Abstract
Flumazenil is an allosteric modulator of the γ-aminobutyric acid-A receptor (GABAAR) benzodiazepine binding site that could normalize neuronal signaling and improve motor impairments in Parkinson’s disease (PD). Little is known about how regional GABAAR availability affects motor symptoms. We investigated the relationship between regional availability of GABAAR benzodiazepine binding sites and motor impairments in PD. Methods: A total of 11 Patients with PD (males; mean age 69.0 ± 4.6 years; Hoehn and Yahr stages 2–3) underwent [11C]flumazenil GABAAR benzodiazepine binding site and [11C]dihydrotetrabenazine vesicular monoamine transporter type-2 (VMAT2) PET imaging and clinical assessment. Stepwise regression analysis was used to predict regional cerebral correlates of the four cardinal UPDRS motor scores using cortical, striatal, thalamic, and cerebellar flumazenil binding estimates. Thalamic GABAAR availability was selectively associated with axial motor scores (R2 = 0.55, F = 11.0, β = −6.4, p = 0.0009). Multi-ligand analysis demonstrated significant axial motor predictor effects by both thalamic GABAAR availability (R2 = 0.47, β = −5.2, F = 7.2, p = 0.028) and striatal VMAT2 binding (R2 = 0.30, β = −3.9, F = 9.1, p = 0.019; total model: R2 = 0.77, F = 11.9, p = 0.0056). Post hoc analysis demonstrated that thalamic [11C]methyl-4-piperidinyl propionate cholinesterase PET and K1 flow delivery findings were not significant confounders. Findings suggest that reduced thalamic GABAAR availability correlates with worsened axial motor impairments in PD, independent of nigrostriatal degeneration. These findings may augur novel non-dopaminergic approaches to treating axial motor impairments in PD.
1. Introduction
Axial motor impairments represent a significant cause of disability in Parkinson’s disease (PD). Dopaminergic medications are often not efficacious in treating these symptoms [1]. Cholinergic system dysfunction has been implicated in some components of postural instability and gait difficulties in PD, in particular falls and sensory processing during postural control, but not with overall severity of axial motor impairments when accounting for nigrostriatal nerve terminal losses [2,3,4]. Postural control and gait functions are mediated by widespread neural networks that cannot be captured by a simplistic model of single neurotransmitter system changes. There is increasing interest in the dysfunction of co-localized neurotransmitter functions to better understand the complexity of the multisystem nature of the neurodegeneration in PD.
γ-aminobutyric acid (GABA) is the major inhibitory neurotransmitter in the central nervous system. GABA binds to and mediates its effects via post-synaptic ionotropic GABAA receptors (GABAAR) and pre- and post-synaptic metabotropic GABAB receptors [5]. The role of GABA neurotransmission has been little studied in PD, despite the fact that the two major outflows of the basal ganglia, principal neurons of the globus pallidus internus, and of the substantia nigra pars reticulata largely employ inhibitory GABA to connect to areas outside the basal ganglia and that increased GABA activity from these nuclei has been demonstrated previously by both electrophysiologic and mRNA analyses in parkinsonian animal models [6,7,8]. Regional imbalance of the major inhibitory central nervous system transmitter activity may have propagating effects on the neuronal network activity underlying motor impairments in PD [9,10].
Benzodiazepine binding sites are present in a significant subset of cerebral GABAA receptors [11]. Flumazenil is a short-acting intravenously administered silent allosteric modulator of the GABAAR benzodiazepine binding site, which rapidly improves motor impairments in PD, including postural instability and gait difficulties [12,13]. Based on current basal ganglia functional models of PD, flumazenil could affect neuronal signaling at several brain regions; there is, however, a knowledge gap about the relationship between availability of regional cerebral GABAAR benzodiazepine binding sites and specific motor impairments in human PD. There are relatively few in vivo imaging reports regarding the impairment of GABAAR benzodiazepine binding sites in the brain in PD. An in vivo imaging report by Japanese researchers found a correlation between reduced cerebral GABAAR benzodiazepine binding sites in the cortex as determined by [123I]iomazenil single-photon computed emission tomography and greater motor disability in PD but did not report on regions other than the cortex or striatal dopaminergic loss [14,15]. To address in more detail the role of GABAAR benzodiazepine binding sites, we investigated the relationship between in vivo regional cerebral availability of GABAAR benzodiazepine binding sites with [11C]flumazenil PET and motor impairments while accounting for nigrostriatal nerve terminal losses and cholinergic activity in subjects with PD.
2. Materials and Methods
2.1. Subjects and Clinical Test Battery
This cross-sectional study involved the analysis of 11 PD subjects (males, mean age 69.0 ± 4.6 years (SD; range 63–76); mean Mini-Mental State Examination score of 28.4 ± 2.4 (range 22–30); and mean duration of motor disease of 10.5 ± 4.1 years (range 5–15)). Subjects met the United Kingdom Parkinson’s Disease Society Brain Bank clinical diagnostic criteria [16]. Abnormal striatal [11C]dihydrotetrabenazine PET findings were consistent with the diagnosis of PD in all subjects. No subjects had a history of a large artery stroke or other significant intracranial disease. Mean modified Hoehn and Yahr stage was 2.6 ± 0.3 (range 2–3) with 1 subject in stage 2, 5 in stage 2.5 and 5 in stage 3 [17]. No subjects were taking benzodiazepine, (anti)cholinergic or neuroleptic drugs. Nine subjects were taking a combination of dopamine agonist and carbidopa–levodopa medications and two were using carbidopa–levodopa alone. All subjects completed the Unified Parkinson’s Disease Rating Scale (UPDRS) [18]. Subjects were examined and underwent [11C]dihydrotetrabenazine PET imaging in the morning after withholding dopaminergic drugs overnight. Mean motor UPDRS score was 28.3 ± 11.6 (range 10–48). UPDRS motor scores were divided into cardinal motor sub-scores for tremor (items 20 and 21), rigidity (item 22), distal appendicular bradykinesia (items 23–26 and 31), and axial symptoms (items 27–30).
This study was approved by the Institutional Review Boards of Ann Arbor Department of Veterans Affairs Medical Center and the University of Michigan. Written informed consent was obtained from all subjects prior to any research procedures.
2.2. Imaging Techniques
All subjects underwent brain MRI, GABAAR benzodiazepine binding site imaging using [11C]flumazenil PET, and [11C]dihydrotetrabenazine vesicular monoamine transporter type-2 (VMAT2) PET imaging. Acetylcholinesterase PET imaging using the [11C]methyl-4-piperidinyl propionate (PMP) ligand was available in 10 subjects for additional analysis. [11C]flumazenil, [11C]dihydrotetrabenazine and [11C]PMP were prepared as described previously [19,20,21]. A bolus/infusion protocol was used for [11C]flumazenil dynamic PET imaging with intravenous bolus injection containing 40% of the total administered 10 mCi [11C]flumazenil dosage over 15 s, followed by continuous infusion of the remaining tracer at a constant rate for 62 min [19]. A bolus infusion was also used for [11C]dihydrotetrabenazine vesicular monoamine transporter type 2 (VMAT2) dynamic PET imaging with bolus injection of 55% of a 15 mCi dose, while the remaining 45% of the dose was continuously infused over the next 60 min [22]. Dynamic acetylcholinesterase PET scanning was performed for 70 min following an intravenous bolus of 15 mCi [11C]PMP. VMAT2 (used to quantify the degree of nigrostriatal striatal dopaminergic denervation) and acetylcholinesterase PET (used to quantify cholinergic thalamic binding) denervation were used for our post-analysis. The three PET scans were performed as part of a single study with two PET scans on the same day and the third one within days.
MRI was performed on a 3 Tesla Philips Achieva system (Philips, Best, The Netherlands) and PET imaging was performed in 3D imaging mode with an ECAT EXACT HR+ tomograph (Siemens Molecular Imaging, Inc., Knoxville, TN, USA) as previously reported [2].
2.3. Imaging Analysis
All image frames were spatially coregistered within subjects with a rigid-body transformation to reduce the effects of subject motion during the imaging session [23]. Interactive Data Language (IDL version 8.7) image analysis software (Research systems, Inc., Boulder, CO, USA) was used to manually trace volumes of interest on the MRI scan. Traced volumes of interest included the bilateral striatum (putamen and caudate nucleus), thalamus, pons, cerebellum, and neocortex. Neocortical volume of interest definition used semi-automated threshold delineation of the neocortical grey matter signal on the MRI images [24].
[11C]flumazenil distribution volume ratios were estimated using the Logan plot graphical analysis method [25]. The input kinetics for the reference tissue were derived from the pons, where the [11C]flumazenil binding is predominantly accounted for by free and nonspecifically bound radiotracer [26,27]. [11C]dihydrotetrabenazine distribution volume ratios were estimated also using the Logan plot graphical analysis method [25] with the striatal time activity curves as the input function and the total neocortex as reference tissue, a reference region overall low in VMAT2 binding sites, with the assumption that the non-displaceable distribution is uniform across the brain at equilibrium to allow accurate and stable assessment of VMAT2 binding when using the distribution volume ratio [22]. Acetylcholinesterase [11C]PMP hydrolysis rates (k3) were estimated using the striatal volume as the input tissue region [28].
2.4. Statistical Analysis
Stepwise regression analyses were used to predict cortical, striatal, thalamic and cerebellar flumazenil binding estimates from the four cardinal motor UPDRS scores as defined in Section 2.1. Analyses were performed using SAS version 9.3, (SAS institute, Cary, NC, USA). We also performed post hoc confounder analysis for the dopaminergic and cholinergic PET ligands. Post hoc confounder analysis was also performed using the K1 proxy flow images extracted from the flumazenil PET kinetic model. A model was considered significant if its p-value fell below our Bonferroni-adjusted α of 0.0125 (0.05/4 models).
3. Results
3.1. Availability of Regional Cerebral GABAAR Benzodiazepine Binding Sites and UPDRS Motor Scores
Significant findings were present for the thalamic region with axial motor scores as the only significant variable in the model (R2 = 0.55, F = 11.0, β = −6.4, p = 0.0009, significant after correction for multiple testing). Tremor was the only variable that entered the model for the cortex (R2 = 0.42, F = 6.6, β = −4.9, p = 0.03), which was no longer significant after correction for the effects of multiple testing. No cardinal UPDRS motor scores entered the regression models for the striatum or cerebellum.
3.2. Post Hoc Analysis of Thalamic GABAAR Benzodiazepine Binding Site Availability, Acetylcholinesterase Hydrolysis Rate, and VMAT2 and Axial UPDRS Motor Scores
A subgroup of 10 subjects completed all three PET ligand studies. Stepwise regression analysis was used to best predict axial UPDRS motor scores from thalamic GABAAR benzodiazepine binding site availability, thalamic acetylcholinesterase hydrolysis rate, striatal VMAT2 activity, age, and duration of motor disease. The overall model was significant (R2 = 0.77, F = 11.9, p = 0.0056) with significant contributions from both the thalamic GABAAR benzodiazepine binding site availability (R2 = 0.47, β = −5.2, F = 7.2, p = 0.028), and striatal VMAT2 binding (R2 = 0.30, β = −3.9, F = 9.1, p = 0.019). Thalamic acetylcholinesterase hydrolysis rates, age, and duration of disease did not meet the entry criteria for the model.
3.3. Post Hoc Analysis of Thalamic [11C]Flumazenil K1 Flow Effects and Axial UPDRS Motor Scores
Although the above findings show that other neurotransmitters, such as dopamine or acetylcholine were not confounders for our GABAAR findings, we used the K1 proxy flow images from the flumazenil PET kinetic model as an additional step to confirm that neural processes other than the two non-GABAergic neurotransmitters (dopamine and acetylcholine) may not play a significant role. This is because reduced gray matter flow is a marker of the global neurodegenerative process (or global neural integrity) and may be associated with glutamatergic activity (the most common neurotransmitter in the brain). For this purpose, we computed thalamic K1 flow derived from the [11C]flumazenil kinetic model. Results showed no significant effect of thalamic K1 flow measures in the prediction of axial motor UPDRS scores (F = 2.92, β = −2.6, p = 0.13). Furthermore, entering the thalamic K1 flow measure together with the thalamic [11C]flumazenil receptor binding measure not only failed to show a significant effect for the thalamic K1 flow measure but actually further strengthened the effect of the [11C]flumazenil receptor binding measure in the prediction of axial motor scores (F = 20.3, β = −6.7, p = 0.0028; total model F = 8.9, p = 0.009, Figure 1).
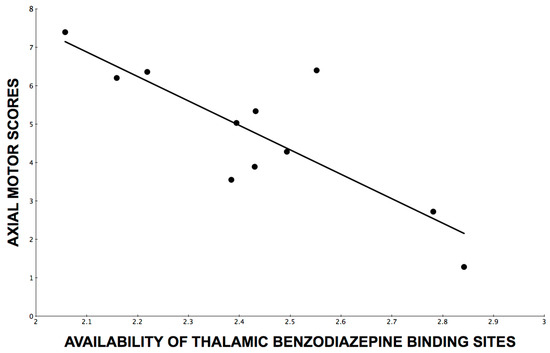
Figure 1.
Line of best fit through a scatterplot of axial motor impairment scores over availability of thalamic benzodiazepine binding sites as assessed by [11C]flumazenil PET distribution volume ratios.
4. Discussion
The thalamus is a key structure involved in motor control. The thalamus receives inhibitory inputs from the basal ganglia and excitatory signals from the cerebellum and cortex. Additional modulation in the form of monoaminergic and cholinergic signaling acts on the thalamus. Together, inputs to the thalamus act to modulate information received from the cortical regions resulting in motor control [29]. Our findings indicate that decreased availability of thalamic GABAAR benzodiazepine binding sites, reflecting increased GABAergic activity, is correlated with increased axial motor impairments in PD, independent of the degree of nigrostriatal degeneration. This may be compatible with the postulated basal ganglia model that the dopamine-denervated striatal nuclei provide inhibitory control over the globus pallidus internus and the substantia nigra pars reticulata [30], effectively “releasing” the tonic GABAergic inhibition mediated by the output structures of the basal ganglia [31]. As such, any dopaminergic hypoactivity within the striatum would therefore lead to a relative increase in inhibitory outflow from the basal ganglia [32]. Consequently, the subthalamic nucleus sends strong excitatory efferent signals to the globus pallidus internus and the substantia nigra pars reticulata, meaning that any increase in the firing rate of subthalamic nucleus neurons leads directly to an increased firing rate within globus pallidus internus and the substantia nigra pars reticulata neurons, in turn inhibiting the thalamic and brainstem structures resulting in mobility disturbances in PD [33]. In short, the dopaminergic hypoactivity in the striatum results in a comparative abundance of GABAergic inhibitory outputs from the basal ganglia to the thalamic region that ultimately leads to increased axial impairment in PD.
Indeed, recent research supports this model; one study demonstrated a correlation between increased GABA in the basal ganglia and axial motor impairment in PD [34], and another study demonstrated that GABAAR antagonism restored dopaminergic firing in the striatum and improved motor symptoms in mouse models [35]. There is also evidence that direct thalamic pathology may contribute to the pathophysiology of motor impairments in PD. For example, a post-mortem study demonstrated a 30–50% loss of cells in the center-median/parafascicular complex, which normally provides important glutaminergic feedback from the thalamus to the putamen [29]. [11C]flumazenil binding site densities may serve as an indicator of synaptic neuropil integrity or may be an indicator of a disease-specific disturbance at the GABAA receptor level. The former rationale is derived from the fact that GABA receptors are expressed on virtually all cortical and subcortical synaptic terminals. We performed a post hoc analysis to test the possibility that thalamic [11C]flumazenil GABAAR benzodiazepine binding site availability findings may be confounded by loss of neuronal integrity. For this purpose, we computed thalamic K1 flow measures derived from the [11C]flumazenil kinetic model but did not find a significant effect. Similar findings have been reported for the [123I]iomazenil single-photon computed emission tomography studies in PD where the authors also found an association between reduced cerebral GABAAR benzodiazepine binding site availability and increased motor disability in PD, which could not be explained by flow or perfusion data, suggesting a specific alteration of GABAA receptors rather than of generalized synaptic or neuronal integrity [15].
These findings support our previous reports on in vivo imaging studies that demonstrated correlations between cholinergic innervation changes and posture, falls, and sensory processing during postural changes [3,24,36,37]. These findings agree with pharmacological studies, showing a benefit of cholinesterase drug treatment and a reduction in falls in subjects with PD with no significant changes in parkinsonian motor rating scores [38,39].
Our results also showed an independent effect for the integrity of nigrostriatal dopaminergic nerve terminals and axial motor impairments in PD. Although axial motor impairments are relatively refractory to dopaminergic treatments, a subset of these impairments are or remain responsive to these drugs [40]. Furthermore, there is emerging evidence for GABA and dopamine co-releasing neurotransmission from substantia nigra pars compacta (SNpc) and ventral tegmental area dopaminergic neurons [41,42]. Animal models of PD suggest that dopaminergic activity in the SNpc may be inhibited due to aberrant tonic inhibition, thought to be the result of excessive astrocytic GABA, leading to further imbalance between dopamine and GABA [43]. These observations illustrate the intricate interplay between these two major neurotransmitter changes and how it may be derailed by nigrostriatal denervation [8].
Our findings of an association between motor impairments and altered GABAAR availability may not be limited to Lewy body parkinsonism but may potentially also apply to other types of parkinsonian disorders. For example, a [11C]flumazenil GABAAR benzodiazepine binding site PET study in patients with vascular parkinsonism with and without gait disturbances found that striatal [11C]flumazenil uptake was inversely correlated with the motor UPDRS scores and [11C]flumazenil binding reductions were associated with the presence of gait disturbance [44]. However, comparisons of these findings and our present results are limited as—at least pure—vascular parkinsonism would not manifest with nigrostriatal degeneration [45], which, inherent to its dysfunction, would lead to a relative increase in inhibitory outflow from the basal ganglia [32].
As previously stated, axial symptoms of PD are often resistant to treatment with dopaminergic replacement therapy [1]. Additionally, treatment with deep brain stimulation (DBS), which commonly targets the subthalamic nucleus and globus pallidus internus, fails to alleviate axial symptoms and may worsen axial disability. Subthalamic nucleus DBS, specifically, has been correlated with greater axial impairment post-surgery [46]. This is in line with our findings as increased firing of excitatory efferents from the subthalamic nucleus may lead to increased inhibition of the thalamic and brainstem regions. Alternative treatments to address the relative hyper GABAergic activity in these regions may lead to breakthroughs in the treatment of axial impairment.
Our findings augur GABAAR benzodiazepine binding site allosteric modulator drug treatment approaches to manage axial motor impairments in PD. Flumazenil is a fused imidazobenzodiazepine, which serves therapeutically as a GABAAR benzodiazepine binding site blocker [47]. Ondo and Hunter reported findings of single-dose (0.5 mg) intravenous flumazenil administration in eight PD patients and found significant improvements in total UPDRS motor scores, where the axial motor UPDRS sub-score tended to account for most of this improvement [12]. These flumazenil treatment data are compatible with the more selective association of reduced GABAAR benzodiazepine binding site availability and axial motor scores in our study.
Limitations of this study include the size and homogeneity of our sample. Because the patients were recruited from a veteran’s hospital, all the participants were male. This, in combination with the limited sample size, could influence the generalizability of the present findings with specific axial motor impairments, such as falls or freezing of gait. In addition, research into specific subtypes of axial impairments, such as patients with falls or freezing of gait, may lead to symptom-specific findings of GABAAR benzodiazepine binding site availability. Another limitation is the lack of a normal control or active disease control group to allow for the investigation of differential effects from normal aging or disease-specific effects of PD. Further studies based on a larger, more diverse study population, preferably with longitudinal follow up, are needed to more thoroughly investigate the role of GABAAR benzodiazepine binding site availability in the neural network underlying motor impairments in PD.
We conclude that thalamic GABAAR benzodiazepine binding site availability is inversely correlated with axial motor impairments in PD, independent from the degree of nigrostriatal degeneration. These findings may augur novel non-dopaminergic approaches to treating axial motor impairments in PD.
Author Contributions
Conceptualization, N.I.B.; methodology, N.I.B., K.A.F. and P.J.H.S.; software, P.K.; validation, N.I.B.; formal analysis, N.I.B. and P.K.; investigation, N.I.B.; resources, N.I.B.; data curation, P.K.; writing—original draft preparation, N.I.B., J.B. and R.V.; writing—review and editing, N.I.B., P.K., J.B., R.P., S.R. and R.V.; visualization, N.I.B.; supervision, N.I.B.; project administration, N.I.B.; funding acquisition, N.I.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Michael J. Fox Foundation, Department of Veterans Affairs (grant number I01 RX000317), and the National Institutes of Health (grant numbers P01 NS015655 and RO1 NS070856).
Institutional Review Board Statement
This study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Boards of the Ann Arbor Department of Veterans Affairs Medical Center and the University of Michigan (HUM00130361, 14 September 2017).
Informed Consent Statement
Informed consent was obtained from all subjects involved in this study.
Data Availability Statement
The data that support the findings of this study are available on reasonable request from the corresponding author. The data are not publicly available due to their containing information that could compromise the privacy of research participants.
Acknowledgments
The authors thank all patients for their time commitment, and research assistants, PET technologists, cyclotron operators, and chemists, for their assistance with this study.
Conflicts of Interest
N.I.B. receives research support from the National Institutes of Health, Michael J. Fox Foundation, and Ann Arbor Department of Veterans Affairs. K.A.F. receives research support from the National Institutes of Health, GE Healthcare, and AVID Radiopharmaceuticals. K.A.F. is a consultant for AVID Radiopharmaceuticals, MIMVista, Bayer-Schering, and GE healthcare, and holds equity (common stock) in GE, Bristol-Myers, Merck, and Novo-Nordisk. The funders had no role in the design of this study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Bloem, B.R.; Steijns, J.A.; Smits-Engelsman, B.C. An update on falls. Curr. Opin. Neurol. 2003, 16, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Bohnen, N.I.; Muller, M.L.; Kotagal, V.; Koeppe, R.A.; Kilbourn, M.R.; Gilman, S.; Albin, R.L.; Frey, K.A. Heterogeneity of cholinergic denervation in Parkinson’s disease without dementia. J. Cereb. Blood Flow Metab. 2012, 32, 1609–1617. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.L.; Albin, R.L.; Kotagal, V.; Koeppe, R.A.; Scott, P.J.; Frey, K.A.; Bohnen, N.I. Thalamic cholinergic innervation and postural sensory integration function in Parkinson’s disease. Brain 2013, 136, 3282–3289. [Google Scholar] [CrossRef] [PubMed]
- Roytman, S.; Paalanen, R.; Griggs, A.; David, S.; Pongmala, C.; Koeppe, R.A.; Scott, P.J.H.; Marusic, U.; Kanel, P.; Bohnen, N.I. Cholinergic system correlates of postural control changes in Parkinson’s disease freezers. Brain 2023, 146, 3243–3257. [Google Scholar] [CrossRef] [PubMed]
- Bowery, N.G.; Bettler, B.; Froestl, W.; Gallagher, J.P.; Marshall, F.; Raiteri, M.; Bonner, T.I.; Enna, S.J. International Union of Pharmacology. XXXIII. Mammalian gamma-aminobutyric acid(B) receptors: Structure and function. Pharmacol. Rev. 2002, 54, 247–264. [Google Scholar] [CrossRef] [PubMed]
- Filion, M.; Tremblay, L. Abnormal spontaneous activity of globus pallidus neurons in monkeys with MPTP-induced parkinsonism. Brain Res. 1991, 547, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Vila, M.; Levy, R.; Herrero, M.T.; Ruberg, M.; Faucheux, B.; Obeso, J.A.; Agid, Y.; Hirsch, E.C. Consequences of nigrostriatal denervation on the functioning of the basal ganglia in human and nonhuman primates: An in situ hybridization study of cytochrome oxidase subunit I mRNA. J. Neurosci. 1997, 17, 765–773. [Google Scholar] [CrossRef]
- Borgkvist, A.; Avegno, E.M.; Wong, M.Y.; Kheirbek, M.A.; Sonders, M.S.; Hen, R.; Sulzer, D. Loss of Striatonigral GABAergic Presynaptic Inhibition Enables Motor Sensitization in Parkinsonian Mice. Neuron 2015, 87, 976–988. [Google Scholar] [CrossRef]
- Boecker, H. Imaging the role of GABA in movement disorders. Curr. Neurol. Neurosci. Rep. 2013, 13, 385. [Google Scholar] [CrossRef]
- Boccalaro, I.L.; Schwerdel, C.; Cristia-Lara, L.; Fritschy, J.M.; Rubi, L. Dopamine depletion induces neuron-specific alterations of GABAergic transmission in the mouse striatum. Eur. J. Neurosci. 2020, 52, 3353–3374. [Google Scholar] [CrossRef]
- Sieghart, W.; Sperk, G. Subunit composition, distribution and function of GABA(A) receptor subtypes. Curr. Top Med. Chem. 2002, 2, 795–816. [Google Scholar] [CrossRef]
- Ondo, W.G.; Hunter, C. Flumazenil, a GABA antagonist, may improve features of Parkinson’s disease. Mov. Disord. 2003, 18, 683–685. [Google Scholar] [CrossRef] [PubMed]
- Ondo, W.G.; Silay, Y.S. Intravenous flumazenil for Parkinson’s disease: A single dose, double blind, placebo controlled, cross-over trial. Mov. Disord. 2006, 21, 1614–1617. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, K.; Tachibana, H.; Sugita, M.; Fukuchi, M. Impairment of benzodiazepine receptor in Parkinson’s disease evaluated by 123I-iomazenil SPECT. Kaku Igaku 1996, 33, 391–397. [Google Scholar] [PubMed]
- Kawabata, K.; Tachibana, H. Evaluation of benzodiazepine receptor in the cerebral cortex of Parkinson’s disease using 123I-iomazenil SPECT. Nihon Rinsho 1997, 55, 244–248. [Google Scholar] [PubMed]
- Hughes, A.J.; Daniel, S.E.; Kilford, L.; Lees, A.J. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: A clinico-pathological study of 100 cases. J. Neurol. Neurosurg. Psychiatry 1992, 55, 181–184. [Google Scholar] [CrossRef]
- Goetz, C.G.; Poewe, W.; Rascol, O.; Sampaio, C.; Stebbins, G.T.; Counsell, C.; Giladi, N.; Holloway, R.G.; Moore, C.G.; Wenning, G.K.; et al. Movement Disorder Society Task Force report on the Hoehn and Yahr staging scale: Status and recommendations. Mov. Disord. 2004, 19, 1020–1028. [Google Scholar] [CrossRef]
- Fahn, S. Unified Parkinson’s disease rating scale. Recent Dev. Park. Dis. 1987, 0, 153–163. [Google Scholar]
- Frey, K.A.; Holthoff, V.A.; Koeppe, R.A.; Jewett, D.M.; Kilbourn, M.R.; Kuhl, D.E. Parametric in vivo imaging of benzodiazepine receptor distribution in human brain. Ann. Neurol. 1991, 30, 663–672. [Google Scholar] [CrossRef]
- Jewett, D.M.; Kilbourn, M.R.; Lee, L.C. A simple synthesis of [11C]dihydrotetrabenazine (DTBZ). Nucl. Med. Biol. 1997, 24, 197–199. [Google Scholar] [CrossRef]
- Shao, X.; Hoareau, R.; Runkle, A.C.; Tluczek, L.J.M.; Hockley, B.G.; Henderson, B.D.; Scott, P.J.H. Highlighting the versatility of the Tracerlab synthesis modules. Part 2: Fully automated production of [11C]-labeled radiopharmaceuticals using a Tracerlab FXC-Pro. J. Label. Compd. Radiopharm. 2011, 54, 819–838. [Google Scholar] [CrossRef]
- Koeppe, R.A.; Frey, K.A.; Kuhl, D.E.; Kilbourn, M.R. Assessment of extrastriatal vesicular monoamine transporter binding site density using stereoisomers of [11C]dihydrotetrabenazine. J. Cereb. Blood Flow Metab. 1999, 19, 1376–1384. [Google Scholar] [CrossRef]
- Minoshima, S.; Koeppe, R.A.; Fessler, J.A.; Mintun, M.A.; Berger, K.L.; Taylor, S.F.; Kuhl, D.E. Integrated and Automated Data-Analysis Method for Neuronal Activation Studies Using O-15-Water Pet. Int. Congr. Ser. 1993, 1030, 409–417. [Google Scholar]
- Bohnen, N.I.; Frey, K.A.; Studenski, S.; Kotagal, V.; Koeppe, R.A.; Constantine, G.M.; Scott, P.J.; Albin, R.L.; Muller, M.L. Extra-nigral pathological conditions are common in Parkinson’s disease with freezing of gait: An in vivo positron emission tomography study. Mov. Disord. 2014, 29, 1118–1124. [Google Scholar] [CrossRef] [PubMed]
- Logan, J.; Fowler, J.S.; Volkow, N.D.; Wang, G.J.; Ding, Y.S.; Alexoff, D.L. Distribution volume ratios without blood sampling from graphical analysis of PET data. J. Cereb. Blood Flow Metab. 1996, 16, 834–840. [Google Scholar] [CrossRef]
- Millet, P.; Graf, C.; Buck, A.; Walder, B.; Ibanez, V. Evaluation of the reference tissue models for PET and SPECT benzodiazepine binding parameters. Neuroimage 2002, 17, 928–942. [Google Scholar] [CrossRef] [PubMed]
- Odano, I.; Halldin, C.; Karlsson, P.; Varrone, A.; Airaksinen, A.J.; Krasikova, R.N.; Farde, L. [18F]flumazenil binding to central benzodiazepine receptor studies by PET—Quantitative analysis and comparisons with [11C]flumazenil. Neuroimage 2009, 45, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Nagatsuka Si, S.; Fukushi, K.; Shinotoh, H.; Namba, H.; Iyo, M.; Tanaka, N.; Aotsuka, A.; Ota, T.; Tanada, S.; Irie, T. Kinetic analysis of [(11)C]MP4A using a high-radioactivity brain region that represents an integrated input function for measurement of cerebral acetylcholinesterase activity without arterial blood sampling. J. Cereb. Blood Flow Metab. 2001, 21, 1354–1366. [Google Scholar] [CrossRef]
- Halliday, G.M. Thalamic changes in Parkinson’s disease. Park. Relat. Disord. 2009, 15 (Suppl. S3), S152–S155. [Google Scholar] [CrossRef]
- Nambu, A. A new dynamic model of the cortico-basal ganglia loop. Prog. Brain Res. 2004, 143, 461–466. [Google Scholar] [CrossRef]
- Takakusaki, K.; Habaguchi, T.; Ohtinata-Sugimoto, J.; Saitoh, K.; Sakamoto, T. Basal ganglia efferents to the brainstem centers controlling postural muscle tone and locomotion: A new concept for understanding motor disorders in basal ganglia dysfunction. Neuroscience 2003, 119, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.J.; Barker, R.A. A pathophysiological model of freezing of gait in Parkinson’s disease. Park. Relat. Disord. 2009, 15, 333–338. [Google Scholar] [CrossRef]
- Lewis, S.J.; Shine, J.M. The Next Step: A Common Neural Mechanism for Freezing of Gait. Neuroscientist 2016, 22, 72–82. [Google Scholar] [CrossRef]
- O’Gorman Tuura, R.L.; Baumann, C.R.; Baumann-Vogel, H. Beyond Dopamine: GABA, Glutamate, and the Axial Symptoms of Parkinson Disease. Front. Neurol. 2018, 9, 806. [Google Scholar] [CrossRef]
- Emmanouilidou, E.; Minakaki, G.; Keramioti, M.V.; Xylaki, M.; Balafas, E.; Chrysanthou-Piterou, M.; Kloukina, I.; Vekrellis, K. GABA transmission via ATP-dependent K+ channels regulates alpha-synuclein secretion in mouse striatum. Brain 2016, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Bohnen, N.I.; Kanel, P.; Zhou, Z.; Koeppe, R.A.; Frey, K.A.; Dauer, W.T.; Albin, R.L.; Müller, M.L. Cholinergic system changes of falls and freezing of gait in Parkinson’s disease. Ann. Neurol. 2019, 85, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Bohnen, N.I.; Muller, M.L.; Koeppe, R.A.; Studenski, S.A.; Kilbourn, M.A.; Frey, K.A.; Albin, R.L. History of falls in Parkinson disease is associated with reduced cholinergic activity. Neurology 2009, 73, 1670–1676. [Google Scholar] [CrossRef]
- Chung, K.A.; Lobb, B.M.; Nutt, J.G.; Horak, F.B. Effects of a central cholinesterase inhibitor on reducing falls in Parkinson disease. Neurology 2010, 75, 1263–1269. [Google Scholar] [CrossRef]
- Henderson, E.J.; Lord, S.R.; Brodie, M.A.; Gaunt, D.M.; Lawrence, A.D.; Close, J.C.; Whone, A.L.; Ben-Shlomo, Y. Rivastigmine for gait stability in patients with Parkinson’s disease (ReSPonD): A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol. 2016, 15, 249–258. [Google Scholar] [CrossRef]
- Bohnen, N.I.; Cham, R. Postural control, gait, and dopamine functions in parkinsonian movement disorders. Clin. Geriatr. Med. 2006, 22, 797–812. [Google Scholar] [CrossRef]
- Tritsch, N.X.; Ding, J.B.; Sabatini, B.L. Dopaminergic neurons inhibit striatal output through non-canonical release of GABA. Nature 2012, 490, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Tritsch, N.X.; Oh, W.J.; Gu, C.; Sabatini, B.L. Midbrain dopamine neurons sustain inhibitory transmission using plasma membrane uptake of GABA, not synthesis. Elife 2014, 3, e01936. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.Y.; Nam, M.H.; Yoon, H.H.; Kim, J.; Hwang, Y.J.; Won, W.; Woo, D.H.; Lee, J.A.; Park, H.J.; Jo, S.; et al. Aberrant Tonic Inhibition of Dopaminergic Neuronal Activity Causes Motor Symptoms in Animal Models of Parkinson’s Disease. Curr. Biol. 2020, 30, 276–291.e279. [Google Scholar] [CrossRef] [PubMed]
- Ihara, M.; Tomimoto, H.; Ishizu, K.; Yoshida, H.; Sawamoto, N.; Hashikawa, K.; Fukuyama, H. Association of vascular parkinsonism with impaired neuronal integrity in the striatum. J. Neural Transm. 2007, 114, 577–584. [Google Scholar] [CrossRef]
- Gerschlager, W.; Bencsits, G.; Pirker, W.; Bloem, B.R.; Asenbaum, S.; Prayer, D.; Zijlmans, J.C.; Hoffmann, M.; Brucke, T. [123I]beta-CIT SPECT distinguishes vascular parkinsonism from Parkinson’s disease. Mov. Disord. 2002, 17, 518–523. [Google Scholar] [CrossRef]
- Zampogna, A.; Cavallieri, F.; Bove, F.; Suppa, A.; Castrioto, A.; Meoni, S.; Pélissier, P.; Schmitt, E.; Bichon, A.; Lhommée, E.; et al. Axial impairment and falls in Parkinson’s disease: 15 years of subthalamic deep brain stimulation. NPJ Park. Dis. 2022, 8, 121. [Google Scholar] [CrossRef]
- Haefely, W.; Hunkeler, W. The story of flumazenil. Eur. J. Anaesthesiol. Suppl. 1988, 2, 3–13. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).