Google Scholar] [CrossRef]
- Gray, J.; Yeo, G.S.H.; Hung, C.; Keogh, J.; Clayton, P.; Banerjee, K.; McAulay, A.; O'Rahilly, S.; Farooqi, S. Functional characterization of human NTRK2 mutations identified in patients with severe early-onset obesity. Int. J. Obes. 2007, 31, 359–364. [Google Scholar] [CrossRef]
- Rios, M. BDNF and the central control of feeding: Accidental bystander or essential player? Trends Neurosci. 2013, 36, 83–90. [Google Scholar] [CrossRef]
- Sandrini, L.; Di Minno, A.; Amadio, P.; Ieraci, A.; Tremoli, E.; Barbieri, S.S. Association between Obesity and Circulating Brain-Derived Neurotrophic Factor (BDNF) Levels: Systematic Review of Literature and Meta-Analysis. Int. J. Mol. Sci. 2018, 19, 2281. [Google Scholar] [CrossRef]
- Si, J.; Zhang, H.; Zhu, L.; Chen, A. The Relationship between Overweight/Obesity and Executive Control in College Students: The Mediating Effect of BDNF and 5-HT. Life 2021, 11, 313. [Google Scholar] [CrossRef]
- Keeler, J.L.; Robinson, L.; Keeler-Schäffeler, R.; Dalton, B.; Treasure, J.; Himmerich, H. Growth factors in anorexia nervosa: A systematic review and meta-analysis of cross-sectional and longitudinal data. World J. Biol. Psychiatry 2022, 23, 582–600. [Google Scholar] [CrossRef]
- Charlson, F.J.; Ferrari, A.J.; Santomauro, D.; Diminic, S.; Stockings, E.; Scott, J.G.; McGrath, J.; A Whiteford, H. Global Epidemiology and Burden of Schizophrenia: Findings From the Global Burden of Disease Study 2016. Schizophr. Bull. 2018, 44, 1195–1203. [Google Scholar] [CrossRef]
- Peng, S.; Li, W.; Lv, L.; Zhang, Z.; Zhan, X. BDNF as a biomarker in diagnosis and evaluation of treatment for schizophrenia and depression. Discov. Med. 2018, 26, 127–136. [Google Scholar]
- Nieto, R.; Kukuljan, M.; Silva, H. BDNF and Schizophrenia: From Neurodevelopment to Neuronal Plasticity, Learning, and Memory. Front. Psychiatry 2013, 4, 45. [Google Scholar] [CrossRef]
- Green, M.J.; Matheson, S.L.; Shepherd, A.; Weickert, C.S.; Carr, V.J. Brain-derived neurotrophic factor levels in schizophrenia: A systematic review with meta-analysis. Mol. Psychiatry 2011, 16, 960–972. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Ye, F.; Liu, C.; Tang, X.; Li, J.; Dong, H.; Sha, W.; Zhang, X. Cognitive impairment in first-episode drug-naïve patients with schizophrenia: Relationships with serum concentrations of brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor. Prog. Neuro-Psychopharmacology Biol. Psychiatry 2017, 76, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.O.; Kramer, S.; Hofman, N.; Flynn, J.; Hansen, M.; Martin, V.; Pillai, A.; Buckley, P.F. A Meta-Analysis of Brain-Derived Neurotrophic Factor Effects on Brain Volume in Schizophrenia: Genotype and Serum Levels. Neuropsychobiology 2021, 80, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, T.; Matsuda, Y.; Matsuoka, K.; Yasuno, F.; Ikebuchi, E.; Kameda, H.; Taoka, T.; Miyasaka, T.; Kichikawa, K.; Kishimoto, T. Computer-assisted cognitive remediation therapy increases hippocampal volume in patients with schizophrenia: A randomized controlled trial. BMC Psychiatry 2018, 18, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, S.; Fisher, M.; Holland, C.; Shelly, W.; Wolkowitz, O.; Mellon, S.H. Is Serum Brain-Derived Neurotrophic Factor a Biomarker for Cognitive Enhancement in Schizophrenia? Biol. Psychiatry 2009, 66, 549–553. [Google Scholar] [CrossRef]
- Zheng, W.; Tong, G.; Ungvari, G.S.; Ng, C.H.; Chiu, H.F.; Xiang, Y.-Q.; Cao, X.-L.; Liu, Z.-R.; Meng, L.-R.; Gazdag, G.; et al. Memory Impairment Following Electroconvulsive Therapy in Chinese Patients with Schizophrenia: Meta-Analysis of Randomized Controlled Trials. Perspect. Psychiatr. Care 2018, 54, 107–114. [Google Scholar] [CrossRef]
- Shahin, O.; Gohar, S.M.; Ibrahim, W.; El-Makawi, S.M.; Fakher, W.; Taher, D.B.; Samie, M.A.; Khalil, M.A.; Saleh, A.A. Brain-Derived neurotrophic factor (BDNF) plasma level increases in patients with resistant schizophrenia treated with electroconvulsive therapy (ECT). Int. J. Psychiatry Clin. Pract. 2022, 26, 370–375. [Google Scholar] [CrossRef]
- Li, J.; Zhang, X.; Tang, X.; Xiao, W.; Ye, F.; Sha, W.; Jia, Q. Neurotrophic factor changes are essential for predict electroconvulsive therapy outcome in schizophrenia. Schizophr. Res. 2020, 218, 295–297. [Google Scholar] [CrossRef]
- Gustavsson, A.; Norton, N.; Fast, T.; Frölich, L.; Georges, J.; Holzapfel, D.; Kirabali, T.; Krolak-Salmon, P.; Rossini, P.M.; Ferretti, M.T.; et al. Global estimates on the number of persons across the Alzheimer's disease continuum. Alzheimer's Dement. 2022. [Google Scholar] [CrossRef]
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef]
- Gao, L.; Zhang, Y.; Sterling, K.; Song, W. Brain-derived neurotrophic factor in Alzheimer’s disease and its pharmaceutical potential. Transl. Neurodegener. 2022, 11, 1–34. [Google Scholar] [CrossRef]
- Qin, X.-Y.; Cao, C.; Cawley, N.X.; Liu, T.-T.; Yuan, J.; Loh, Y.P.; Cheng, Y. Decreased peripheral brain-derived neurotrophic factor levels in Alzheimer’s disease: A meta-analysis study (N=7277). Mol. Psychiatry 2017, 22, 312–320. [Google Scholar] [CrossRef]
- Ng, T.K.S.; Ho, C.S.H.; Tam, W.W.S.; Kua, E.H.; Ho, R.C.-M. Decreased Serum Brain-Derived Neurotrophic Factor (BDNF) Levels in Patients with Alzheimer’s Disease (AD): A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2019, 20, 257. [Google Scholar] [CrossRef]
- Du, Y.; Wu, H.-T.; Qin, X.-Y.; Cao, C.; Liu, Y.; Cao, Z.-Z.; Cheng, Y. Postmortem Brain, Cerebrospinal Fluid, and Blood Neurotrophic Factor Levels in Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Mol. Neurosci. 2018, 65, 289–300. [Google Scholar] [CrossRef]
- Xie, B.; Zhou, H.; Liu, W.; Yu, W.; Liu, Z.; Jiang, L.; Zhang, R.; Cui, D.; Shi, Z.; Xu, S. Evaluation of the diagnostic value of peripheral BDNF levels for Alzheimer’s disease and mild cognitive impairment: Results of a meta-analysis. Int. J. Neurosci. 2020, 130, 218–230. [Google Scholar] [CrossRef]
- Azman, K.F.; Zakaria, R. Recent Advances on the Role of Brain-Derived Neurotrophic Factor (BDNF) in Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 6827. [Google Scholar] [CrossRef]
- Walsh, S.; Merrick, R.; Milne, R.; Brayne, C. Aducanumab for Alzheimer’s disease? BMJ 2021, 374, n1682. [Google Scholar] [CrossRef]
- Numakawa, T.; Odaka, H. Brain-Derived Neurotrophic Factor Signaling in the Pathophysiology of Alzheimer’s Disease: Beneficial Effects of Flavonoids for Neuroprotection. Int. J. Mol. Sci. 2021, 22, 5719. [Google Scholar] [CrossRef] [PubMed]
- Kou, X.; Chen, D.; Chen, N. The Regulation of microRNAs in Alzheimer's Disease. Front. Neurol. 2020, 11, 288. [Google Scholar] [CrossRef]
- Lee, C.Y.; Ryu, I.S.; Ryu, J.-H.; Cho, H.-J. miRNAs as Therapeutic Tools in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 13012. [Google Scholar] [CrossRef]
- Keifer, J.; Zheng, Z.; Ambigapathy, G. A MicroRNA-BDNF Negative Feedback Signaling Loop in Brain: Implications for Alzheimer’s Disease. Microrna 2015, 4, 101–108. [Google Scholar] [CrossRef]
- Jiménez, Y.K.; Loria, F.; Corona, J.C.; Díaz-Nido, J. Gene Transfer of Brain-derived Neurotrophic Factor (BDNF) Prevents Neurodegeneration Triggered by FXN Deficiency. Mol. Ther. 2016, 24, 877–889. [Google Scholar] [CrossRef] [PubMed]
- Lopes, C.D.; Gonçalves, N.P.; Gomes, C.P.; Saraiva, M.J.; Pêgo, A.P. BDNF gene delivery mediated by neuron-targeted nanoparticles is neuroprotective in peripheral nerve injury. Biomaterials 2017, 121, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Tuszynski, M. A Clinical Trial of AAV2-BDNF Gene Therapy in Early Alzheimer’s Disease and Mild Cognitive Impairment. Available online: https://clinicaltrials.gov/ct2/show/NCT05040217 (accessed on 7 October 2021).


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gliwińska, A.; Czubilińska-Łada, J.; Więckiewicz, G.; Świętochowska, E.; Badeński, A.; Dworak, M.; Szczepańska, M. The Role of Brain-Derived Neurotrophic Factor (BDNF) in Diagnosis and Treatment of Epilepsy, Depression, Schizophrenia, Anorexia Nervosa and Alzheimer’s Disease as Highly Drug-Resistant Diseases: A Narrative Review. Brain Sci. 2023, 13, 163. https://doi.org/10.3390/brainsci13020163
Gliwińska A, Czubilińska-Łada J, Więckiewicz G, Świętochowska E, Badeński A, Dworak M, Szczepańska M. The Role of Brain-Derived Neurotrophic Factor (BDNF) in Diagnosis and Treatment of Epilepsy, Depression, Schizophrenia, Anorexia Nervosa and Alzheimer’s Disease as Highly Drug-Resistant Diseases: A Narrative Review. Brain Sciences. 2023; 13(2):163. https://doi.org/10.3390/brainsci13020163
Chicago/Turabian StyleGliwińska, Aleksandra, Justyna Czubilińska-Łada, Gniewko Więckiewicz, Elżbieta Świętochowska, Andrzej Badeński, Marta Dworak, and Maria Szczepańska. 2023. "The Role of Brain-Derived Neurotrophic Factor (BDNF) in Diagnosis and Treatment of Epilepsy, Depression, Schizophrenia, Anorexia Nervosa and Alzheimer’s Disease as Highly Drug-Resistant Diseases: A Narrative Review" Brain Sciences 13, no. 2: 163. https://doi.org/10.3390/brainsci13020163
APA StyleGliwińska, A., Czubilińska-Łada, J., Więckiewicz, G., Świętochowska, E., Badeński, A., Dworak, M., & Szczepańska, M. (2023). The Role of Brain-Derived Neurotrophic Factor (BDNF) in Diagnosis and Treatment of Epilepsy, Depression, Schizophrenia, Anorexia Nervosa and Alzheimer’s Disease as Highly Drug-Resistant Diseases: A Narrative Review. Brain Sciences, 13(2), 163. https://doi.org/10.3390/brainsci13020163