
Role of SARS-CoV-2 in Modifying Neurodegenerative Processes in Parkinson’s Disease: A Narrative Review


Abstract
:
1. Introduction
1.1. COVID-19 Pandemic
1.2. Neurological Mayhem during SARS-CoV-2 Infection
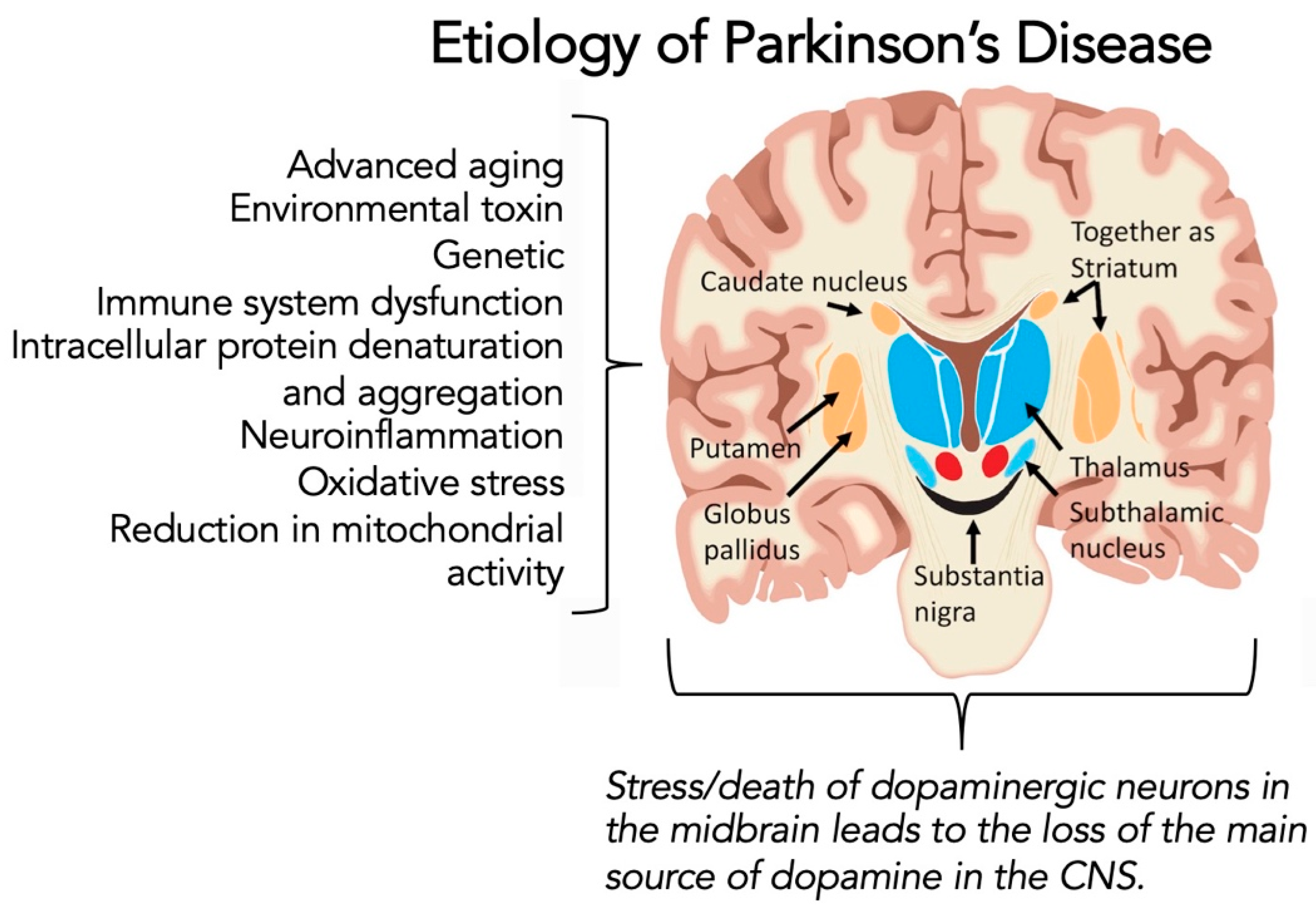
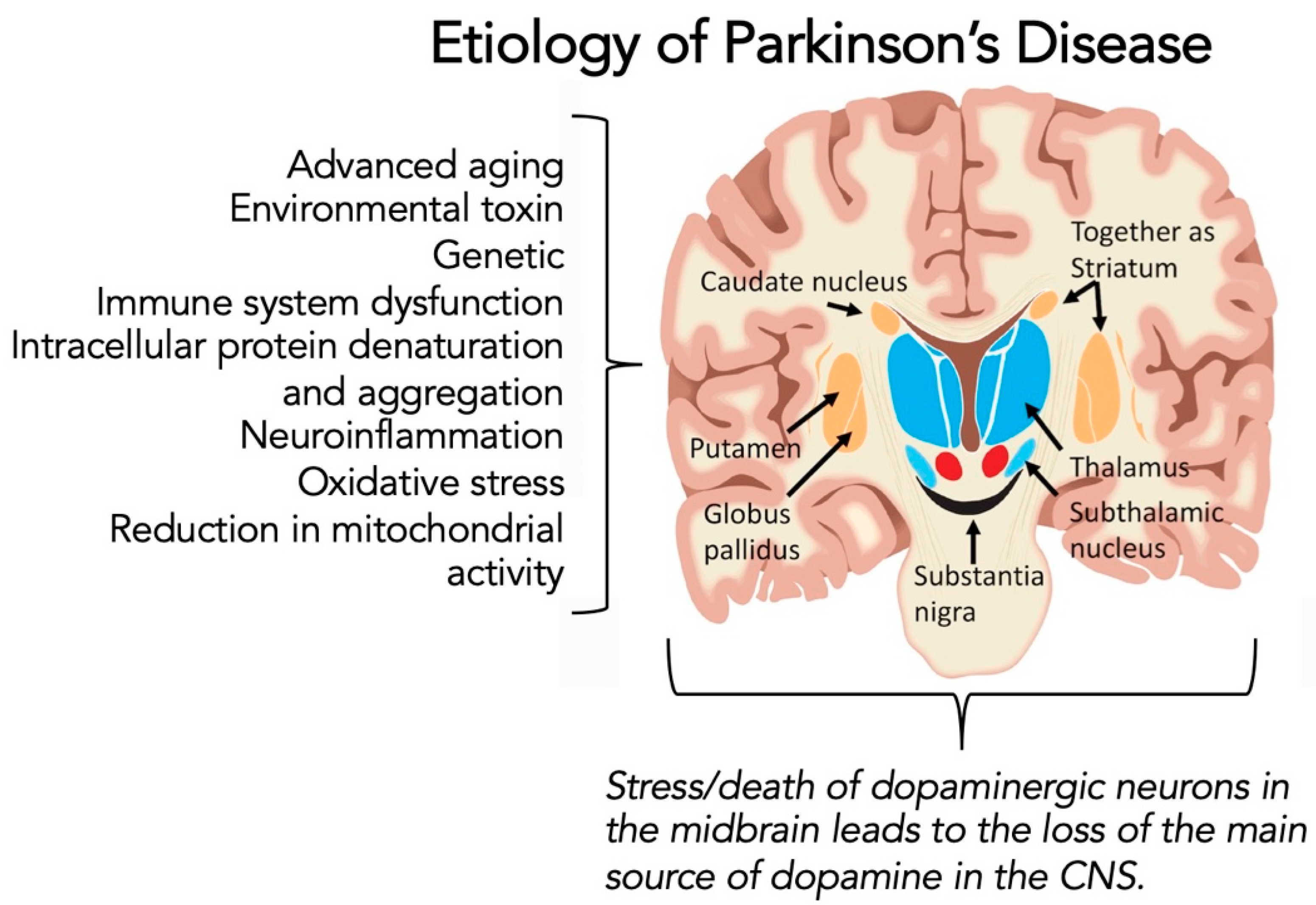
1.3. Parkinson’s Disease
1.4. Potential Link between COVID-19 Infection and Parkinson’s Diseases
- the older age of susceptibility for both severe COVID-19 infection and developing PD;
- the intense pathophysiology of the pro-inflammatory response of the host to SARS-CoV-2;
- the history of other viruses (including coronaviruses) invading the CNS and promoting neurodegenerative disorders;
- the growing evidence that SARS-CoV-2 has a neurotropic potential;
- and, the circumstantial reports in the literature of worsening of symptoms in persons previously diagnosed with PD.
2. COVID-19 Infection
2.1. Structure and Function of SARS-CoV-2
2.2. Role of the Spike Protein
2.3. Angiotensin-Converting Enzyme 2 (ACE2) Receptor
2.4. Pathophysiology
2.5. Cytokine Storm Syndrome
3. COVID-19, Neural Entry, and the Functional Role in Neurodegeneration
3.1. Neurological Interactions of SARS-CoV-2
3.2. Hyperinflammation
3.3. Evidence of SARS-CoV-2 in Cerebrospinal Fluid (CSF)
3.4. In Vitro Evidence for an Interaction between SARS-CoV-2, α-Synuclein from Parkinson’s Disease, and Neuronal Cells
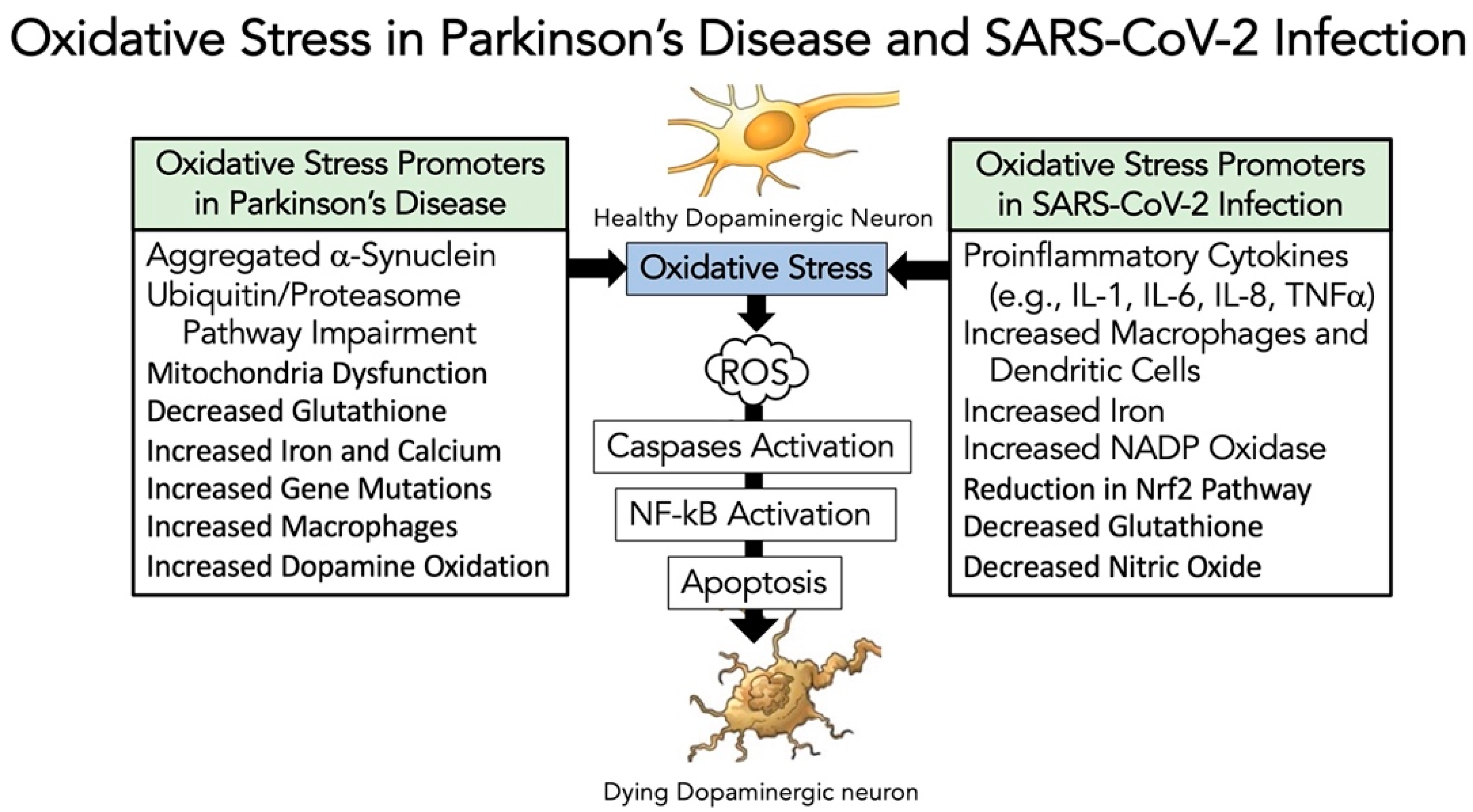
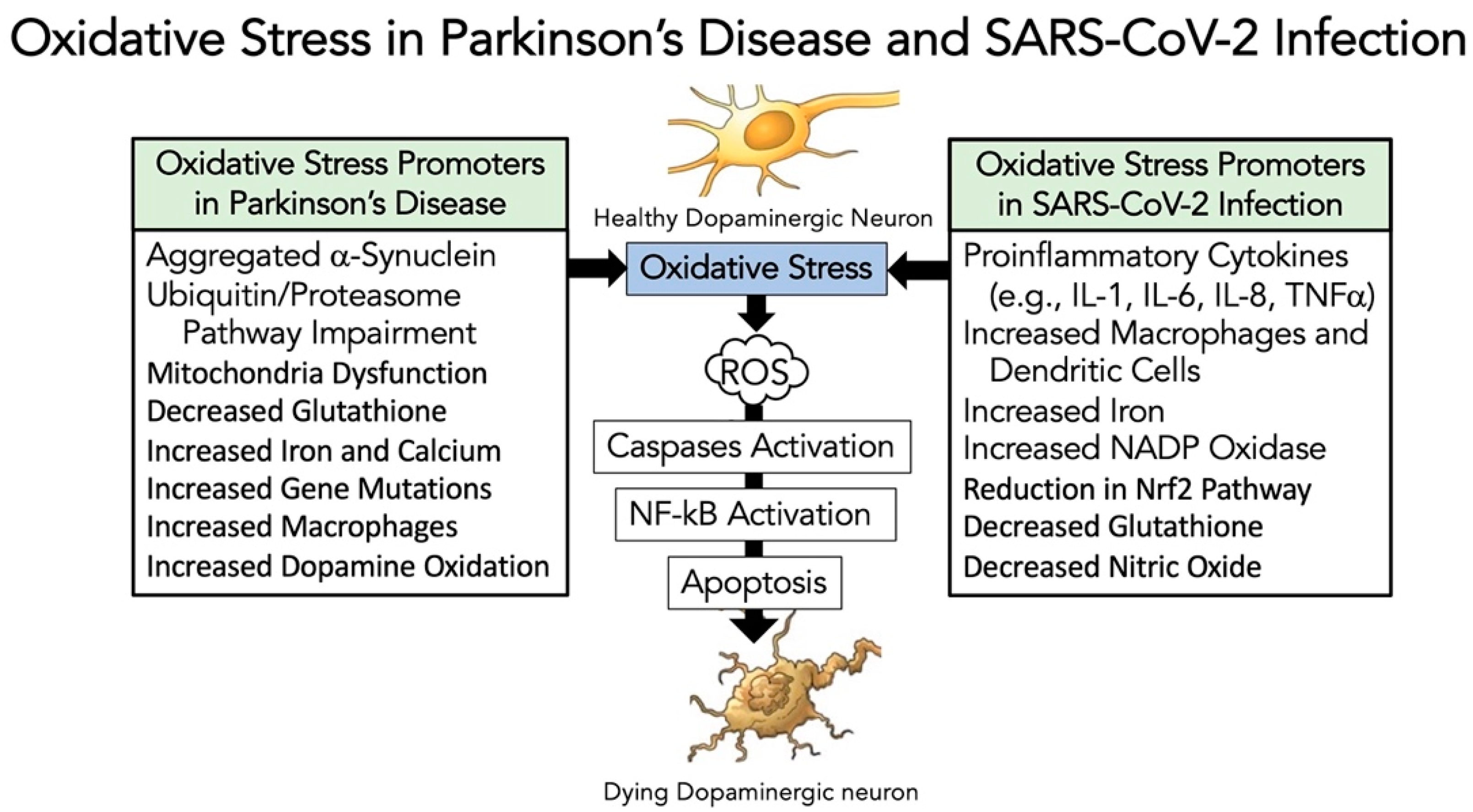
3.5. Evidence That SARS-CoV-2 Infection Enhances Oxidative-Stress Induced Parkinsonism Models
4. The Altered Immune System in Older Adults and Curious Roles for the Immune System in Parkinson’s Disease and COVID-19
4.1. Overview of the Immune System in Health and Disease
4.2. Cytokines in Inflammation, Immunity and COVID-19 Infection
4.3. The Defective Immune System in Older Adults in the Absence and Presence of SARS-CoV-2 Infection
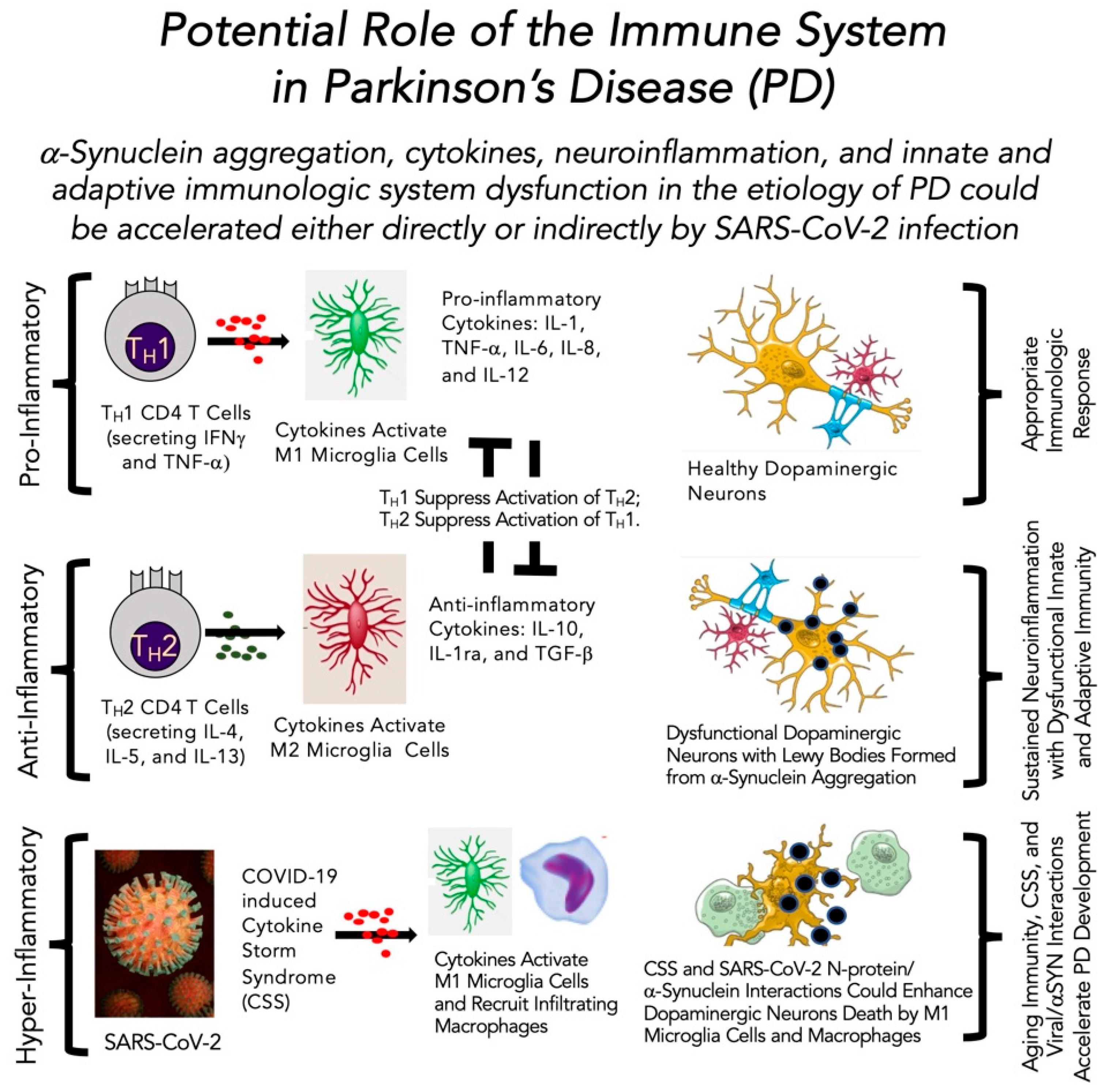
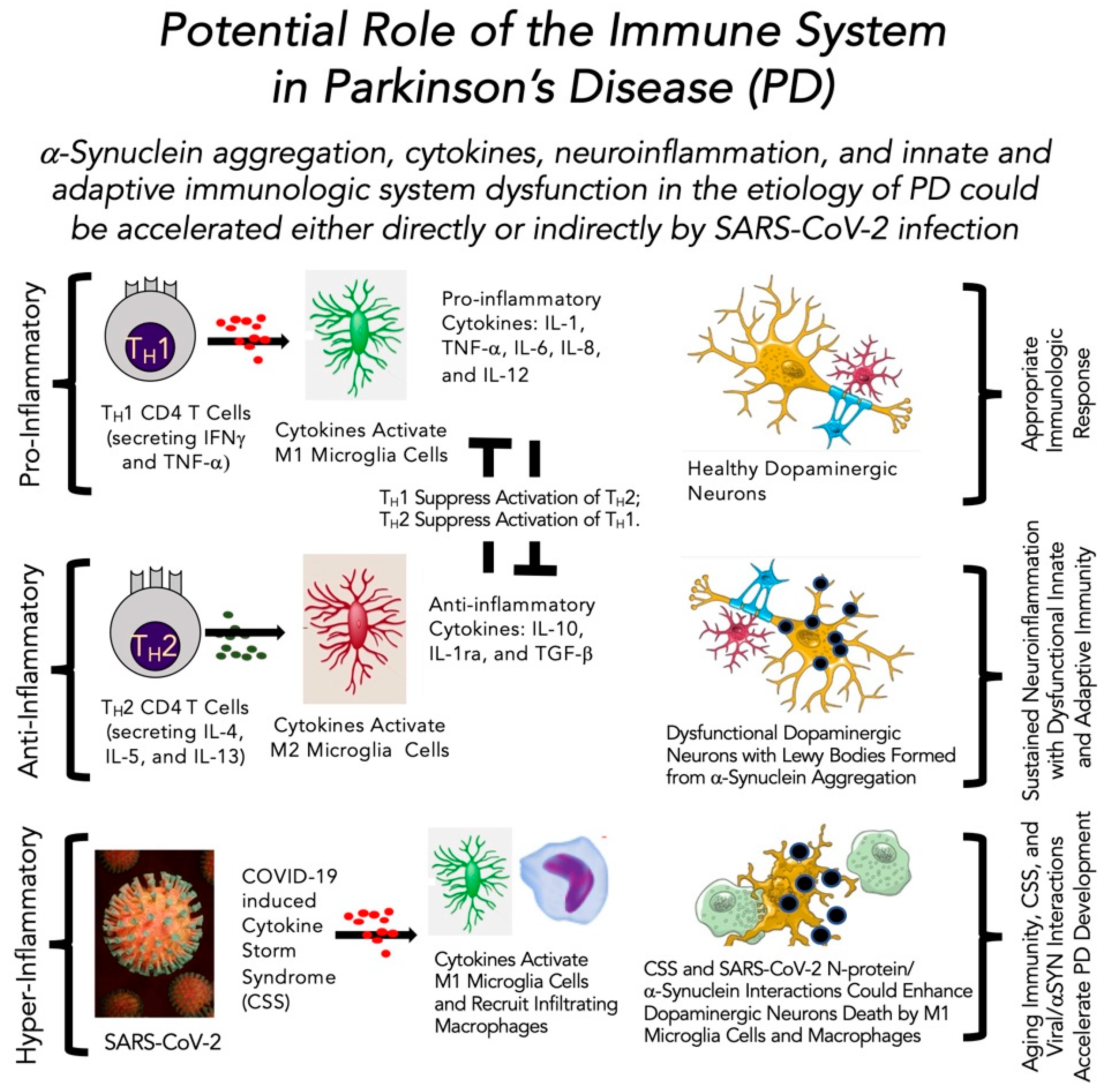
4.4. Role of the Immune System in Parkinson’s Disease in the Absence and Presence of SARS-CoV-2 Infection
5. Evidence That COVID-19 May Promote, Support or Accelerate Parkinson’s Disease
5.1. Historical Perspective
5.2. Hypotheses
5.3. Viral Invasion Linked Inflammation and Immunity
5.4. Neuropathological Alterations Caused by SARS-CoV-2
5.5. Existing Link with Neurological Consequences
6. Evidence Linking SARS-CoV-2, Neuroinflammation, and Atypical Parkinsonism
6.1. Atypical Parkinsonism
6.2. SARS-CoV-2 and Neuroinflammation in Atypical Parkinsonism
7. Evidence That PD May Worsen SARS-CoV-2 Infection Symptoms and Outcomes
8. Does SARS-CoV-2 Modify Neurodegenerative Processes in Parkinson’s Disease?
8.1. Theories That SARS-CoV-2 Promotes the Development of Parkinson’s Disease: Pros and Cons
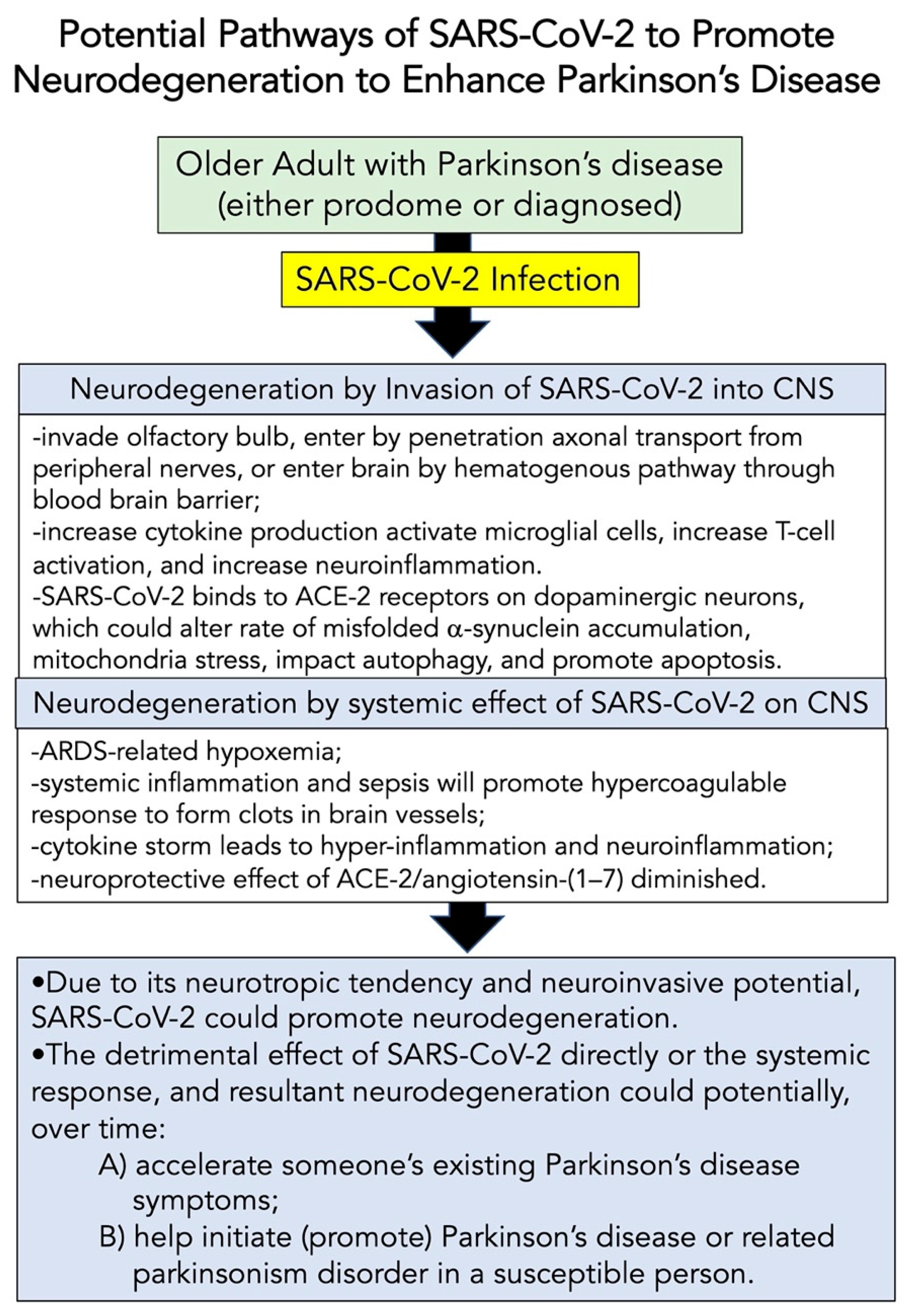
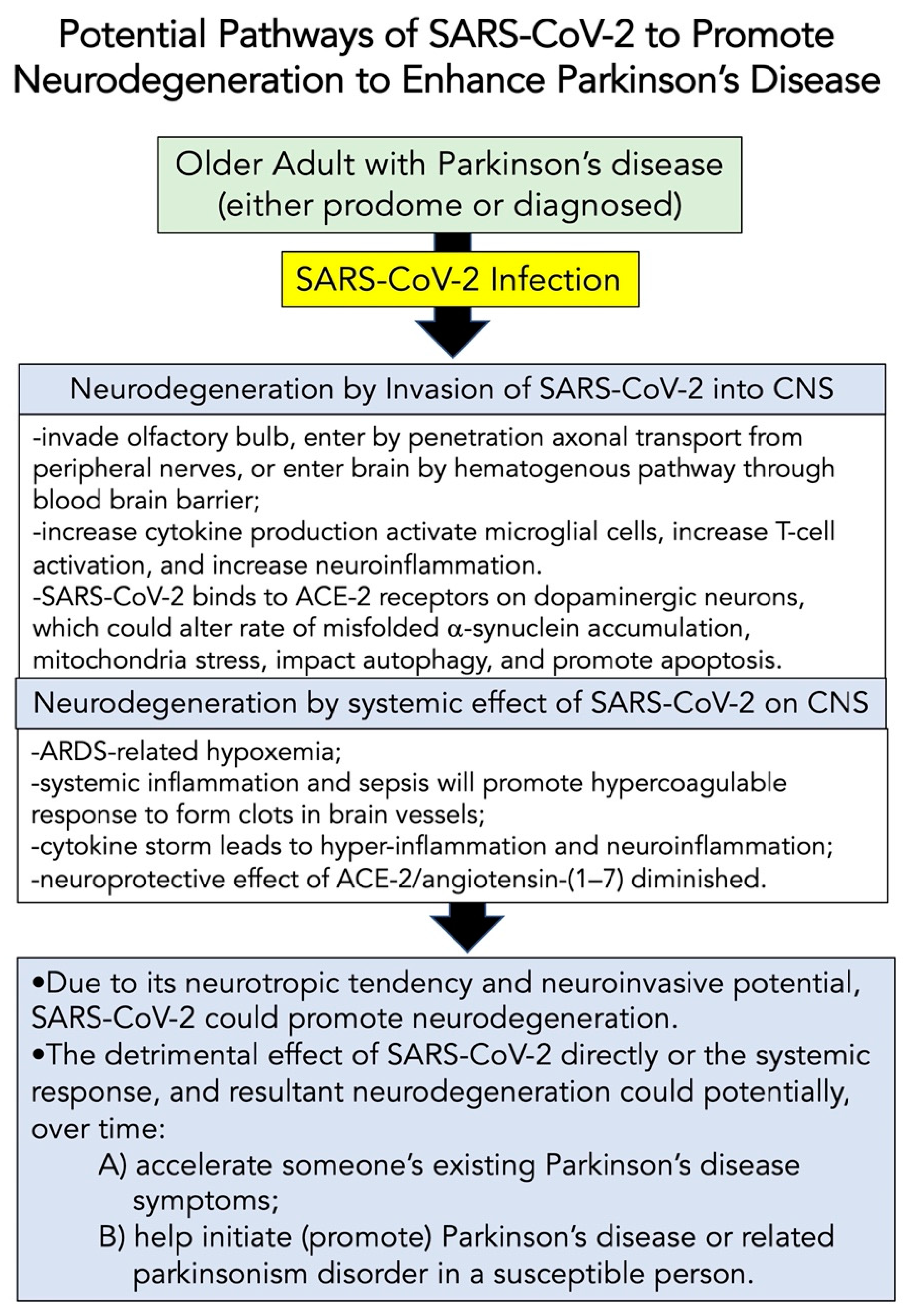
8.2. Potential Pathways of SARS-CoV-2 to Modify Neurodegeneration
8.3. Future Directions
8.4. Limitations
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, transmission, diagnosis, and treatment of coronavirus disease 2019 (COVID-19): A review. JAMA 2020, 324, 782–793. [Google Scholar] [CrossRef] [PubMed]
- Polak, S.B.; Van Gool, I.C.; Cohen, D.; Jan, H.; van Paassen, J. A systematic review of pathological findings in COVID-19: A pathophysiological timeline and possible mechanisms of disease progression. Mod. Pathol. 2020, 33, 2128–2138. [Google Scholar] [CrossRef] [PubMed]
- Yuki, K.; Fujiogi, M.; Koutsogiannaki, S. COVID-19 pathophysiology: A review. Clin. Immunol. 2020, 215, 108427. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Guo, J.; Wang, C.; Luo, F.; Yu, X.; Zhang, W.; Li, J.; Zhao, D.; Xu, D.; Gong, Q. Clinical characteristics and intrauterine vertical transmission potential of COVID-19 infection in nine pregnant women: A retrospective review of medical records. Lancet 2020, 395, 809–815. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Deng, L.; Zhang, L.; Cai, Y.; Cheung, C.W.; Xia, Z. Review of the clinical characteristics of coronavirus disease 2019 (COVID-19). J. Gen. Intern. Med. 2020, 35, 1549. [Google Scholar] [CrossRef] [Green Version]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef]
- Richardson, S.; Hirsch, J.S.; Narasimhan, M.; Crawford, J.M.; McGinn, T.; Davidson, K.W.; Barnaby, D.P.; Becker, L.B.; Chelico, J.D.; Cohen, S.L. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City area. JAMA 2020, 323, 2052–2059. [Google Scholar] [CrossRef]
- World Health Organization. COVID-19 Weekly Epidemiological Update, Edition 58, 21 September; World Health Organization: Geneva, Switzerland, 2021.
- Chen, X.; Laurent, S.; Onur, O.A.; Kleineberg, N.N.; Fink, G.R.; Schweitzer, F.; Warnke, C. A systematic review of neurological symptoms and complications of COVID-19. J. Neurol. 2021, 268, 392–402. [Google Scholar] [CrossRef]
- Russell, B.; Moss, C.; Rigg, A.; Hopkins, C.; Papa, S.; Van Hemelrijck, M. Anosmia and ageusia are emerging as symptoms in patients with COVID-19: What does the current evidence say? Ecancermedicalscience 2020, 14, ed98. [Google Scholar] [CrossRef] [Green Version]
- Lahiri, D.; Ardila, A. COVID-19 Pandemic: A Neurological Perspective. Cureus 2020, 12, e7889. [Google Scholar] [CrossRef]
- Yachou, Y.; El Idrissi, A.; Belapasov, V.; Benali, S.A. Neuroinvasion, neurotropic, and neuroinflammatory events of SARS-CoV-2: Understanding the neurological manifestations in COVID-19 patients. Neurol. Sci. 2020, 41, 2657–2669. [Google Scholar] [CrossRef] [PubMed]
- Pezzini, A.; Padovani, A. Lifting the mask on neurological manifestations of COVID-19. Nat. Rev. Neurol. 2020, 16, 636–644. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Huang, S.; Yin, L. The cytokine storm and COVID-19. J. Med. Virol. 2021, 93, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Vaninov, N. In the eye of the COVID-19 cytokine storm. Nat. Rev. Immunol. 2020, 20, 277. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.; Lang, A. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Ahlskog, J.E. The New Parkinson’s Disease Treatment Book: Partnering with Your Doctor to Get the Most from Your Medications; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Santens, P.; Boon, P.; Van Roost, D.; Caemaert, J. The pathophysiology of motor symptoms in Parkinson’s disease. Acta Neurol Belg. 2003, 103, 103. [Google Scholar]
- Jankovic, J. Motor fluctuations and dyskinesias in Parkinson’s disease: Clinical manifestations. Mov. Disord. 2005, 20, S11–S16. [Google Scholar] [CrossRef]
- Ferrazzoli, D.; Ortelli, P.; Cucca, A.; Bakdounes, L.; Canesi, M.; Volpe, D. Motor-cognitive approach and aerobic training: A synergism for rehabilitative intervention in Parkinson’s disease. Neurodegener. Dis. Manag. 2020, 10, 41–55. [Google Scholar] [CrossRef] [Green Version]
- Berganzo, K.; Tijero, B.; Gonzalez-Eizaguirre, A.; Somme, J.; Lezcano, E.; Gabilondo, I.; Fernandez, M.; Zarranz, J.; Gómez-Esteban, J. Motor and non-motor symptoms of Parkinson’s disease and their impact on quality of life and on different clinical subgroups. Neurología 2016, 31, 585–591. [Google Scholar] [CrossRef]
- Hall, M.-F.E.; Church, F.C. Integrative Medicine and Health Therapy for Parkinson Disease. Top. Geriatr. Rehabil. 2020, 36, 176–186. [Google Scholar] [CrossRef]
- Crowley, E.K.; Nolan, Y.M.; Sullivan, A.M. Exercise as a therapeutic intervention for motor and non-motor symptoms in Parkinson’s disease: Evidence from rodent models. Prog. Neurobiol. 2019, 172, 2–22. [Google Scholar] [CrossRef] [PubMed]
- Carapellotti, A.M.; Stevenson, R.; Doumas, M. The efficacy of dance for improving motor impairments, non-motor symptoms, and quality of life in Parkinson’s disease: A systematic review and meta-analysis. PLoS ONE 2020, 15, e0236820. [Google Scholar] [CrossRef] [PubMed]
- Amara, A.W.; Memon, A.A. Effects of exercise on non-motor symptoms in Parkinson’s disease. Clin. Ther. 2018, 40, 8–15. [Google Scholar] [CrossRef] [Green Version]
- Church, F.C. Treatment Options for Motor and Non-Motor Symptoms of Parkinson’s Disease. Biomolecules 2021, 11, 612. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Okun, M.S. Diagnosis and treatment of Parkinson disease: A review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef]
- Simon, D.K.; Tanner, C.M.; Brundin, P. Parkinson Disease Epidemiology, Pathology, Genetics, and Pathophysiology. Clin. Geriatr. Med. 2020, 36, 1–12. [Google Scholar] [CrossRef]
- Bandres-Ciga, S.; Diez-Fairen, M.; Kim, J.J.; Singleton, A.B. Genetics of Parkinson’s disease: An introspection of its journey towards precision medicine. Neurobiol. Dis. 2020, 137, 104782. [Google Scholar] [CrossRef]
- van der Kolk, N.M.; de Vries, N.M.; Kessels, R.P.; Joosten, H.; Zwinderman, A.H.; Post, B.; Bloem, B.R. Effectiveness of home-based and remotely supervised aerobic exercise in Parkinson’s disease: A double-blind, randomised controlled trial. Lancet Neurol. 2019, 18, 998–1008. [Google Scholar] [CrossRef] [Green Version]
- Johansson, M.E.; Cameron, I.G.; Van der Kolk, N.M.; de Vries, N.M.; Klimars, E.; Toni, I.; Bloem, B.R.; Helmich, R.C. Aerobic Exercise Alters Brain Function and Structure in Parkinson’s Disease: A Randomized Controlled Trial. Ann. Neurol. 2022, 91, 203–216. [Google Scholar] [CrossRef]
- Connolly, B.S.; Lang, A.E. Pharmacological treatment of Parkinson disease: A review. JAMA 2014, 311, 1670–1683. [Google Scholar] [CrossRef] [PubMed]
- Hribar, C.A.; Cobbold, P.H.; Church, F.C. Potential Role of Vitamin D in the Elderly to Resist COVID-19 and to Slow Progression of Parkinson’s Disease. Brain Sci. 2020, 10, 284. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.-F.E.; Church, F.C. Exercise for older adults improves the quality of life in Parkinson’s disease and potentially enhances the immune response to COVID-19. Brain Sci. 2020, 10, 612. [Google Scholar] [CrossRef] [PubMed]
- Bliss, R.R.; Church, F.C. Golf as a Physical Activity to Potentially Reduce the Risk of Falls in Older Adults with Parkinson’s Disease. Sports 2021, 9, 72. [Google Scholar] [CrossRef]
- Rothstein, A.; Oldridge, O.; Schwennesen, H.; Do, D.; Cucchiara, B.L. Acute cerebrovascular events in hospitalized COVID-19 patients. Stroke 2020, 51, e219–e222. [Google Scholar] [CrossRef]
- Tancheva, L.; Petralia, M.C.; Miteva, S.; Dragomanova, S.; Solak, A.; Kalfin, R.; Lazarova, M.; Yarkov, D.; Ciurleo, R.; Cavalli, E. Emerging neurological and psychobiological aspects of COVID-19 infection. Brain Sci. 2020, 10, 852. [Google Scholar] [CrossRef]
- Fotuhi, M.; Mian, A.; Meysami, S.; Raji, C.A. Neurobiology of COVID-19. J. Alzheimer’s Dis. 2020, 76, 3–19. [Google Scholar] [CrossRef]
- Sainz-Amo, R.; Baena-Álvarez, B.; Pareés, I.; Sánchez-Díez, G.; Pérez-Torre, P.; López-Sendón, J.; Fanjul-Arbos, S.; Monreal, E.; Corral-Corral, I.; García-Barragán, N. COVID-19 in Parkinson’s disease: What holds the key? J. Neurol. 2021, 268, 2666–2670. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Li, Q.; Melino, S.; Melino, G.; Shi, Y. Can COVID-19 pandemic boost the epidemic of neurodegenerative diseases? Biol. Direct 2020, 15, 1–8. [Google Scholar] [CrossRef]
- Stefano, G.B.; Büttiker, P.; Weissenberger, S.; Martin, A.; Ptacek, R.; Kream, R.M. The Pathogenesis of Long-Term Neuropsychiatric COVID-19 and the Role of Microglia, Mitochondria, and Persistent Neuroinflammation: A Hypothesis. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2021, 27, e933015-1–e933015-4. [Google Scholar] [CrossRef]
- Nath, A.; Smith, B. Neurological issues during COVID-19: An overview. Neurosci. Lett. 2021, 742, 135533. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Chen, C.; Dong, X.-P. Impact of COVID-19 pandemic on patients with neurodegenerative diseases. Front. Aging Neurosci. 2021, 13, 173. [Google Scholar] [CrossRef] [PubMed]
- Chiappelli, F. Towards neuro-covid-19. Bioinformation 2020, 16, 288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jasti, M.; Nalleballe, K.; Dandu, V.; Onteddu, S. A review of pathophysiology and neuropsychiatric manifestations of COVID-19. J. Neurol. 2021, 268, 2007–2012. [Google Scholar] [CrossRef] [PubMed]
- Rabi, F.A.; Al Zoubi, M.S.; Kasasbeh, G.A.; Salameh, D.M.; Al-Nasser, A.D. SARS-CoV-2 and coronavirus disease 2019: What we know so far. Pathogens 2020, 9, 231. [Google Scholar] [CrossRef] [PubMed]
- Bosch, B.J.; Van der Zee, R.; De Haan, C.A.; Rottier, P.J. The coronavirus spike protein is a class I virus fusion protein: Structural and functional characterization of the fusion core complex. J. Virol. 2003, 77, 8801–8811. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [Green Version]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Belouzard, S.; Millet, J.K.; Licitra, B.N.; Whittaker, G.R. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 2012, 4, 1011–1033. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, X.; Chen, J.; Zhang, H.; Deng, A. Association of renin-angiotensin system inhibitors with severity or risk of death in patients with hypertension hospitalized for coronavirus disease 2019 (COVID-19) infection in Wuhan, China. JAMA Cardiol. 2020, 5, 825–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosso, M.; Thanaraj, T.A.; Abu-Farha, M.; Alanbaei, M.; Abubaker, J.; Al-Mulla, F. The two faces of ACE2: The role of ACE2 receptor and its polymorphisms in hypertension and COVID-19. Mol. Ther.-Methods Clin. Dev. 2020, 18, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.K.; Velkoska, E.; Burrell, L.M. Emerging markers in cardiovascular disease: Where does angiotensin-converting enzyme 2 fit in? Clin. Exp. Pharmacol. Physiol. 2013, 40, 551–559. [Google Scholar] [CrossRef]
- Gu, J.; Korteweg, C. Pathology and pathogenesis of severe acute respiratory syndrome. Am. J. Pathol. 2007, 170, 1136–1147. [Google Scholar] [CrossRef] [Green Version]
- Mao, L.; Jin, H.; Wang, M.; Hu, Y.; Chen, S.; He, Q.; Chang, J.; Hong, C.; Zhou, Y.; Wang, D. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol. 2020, 77, 683–690. [Google Scholar] [CrossRef] [Green Version]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Ramos-Casals, M.; Brito-Zerón, P.; López-Guillermo, A.; Khamashta, M.A.; Bosch, X. Adult haemophagocytic syndrome. Lancet 2014, 383, 1503–1516. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Meftahi, G.H.; Jangravi, Z.; Sahraei, H.; Bahari, Z. The possible pathophysiology mechanism of cytokine storm in elderly adults with COVID-19 infection: The contribution of “inflame-aging”. Inflamm. Res. 2020, 69, 825–839. [Google Scholar] [CrossRef]
- Baig, A.M.; Khaleeq, A.; Ali, U.; Syeda, H. Evidence of the COVID-19 virus targeting the CNS: Tissue distribution, host–virus interaction, and proposed neurotropic mechanisms. ACS Chem. Neurosci. 2020, 11, 995–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Netland, J.; Meyerholz, D.K.; Moore, S.; Cassell, M.; Perlman, S. Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J. Virol. 2008, 82, 7264–7275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oxley, T.J.; Mocco, J.; Majidi, S.; Kellner, C.P.; Shoirah, H.; Singh, I.P.; De Leacy, R.A.; Shigematsu, T.; Ladner, T.R.; Yaeger, K.A. Large-vessel stroke as a presenting feature of Covid-19 in the young. N. Engl. J. Med. 2020, 382, e60. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Bai, W.Z.; Hashikawa, T. The neuroinvasive potential of SARS-CoV2 may play a role in the respiratory failure of COVID-19 patients. J. Med. Virol. 2020, 92, 552–555. [Google Scholar] [CrossRef] [PubMed]
- Umapathi, T.; Kor, A.C.; Venketasubramanian, N.; Lim, C.T.; Pang, B.C.; Yeo, T.T.; Lee, C.C.; Lim, P.L.; Ponnudurai, K.; Chuah, K.L. Large artery ischaemic stroke in severe acute respiratory syndrome (SARS). J. Neurol. 2004, 251, 1227–1231. [Google Scholar] [CrossRef] [Green Version]
- Rossi, A. Imaging of acute disseminated encephalomyelitis. Neuroimaging Clin. North Am. 2008, 18, 149–161. [Google Scholar] [CrossRef]
- Wohleb, E.S.; McKim, D.B.; Sheridan, J.F.; Godbout, J.P. Monocyte trafficking to the brain with stress and inflammation: A novel axis of immune-to-brain communication that influences mood and behavior. Front. Neurosci. 2015, 8, 447. [Google Scholar] [CrossRef]
- Dantzer, R. Neuroimmune interactions: From the brain to the immune system and vice versa. Physiol. Rev. 2018, 98, 477–504. [Google Scholar] [CrossRef]
- Oktar, A.Ç.; Göçgün, N.; Yaşgüçlükal, M.A.; Bayar, M.D.; Alaçam, S.; Karabulut, N.; Duman, E.B.; Şişman, B.; Baştan, B.; Balcı, B.P. COVID-19-associated Isolated Cortical Vein Thrombosis: Detection of SARS-CoV-2 in CSF. Neurohospitalist 2022. [Google Scholar] [CrossRef]
- Tee, T.Y.; Thabit, A.A.M.; Khoo, C.S.; Shahrom, H.M.; Chan, E.Z.; Marzukie, M.M.; Kamaruddin, Z.A.C.; Thayan, R.; Chidambaram, S.K. Acute encephalitis associated with SARS-CoV-2 confirmed in cerebrospinal fluid: First case in Malaysia. J. Clin. Neurol. 2021, 17, 490. [Google Scholar] [CrossRef]
- Jarius, S.; Pache, F.; Körtvelyessy, P.; Jelčić, I.; Stettner, M.; Franciotta, D.; Keller, E.; Neumann, B.; Ringelstein, M.; Senel, M. Cerebrospinal fluid findings in COVID-19: A multicenter study of 150 lumbar punctures in 127 patients. J. Neuroinflamm. 2022, 19, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Domingues, R.B.; Leite, F.B.V.d.M.; Senne, C. Cerebrospinal fluid analysis in patients with COVID-19-associated central nervous system manifestations: A systematic review. Arq. Neuro-Psiquiatr. 2022, 80, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.; Frontera, J.; Placantonakis, D.G.; Lighter, J.; Galetta, S.; Balcer, L.; Melmed, K.R. Cerebrospinal fluid in COVID-19: A systematic review of the literature. J. Neurol. Sci. 2021, 421, 117316. [Google Scholar] [CrossRef] [PubMed]
- Paniz-Mondolfi, A.; Bryce, C.; Grimes, Z.; Gordon, R.E.; Reidy, J.; Lednicky, J.; Sordillo, E.M.; Fowkes, M. Central nervous system involvement by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). J. Med. Virol. 2020, 92, 699–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Miranda-Saksena, M.; Saksena, N.K. Viruses and neurodegeneration. Virol. J. 2013, 10, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semerdzhiev, S.A.; Fakhree, M.A.A.; Segers-Nolten, I.; Blum, C.; Claessens, M.M.A.E. Interactions between SARS-CoV-2 N-Protein and α-Synuclein Accelerate Amyloid Formation. ACS Chem. Neurosci. 2021. [Google Scholar] [CrossRef]
- Lücking, C. Alpha-synuclein and Parkinson’s disease. Cell. Mol. Life Sci. CMLS 2000, 57, 1894–1908. [Google Scholar] [CrossRef]
- EI-Agnaf, O.; Irvine, G. Aggregation and neurotoxicity of α-synuclein and related peptides. Biochem. Soc. Trans. 2002, 30, 559–565. [Google Scholar] [CrossRef]
- Jenner, P. Oxidative stress in Parkinson’s disease. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2003, 53, S26–S38. [Google Scholar] [CrossRef]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef] [Green Version]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Parkinson’s Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannan, K.; Jain, S.K. Oxidative stress and apoptosis. Pathophysiology 2000, 7, 153–163. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z.-G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chernyak, B.; Popova, E.; Prikhodko, A.; Grebenchikov, O.; Zinovkina, L.; Zinovkin, R. COVID-19 and oxidative stress. Biochemistry 2020, 85, 1543–1553. [Google Scholar] [CrossRef]
- Laforge, M.; Elbim, C.; Frère, C.; Hémadi, M.; Massaad, C.; Nuss, P.; Benoliel, J.-J.; Becker, C. Tissue damage from neutrophil-induced oxidative stress in COVID-19. Nat. Rev. Immunol. 2020, 20, 515–516. [Google Scholar] [CrossRef] [PubMed]
- Beltrán-García, J.; Osca-Verdegal, R.; Pallardó, F.V.; Ferreres, J.; Rodríguez, M.; Mulet, S.; Sanchis-Gomar, F.; Carbonell, N.; García-Giménez, J.L. Oxidative stress and inflammation in COVID-19-associated sepsis: The potential role of anti-oxidant therapy in avoiding disease progression. Antioxidants 2020, 9, 936. [Google Scholar] [CrossRef]
- Suhail, S.; Zajac, J.; Fossum, C.; Lowater, H.; McCracken, C.; Severson, N.; Laatsch, B.; Narkiewicz-Jodko, A.; Johnson, B.; Liebau, J. Role of oxidative stress on SARS-CoV (SARS) and SARS-CoV-2 (COVID-19) infection: A review. Protein J. 2020, 39, 644–656. [Google Scholar] [CrossRef]
- Chaudhry, Z.L.; Klenja, D.; Janjua, N.; Cami-Kobeci, G.; Ahmed, B.Y. COVID-19 and Parkinson’s disease: Shared inflammatory pathways under oxidative stress. Brain Sci. 2020, 10, 807. [Google Scholar] [CrossRef]
- Musgrove, R.E.; Helwig, M.; Bae, E.-J.; Aboutalebi, H.; Lee, S.-J.; Ulusoy, A.; Di Monte, D.A. Oxidative stress in vagal neurons promotes parkinsonian pathology and intercellular α-synuclein transfer. J. Clin. Investig. 2019, 129, 3738–3753. [Google Scholar] [CrossRef]
- Smeyne, R.J.; Eells, J.; Chatterjee, D.; Byrne, M.; Akula, S.M.; Sriramula, S.; O’Rourke, D.P.; Schmidt, P. COVID-19 infection enhances susceptibility to oxidative-stress induced parkinsonism. bioRxiv, 2022; manuscript submitted for review. [Google Scholar]
- Fernández-Castañeda, A.; Lu, P.; Geraghty, A.C.; Song, E.; Lee, M.-H.; Wood, J.; Yalcin, B.; Taylor, K.R.; Dutton, S.; Acosta-Alvarez, L. Mild respiratory SARS-CoV-2 infection can cause multi-lineage cellular dysregulation and myelin loss in the brain. bioRxiv 2022. [Google Scholar] [CrossRef]
- Gibson, E.M.; Nagaraja, S.; Ocampo, A.; Tam, L.T.; Wood, L.S.; Pallegar, P.N.; Greene, J.J.; Geraghty, A.C.; Goldstein, A.K.; Ni, L. Methotrexate chemotherapy induces persistent tri-glial dysregulation that underlies chemotherapy-related cognitive impairment. Cell 2019, 176, 43–55.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geraghty, A.C.; Gibson, E.M.; Ghanem, R.A.; Greene, J.J.; Ocampo, A.; Goldstein, A.K.; Ni, L.; Yang, T.; Marton, R.M.; Paşca, S.P. Loss of adaptive myelination contributes to methotrexate chemotherapy-related cognitive impairment. Neuron 2019, 103, 250–265.e8. [Google Scholar] [CrossRef] [PubMed]
- Monje, M.L.; Vogel, H.; Masek, M.; Ligon, K.L.; Fisher, P.G.; Palmer, T.D. Impaired human hippocampal neurogenesis after treatment for central nervous system malignancies. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2007, 62, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Villeda, S.A.; Luo, J.; Mosher, K.I.; Zou, B.; Britschgi, M.; Bieri, G.; Stan, T.M.; Fainberg, N.; Ding, Z.; Eggel, A. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 2011, 477, 90–94. [Google Scholar] [CrossRef] [Green Version]
- Tiku, V.; Tan, M.W.; Dikic, I. Mitochondrial Functions in Infection and Immunity. Trends Cell Biol. 2020, 30, 263. [Google Scholar] [CrossRef] [Green Version]
- Ferger, A.I.; Campanelli, L.; Reimer, V.; Muth, K.N.; Merdian, I.; Ludolph, A.C.; Witting, A. Effects of mitochondrial dysfunction on the immunological properties of microglia. J. Neuroinflamm. 2010, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Syed, G.H.; Kim, S.J.; Siddiqui, A. Mitochondrial dynamics and viral infections: A close nexus. BBA-Mol. Cell Res. 2015, 1853, 2822. [Google Scholar] [CrossRef] [Green Version]
- Harry, G.J.; Childers, G.; Giridharan, S.; Hernandes, I.L. An association between mitochondria and microglia effector function. What do we think we know? Neuroimmunol. Neuroinflamm. 2020, 7, 150. [Google Scholar] [CrossRef]
- Singh, K.K.; Chaubey, G.; Chen, J.Y.; Suravajhala, P. Decoding SARS-CoV-2 hijacking of host mitochondria in COVID-19 pathogenesis. Am. J. Physiol. Cell Physiol. 2020, 319, C258. [Google Scholar] [CrossRef]
- Shi, T.T.; Yang, F.Y.; Liu, C.; Cao, X.; Lu, J.; Zhang, X.L.; Yuan, M.X.; Chen, C.; Yang, J.K. Angiotensin-converting enzyme 2 regulates mitochondrial function in pancreatic beta-cells. Biochem. Biophys. Res. Commun. 2018, 495, 860. [Google Scholar] [CrossRef] [PubMed]
- Bordt, E.A.; Polster, B.M. NADPH oxidase-and mitochondria-derived reactive oxygen species in proinflammatory microglial activation: A bipartisan affair? Free Radic. Biol. Med. 2014, 76, 34–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clough, E.; Inigo, J.; Chandra, D.; Chaves, L.; Reynolds, J.L.; Aalinkeel, R.; Schwartz, S.A.; Khmaladze, A.; Mahajan, S.D. Mitochondrial dynamics in SARS-CoV-2 spike protein treated human Microglia: Implications for Neuro-COVID. J. Neuroimmune Pharmacol. 2021, 16, 770. [Google Scholar] [CrossRef] [PubMed]
- Pliss, A.; Kuzmin, A.N.; Prasad, P.N.; Mahajan, S.D. Mitochondrial Dysfunction: A Prelude to Neuropathogenesis of SARS-CoV-2. ACS Chem. Neurosci. 2022, 13, 308–312. [Google Scholar] [CrossRef]
- Valenzuela, R.; Rodriguez-Perez, A.I.; Costa-Besada, M.A.; Rivas-Santisteban, R.; Garrido-Gil, P.; Lopez-Lopez, A.; Navarro, G.; Lanciego, J.L.; Franco, R.; Labandeira-Garcia, J.L. An ACE2/Mas-related receptor MrgE axis in dopaminergic neuron mitochondria. Redox Biol. 2021, 46, 102078. [Google Scholar] [CrossRef]
- Labandeira-Garcia, J.L.; Valenzuela, R.; Costa-Besada, M.A.; Villar-Cheda, B.; Rodriguez-Perez, A.I. The intracellular renin-angiotensin system: Friend or foe. Some light from the dopaminergic neurons. Prog. Neurobiol. 2021, 199, 101919. [Google Scholar] [CrossRef]
- Chen, R.; Wang, K.; Yu, J.; Howard, D.; French, L.; Chen, Z.; Wen, C.; Xu, Z. The Spatial and Cell-Type Distribution of SARS-CoV-2 Receptor ACE2 in the Human and Mouse Brains. Front. Neurol. 2021, 11, 573095. [Google Scholar] [CrossRef]
- Di Filippo, M.; Chiasserini, D.; Tozzi, A.; Picconi, B.; Calabresi, P. Mitochondria and the link between neuroinflammation and neurodegeneration. J. Alzheimer’s Dis. 2010, 20, S369–S379. [Google Scholar] [CrossRef] [Green Version]
- Abbas, A.K.; Lichtman, A.; Pillai, S. Basic Immunology: Functions and Disorders of the Immune System, 6e: Sae-E-Book; Elsevier India: New Delhi, India, 2019. [Google Scholar]
- Murphy, K.; Weaver, C. Janeway’s immunobiology; Garland Science: New York, NY, USA, 2016. [Google Scholar]
- Becher, B.; Spath, S.; Goverman, J. Cytokine networks in neuroinflammation. Nat. Rev. Immunol. 2017, 17, 49–59. [Google Scholar] [CrossRef]
- Nathan, C.; Sporn, M. Cytokines in context. J. Cell Biol. 1991, 113, 981–986. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C. Role of pro-and anti-inflammatory cytokines during inflammation: Experimental and clinical findings. J. Biol. Regul. Homeost. Agents 1997, 11, 91–103. [Google Scholar] [PubMed]
- Baylis, D.; Bartlett, D.B.; Patel, H.P.; Roberts, H.C. Understanding how we age: Insights into inflammaging. Longev. Healthspan 2013, 2, 8. [Google Scholar] [CrossRef] [PubMed]
- Minciullo, P.L.; Catalano, A.; Mandraffino, G.; Casciaro, M.; Crucitti, A.; Maltese, G.; Morabito, N.; Lasco, A.; Gangemi, S.; Basile, G. Inflammaging and anti-inflammaging: The role of cytokines in extreme longevity. Arch. Immunol. Ther. Exp. 2016, 64, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Vitale, G.; Capri, M.; Salvioli, S. Inflammaging and ‘Garb-aging’. Trends Endocrinol. Metab. 2017, 28, 199–212. [Google Scholar] [CrossRef] [Green Version]
- Akbar, A.N.; Gilroy, D.W. Aging immunity may exacerbate COVID-19. Science 2020, 369, 256–257. [Google Scholar] [CrossRef]
- Bartleson, J.M.; Radenkovic, D.; Covarrubias, A.J.; Furman, D.; Winer, D.A.; Verdin, E. SARS-CoV-2, COVID-19 and the aging immune system. Nat. Aging 2021, 1, 769–782. [Google Scholar] [CrossRef]
- Odoj, K.; Brawek, B.; Asavapanumas, N.; Mojtahedi, N.; Heneka, M.T.; Garaschuk, O. In vivo mechanisms of cortical network dysfunction induced by systemic inflammation. Brain Behav. Immun. 2021, 96, 113–126. [Google Scholar] [CrossRef]
- Lopes-Paciencia, S.; Saint-Germain, E.; Rowell, M.-C.; Ruiz, A.F.; Kalegari, P.; Ferbeyre, G. The senescence-associated secretory phenotype and its regulation. Cytokine 2019, 117, 15–22. [Google Scholar] [CrossRef]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef]
- Gruber, C. Impaired interferon signature in severe COVID-19. Nat. Rev. Immunol. 2020, 20, 353. [Google Scholar] [CrossRef]
- Britanova, O.V.; Putintseva, E.V.; Shugay, M.; Merzlyak, E.M.; Turchaninova, M.A.; Staroverov, D.B.; Bolotin, D.A.; Lukyanov, S.; Bogdanova, E.A.; Mamedov, I.Z. Age-related decrease in TCR repertoire diversity measured with deep and normalized sequence profiling. J. Immunol. 2014, 192, 2689–2698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egorov, E.S.; Kasatskaya, S.A.; Zubov, V.N.; Izraelson, M.; Nakonechnaya, T.O.; Staroverov, D.B.; Angius, A.; Cucca, F.; Mamedov, I.Z.; Rosati, E. The changing landscape of naive T cell receptor repertoire with human aging. Front Immunol 2018, 9, 1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frasca, D.; Diaz, A.; Romero, M.; Landin, A.M.; Blomberg, B.B. Age effects on B cells and humoral immunity in humans. Ageing Res. Rev. 2011, 10, 330–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidler, S.; Zimmermann, H.W.; Bartneck, M.; Trautwein, C.; Tacke, F. Age-dependent alterations of monocyte subsets and monocyte-related chemokine pathways in healthy adults. BMC Immunol. 2010, 11, 30. [Google Scholar] [CrossRef] [Green Version]
- Ortmann, W.; Kolaczkowska, E. Age is the work of art? Impact of neutrophil and organism age on neutrophil extracellular trap formation. Cell Tissue Res. 2018, 371, 473–488. [Google Scholar] [CrossRef] [Green Version]
- Roodveldt, C.; Christodoulou, J.; Dobson, C.M. Immunological features of α-synuclein in Parkinson’s disease. J. Cell. Mol. Med. 2008, 12, 1820–1829. [Google Scholar] [CrossRef]
- Forloni, G.; Bertani, I.; Calella, A.M.; Thaler, F.; Invernizzi, R. α-Synuclein and Parkinson’s disease: Selective neurodegenerative effect of α-synuclein fragment on dopaminergic neurons in vitro and in vivo. Ann. Neurol. 2000, 47, 632–640. [Google Scholar] [CrossRef]
- Harms, A.S.; Ferreira, S.A.; Romero-Ramos, M. Periphery and brain, innate and adaptive immunity in Parkinson’s disease. Acta Neuropathol. 2021, 141, 527–545. [Google Scholar] [CrossRef]
- Mayne, K.; White, J.A.; McMurran, C.E.; Rivera, F.J.; de la Fuente, A.G. Aging and neurodegenerative disease: Is the adaptive immune system a friend or foe? Front. Aging Neurosci. 2020, 12, 305. [Google Scholar] [CrossRef]
- De Virgilio, A.; Greco, A.; Fabbrini, G.; Inghilleri, M.; Rizzo, M.I.; Gallo, A.; Conte, M.; Rosato, C.; Appiani, M.C.; De Vincentiis, M. Parkinson’s disease: Autoimmunity and neuroinflammation. Autoimmun. Rev. 2016, 15, 1005–1011. [Google Scholar] [CrossRef] [Green Version]
- Garretti, F.; Agalliu, D.; Lindestam Arlehamn, C.S.; Sette, A.; Sulzer, D. Autoimmunity in Parkinson’s Disease: The role of α-synuclein-specific T cells. Front. Immunol. 2019, 10, 303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulzer, D.; Alcalay, R.N.; Garretti, F.; Cote, L.; Kanter, E.; Agin-Liebes, J.; Liong, C.; McMurtrey, C.; Hildebrand, W.H.; Mao, X. T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature 2017, 546, 656–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.-S. Aggregated α-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Maguire-Zeiss, K.A.; Giuliano, R.; Prifti, L.; Venkatesh, K.; Federoff, H.J. Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol. Aging 2008, 29, 1690–1701. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-J.; Patel, S.; Lee, S.-J. Intravesicular localization and exocytosis of α-synuclein and its aggregates. J. Neurosci. 2005, 25, 6016–6024. [Google Scholar] [CrossRef]
- Klegeris, A.; Pelech, S.; Giasson, B.I.; Maguire, J.; Zhang, H.; McGeer, E.G.; McGeer, P.L. α-Synuclein activates stress signaling protein kinases in THP-1 cells and microglia. Neurobiol. Aging 2008, 29, 739–752. [Google Scholar] [CrossRef]
- Hou, L.; Bao, X.; Zang, C.; Yang, H.; Sun, F.; Che, Y.; Wu, X.; Li, S.; Zhang, D.; Wang, Q. Integrin CD11b mediates α-synuclein-induced activation of NADPH oxidase through a Rho-dependent pathway. Redox Biol. 2018, 14, 600–608. [Google Scholar] [CrossRef]
- Faber, I.; Pedro, R.P.; Brandao, F.M.; Diógenes, D.; de Carvalho, B.; Fernando, B.M.; Francisco, C. Coronavirus disease 2019 and parkinsonism: A non-post-encephalitic case. Mov Disord 2020, 35, 1721–1722. [Google Scholar] [CrossRef]
- Makhoul, K.; Jankovic, J. Parkinson’s disease after COVID-19. J. Neurol. Sci. 2021, 422, 117331. [Google Scholar] [CrossRef]
- Estrada, E. Cascading from SARS-CoV-2 to Parkinson’s Disease through Protein-Protein Interactions. Viruses 2021, 13, 897. [Google Scholar] [CrossRef]
- Sulzer, D.; Antonini, A.; Leta, V.; Nordvig, A.; Smeyne, R.J.; Goldman, J.E.; Al-Dalahmah, O.; Zecca, L.; Sette, A.; Bubacco, L. COVID-19 and possible links with Parkinson’s disease and parkinsonism: From bench to bedside. NPJ Parkinson’s Dis. 2020, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Amruta, N.; Chastain, W.H.; Paz, M.; Solch, R.J.; Murray-Brown, I.C.; Befeler, J.B.; Gressett, T.E.; Longo, M.T.; Engler-Chiurazzi, E.B.; Bix, G. SARS-CoV-2 mediated neuroinflammation and the impact of COVID-19 in neurological disorders. Cytokine Growth Factor Rev. 2021, 58, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Brundin, P.; Nath, A.; Beckham, J.D. Is COVID-19 a perfect storm for Parkinson’s disease? Trends Neurosci. 2020, 43, 931–933. [Google Scholar] [CrossRef] [PubMed]
- Antonini, A.; Leta, V.; Teo, J.; Chaudhuri, K.R. Outcome of Parkinson’s Disease patients affected by COVID-19. Mov. Disord. 2020. [Google Scholar] [CrossRef]
- Fearon, C.; Fasano, A. Parkinson’s disease and the COVID-19 pandemic. J. Parkinson’s Dis. 2021, 11, 431–444. [Google Scholar] [CrossRef]
- Philippens, I.H.; Boszormenyi, K.P.; Wubben, J.A.; Fagrouch, Z.C.; van Driel, N.; Mayenburg, A.Q.; Lozovagia, D.; Roos, E.; Schurink, B.; Bugiani, M. SARS-CoV-2 causes brain inflammation and induces Lewy body formation in macaques. bioRxiv 2021. [Google Scholar]
- Maiese, A.; Manetti, A.C.; Bosetti, C.; Del Duca, F.; La Russa, R.; Frati, P.; Di Paolo, M.; Turillazzi, E.; Fineschi, V. SARS-CoV-2 and the brain: A review of the current knowledge on neuropathology in COVID-19. Brain Pathol. 2021, 31, e13013. [Google Scholar] [CrossRef]
- Colombo, D.; Falasca, L.; Marchioni, L.; Tammaro, A.; Adebanjo, G.A.R.; Ippolito, G.; Zumla, A.; Piacentini, M.; Nardacci, R.; Del Nonno, F. Neuropathology and Inflammatory Cell Characterization in 10 Autoptic COVID-19 Brains. Cells 2021, 10, 2262. [Google Scholar] [CrossRef]
- Kantonen, J.; Mahzabin, S.; Mäyränpää, M.I.; Tynninen, O.; Paetau, A.; Andersson, N.; Sajantila, A.; Vapalahti, O.; Carpén, O.; Kekäläinen, E. Neuropathologic features of four autopsied COVID-19 patients. Brain Pathol 2020, 30, 1012–1016. [Google Scholar] [CrossRef]
- Matschke, J.; Lütgehetmann, M.; Hagel, C.; Sperhake, J.P.; Schröder, A.S.; Edler, C.; Mushumba, H.; Fitzek, A.; Allweiss, L.; Dandri, M. Neuropathology of patients with COVID-19 in Germany: A post-mortem case series. Lancet Neurol. 2020, 19, 919–929. [Google Scholar] [CrossRef]
- Thakur, K.T.; Miller, E.H.; Glendinning, M.D.; Al-Dalahmah, O.; Banu, M.A.; Boehme, A.K.; Boubour, A.L.; Bruce, S.S.; Chong, A.M.; Claassen, J. COVID-19 neuropathology at columbia university irving medical center/New York presbyterian hospital. Brain 2021, 144, 2696–2708. [Google Scholar] [CrossRef] [PubMed]
- Poloni, T.E.; Medici, V.; Moretti, M.; Visonà, S.D.; Cirrincione, A.; Carlos, A.F.; Davin, A.; Gagliardi, S.; Pansarasa, O.; Cereda, C. COVID-19-related neuropathology and microglial activation in elderly with and without dementia. Brain Pathol. 2021, 31, e12997. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.; Le Quesne, J.; Officer-Jones, L.; Teodòsio, A.; Thaventhiran, J.; Ficken, C.; Goddard, M.; Smith, C.; Menon, D.; Allinson, K. Neuropathological findings in two patients with fatal COVID-19. Neuropathol. Appl. Neurobiol. 2021, 47, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Wierzba-Bobrowicz, T.; Krajewski, P.; Tarka, S.; Acewicz, A.; Felczak, P.; Stępień, T.; Golan, M.P.; Grzegorczyk, M. Neuropathological analysis of the brains of fifty-two patients with COVID-19. Folia Neuropathol. 2021, 59, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Meinhardt, J.; Radke, J.; Dittmayer, C.; Franz, J.; Thomas, C.; Mothes, R.; Laue, M.; Schneider, J.; Brünink, S.; Greuel, S. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat. Neurosci. 2021, 24, 168–175. [Google Scholar] [CrossRef]
- Reichard, R.R.; Kashani, K.B.; Boire, N.A.; Constantopoulos, E.; Guo, Y.; Lucchinetti, C.F. Neuropathology of COVID-19: A spectrum of vascular and acute disseminated encephalomyelitis (ADEM)-like pathology. Acta Neuropathol. 2020, 140, 1–6. [Google Scholar] [CrossRef]
- Rhodes, R.H.; Love, G.L.; Lameira, F.D.S.; Sadough, M.S.; Fox, S.E.; Vander Heide, R.S. Acute endotheliitis (type 3 hypersensitivity vasculitis) in ten COVID-19 autopsy brains. medRxiv 2021. [Google Scholar] [CrossRef]
- Schurink, B.; Roos, E.; Radonic, T.; Barbe, E.; Bouman, C.S.; de Boer, H.H.; de Bree, G.J.; Bulle, E.B.; Aronica, E.M.; Florquin, S. Viral presence and immunopathology in patients with lethal COVID-19: A prospective autopsy cohort study. Lancet Microbe 2020, 1, e290–e299. [Google Scholar] [CrossRef]
- Solomon, I.H.; Normandin, E.; Bhattacharyya, S.; Mukerji, S.S.; Keller, K.; Ali, A.S.; Adams, G.; Hornick, J.L.; Padera Jr, R.F.; Sabeti, P. Neuropathological features of Covid-19. N. Engl. J. Med. 2020, 383, 989–992. [Google Scholar] [CrossRef]
- Bradley, B.T.; Maioli, H.; Johnston, R.; Chaudhry, I.; Fink, S.L.; Xu, H.; Najafian, B.; Deutsch, G.; Lacy, J.M.; Williams, T. Histopathology and ultrastructural findings of fatal COVID-19 infections in Washington State: A case series. Lancet 2020, 396, 320–332. [Google Scholar] [CrossRef]
- Bryce, C.; Grimes, Z.; Pujadas, E.; Ahuja, S.; Beasley, M.B.; Albrecht, R.; Hernandez, T.; Stock, A.; Zhao, Z.; AlRasheed, M.R. Pathophysiology of SARS-CoV-2: The Mount Sinai COVID-19 autopsy experience. Mod. Pathol. 2021, 34, 1456–1467. [Google Scholar] [CrossRef] [PubMed]
- Remmelink, M.; De Mendonça, R.; D’Haene, N.; De Clercq, S.; Verocq, C.; Lebrun, L.; Lavis, P.; Racu, M.-L.; Trépant, A.-L.; Maris, C. Unspecific post-mortem findings despite multiorgan viral spread in COVID-19 patients. Crit. Care 2020, 24, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jaunmuktane, Z.; Mahadeva, U.; Green, A.; Sekhawat, V.; Barrett, N.A.; Childs, L.; Shankar-Hari, M.; Thom, M.; Jäger, H.R.; Brandner, S. Microvascular injury and hypoxic damage: Emerging neuropathological signatures in COVID-19. Acta Neuropathol. 2020, 140, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Hanley, B.; Naresh, K.N.; Roufosse, C.; Nicholson, A.G.; Weir, J.; Cooke, G.S.; Thursz, M.; Manousou, P.; Corbett, R.; Goldin, R. Histopathological findings and viral tropism in UK patients with severe fatal COVID-19: A post-mortem study. Lancet Microbe 2020, 1, e245–e253. [Google Scholar] [CrossRef]
- Fabbri, V.P.; Foschini, M.P.; Lazzarotto, T.; Gabrielli, L.; Cenacchi, G.; Gallo, C.; Aspide, R.; Frascaroli, G.; Cortelli, P.; Riefolo, M. Brain ischemic injury in COVID-19-infected patients: A series of 10 post-mortem cases. Brain Pathol. 2021, 31, 205. [Google Scholar] [CrossRef]
- von Weyhern, C.H.; Kaufmann, I.; Neff, F.; Kremer, M. Early evidence of pronounced brain involvement in fatal COVID-19 outcomes. Lancet 2020, 395, e109. [Google Scholar] [CrossRef]
- Al-Dalahmah, O.; Thakur, K.T.; Nordvig, A.S.; Prust, M.L.; Roth, W.; Lignelli, A.; Uhlemann, A.-C.; Miller, E.H.; Kunnath-Velayudhan, S.; Del Portillo, A. Neuronophagia and microglial nodules in a SARS-CoV-2 patient with cerebellar hemorrhage. Acta Neuropathol. Commun. 2020, 8, 1–7. [Google Scholar] [CrossRef]
- Kirschenbaum, D.; Imbach, L.L.; Ulrich, S.; Rushing, E.J.; Keller, E.; Reimann, R.R.; Frauenknecht, K.B.; Lichtblau, M.; Witt, M.; Hummel, T. Inflammatory olfactory neuropathy in two patients with COVID-19. Lancet 2020, 396, 166. [Google Scholar] [CrossRef]
- Deigendesch, N.; Sironi, L.; Kutza, M.; Wischnewski, S.; Fuchs, V.; Hench, J.; Frank, A.; Nienhold, R.; Mertz, K.D.; Cathomas, G. Correlates of critical illness-related encephalopathy predominate postmortem COVID-19 neuropathology. Acta Neuropathol. 2020, 140, 583–586. [Google Scholar] [CrossRef]
- Cosentino, G.; Todisco, M.; Hota, N.; Della Porta, G.; Morbini, P.; Tassorelli, C.; Pisani, A. Neuropathological findings from COVID-19 patients with neurological symptoms argue against a direct brain invasion of SARS-CoV-2: A critical systematic review. Eur. J. Neurol. 2021, 28, 3856–3865. [Google Scholar] [CrossRef]
- Bulfamante, G.; Bocci, T.; Falleni, M.; Campiglio, L.; Coppola, S.; Tosi, D.; Chiumello, D.; Priori, A. Brainstem neuropathology in two cases of COVID-19: SARS-CoV-2 trafficking between brain and lung. J. Neurol. 2021, 268, 4486–4491. [Google Scholar] [CrossRef] [PubMed]
- Helmich, R.C.; Bloem, B.R. The impact of the COVID-19 pandemic on Parkinson’s disease: Hidden sorrows and emerging opportunities. J. Parkinson’s Dis. 2020, 10, 351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anghelescu, B.A.-M.; Bruno, V.; Martino, D.; Roach, P. Effects of the COVID-19 pandemic on Parkinson’s disease: A single-centered qualitative study. Can. J. Neurol. Sci. 2021, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fasano, A.; Elia, A.E.; Dallocchio, C.; Canesi, M.; Alimonti, D.; Sorbera, C.; Alonso-Canovas, A.; Pezzoli, G. Predictors of COVID-19 outcome in Parkinson’s disease. Parkinsonism Relat. Disord. 2020, 78, 134–137. [Google Scholar] [CrossRef]
- Méndez-Guerrero, A.; Laespada-García, M.I.; Gómez-Grande, A.; Ruiz-Ortiz, M.; Blanco-Palmero, V.A.; Azcarate-Diaz, F.J.; Rábano-Suárez, P.; Álvarez-Torres, E.; de Fuenmayor-Fernández de la Hoz, C.P.; Vega Pérez, D.; et al. Acute hypokinetic-rigid syndrome following SARS-CoV-2 infection. Neurology 2020, 95, e2109–e2118. [Google Scholar] [CrossRef]
- Cohen, M.E.; Eichel, R.; Steiner-Birmanns, B.; Janah, A.; Ioshpa, M.; Bar-Shalom, R.; Paul, J.J.; Gaber, H.; Skrahina, V.; Bornstein, N.M.; et al. A case of probable Parkinson’s disease after SARS-CoV-2 infection. Lancet Neurol. 2020, 19, 804–805. [Google Scholar] [CrossRef]
- Rao, A.R.; Hidayathullah, S.M.; Hegde, K.; Adhikari, P. Parkinsonism: An emerging post COVID sequelae. IDCases 2022, e01388. [Google Scholar] [CrossRef]
- Tolosa, E.; Wenning, G.; Poewe, W. The diagnosis of Parkinson’s disease. Lancet Neurol. 2006, 5, 75–86. [Google Scholar] [CrossRef]
- Deutschländer, A.; Ross, O.; Dickson, D.; Wszolek, Z. Atypical parkinsonian syndromes: A general neurologist’s perspective. Eur. J. Neurol. 2018, 25, 41–58. [Google Scholar] [CrossRef]
- Tolosa, E.; Garrido, A.; Scholz, S.W.; Poewe, W. Challenges in the diagnosis of Parkinson’s disease. Lancet Neurol. 2021, 20, 385–397. [Google Scholar] [CrossRef]
- Stamelou, M.; Hoeglinger, G.U. Atypical parkinsonism: An update. Curr. Opin. Neurol. 2013, 26, 401–405. [Google Scholar] [CrossRef] [PubMed]
- Levin, J.; Kurz, A.; Arzberger, T.; Giese, A.; Höglinger, G.U. The differential diagnosis and treatment of atypical parkinsonism. Dtsch. Ärzteblatt. Int. 2016, 113, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, F.; Marsili, L.; Truong, D.D. Parkinsonism in viral, paraneoplastic, and autoimmune diseases. J. Neurol. Sci. 2022, 433, 120014. [Google Scholar] [CrossRef] [PubMed]
- de Marcaida, J.A.; Lahrmann, J.; Machado, D.; Bluth, L.; Dagostine, M.; Moro-de Casillas, M.; Bortan, E.; Kanchana, S.; Alberts, M. Clinical Characteristics of Coronavirus Disease 2019 (COVID-19) among Patients at a Movement Disorders Center. Geriatrics 2020, 5, 54. [Google Scholar] [CrossRef]
- Cámara, A.; Compta, Y.; Pérez-Soriano, A.; Montagut, N.; Baixauli, M.; Maragall, L.; Ludeña, E.; de Los Reyes, J.C.L.; Peri-Cusi, L.; Fernández, N. Effects of COVID-19 pandemic and lockdown on people with multiple system atrophy participating in a therapeutic education program. Parkinsonism Relat. Disord. 2021, 86, 78–80. [Google Scholar] [CrossRef]
- Hawkes, C.H.; Del Tredici, K.; Braak, H. Parkinson’s disease: A dual-hit hypothesis. Neuropathol. Appl. Neurobiol. 2007, 33, 599–614. [Google Scholar] [CrossRef]
- Alster, P.; Madetko, N.; Koziorowski, D.; Friedman, A. Microglial activation and inflammation as a factor in the pathogenesis of progressive supranuclear palsy (PSP). Front. Neurosci. 2020, 14, 893. [Google Scholar] [CrossRef]
- Alster, P.; Madetko, N.; Koziorowski, D.; Friedman, A. Progressive Supranuclear Palsy—Parkinsonism Predominant (PSP-P)—A Clinical Challenge at the Boundaries of PSP and Parkinson’s Disease (PD). Front. Neurol. 2020, 11, 180. [Google Scholar] [CrossRef]
- Madetko, N.; Migda, B.; Alster, P.; Turski, P.; Koziorowski, D.; Friedman, A. Platelet-to-lymphocyte ratio and neutrophil-tolymphocyte ratio may reflect differences in PD and MSA-P neuroinflammation patterns. Neurol. I Neurochir. Pol. 2022, 196. [Google Scholar] [CrossRef]
- Mosley, R.L.; Benner, E.J.; Kadiu, I.; Thomas, M.; Boska, M.D.; Hasan, K.; Laurie, C.; Gendelman, H.E. Neuroinflammation, oxidative stress, and the pathogenesis of Parkinson’s disease. Clin. Neurosci. Res. 2006, 6, 261–281. [Google Scholar] [CrossRef] [Green Version]
- Tansey, M.G.; Goldberg, M.S. Neuroinflammation in Parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 2010, 37, 510–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, V.; Alquraish, D.; Sarfraz, Z.; Sarfraz, A.; Nagpal, S.; Singh Shrestha, P.; Mukherjee, D.; Guntipalli, P.; Sánchez Velazco, D.F.; Bhatnagar, A. The influence of Coronavirus disease-2019 (COVID-19) On Parkinson’s disease: An updated systematic review. J. Prim. Care Community Health 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Salari, M.; Etemadifar, M.; Zali, A.; Aminzade, Z.; Navalpotro-Gomez, I.; Fateh, S.T. Covid-19 in Parkinson’s Disease treated by drugs or brain stimulation. Neurología 2021. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Long, X.; Zhu, C.; Wang, R.; Hu, S.; Wang, T.; Li, J.; Lin, Z.; Xiong, N. Management of a Parkinson’s disease patient with severe COVID-19 pneumonia. Ther. Adv. Chronic Dis. 2020, 11, 2040622320949423. [Google Scholar] [CrossRef]
- Lau, Y.H.; Lau, K.M.; Ibrahim, N.M. Management of Parkinson’s Disease in the COVID-19 Pandemic and Future Perspectives in the Era of Vaccination. J. Mov. Disord. 2021, 14, 177. [Google Scholar] [CrossRef]
- Scherbaum, R.; Kwon, E.H.; Richter, D.; Bartig, D.; Gold, R.; Krogias, C.; Tönges, L. Clinical Profiles and Mortality of COVID-19 Inpatients with Parkinson’s Disease in Germany. Mov. Disord. 2021, 36, 1049–1057. [Google Scholar] [CrossRef]
- Artusi, C.A.; Romagnolo, A.; Ledda, C.; Zibetti, M.; Rizzone, M.G.; Montanaro, E.; Bozzali, M.; Lopiano, L. COVID-19 and Parkinson’s disease: What we know so far? J. Parkinson’s Dis. 2021, 1–10. [Google Scholar] [CrossRef]
- Artusi, C.A.; Romagnolo, A.; Imbalzano, G.; Marchet, A.; Zibetti, M.; Rizzone, M.G.; Lopiano, L. COVID-19 in Parkinson’s disease: Report on prevalence and outcome. Parkinsonism Relat. Disord. 2020, 80, 7–9. [Google Scholar] [CrossRef]
- Zhai, H.; Lv, Y.; Xu, Y.; Wu, Y.; Zeng, W.; Wang, T.; Cao, X.; Xu, Y. Characteristic of Parkinson’s disease with severe COVID-19: A study of 10 cases from Wuhan. J. Neural. Transm. 2021, 128, 37–48. [Google Scholar] [CrossRef]
- Ghosh, R.; Ray, A.; Roy, D.; Das, S.; Dubey, S.; Benito-León, J. Parkinsonism with akinetic mutism following osmotic demyelination syndrome in a SARS-CoV-2 infected elderly diabetic woman: A case report. Neurologia 2021. [Google Scholar] [CrossRef]
- Ong, T.L.; Nor, K.M.; Yusoff, Y.; Sapuan, S. COVID-19 Associated Acute Necrotizing Encephalopathy Presenting as Parkinsonism and Myorhythmia. J. Mov. Disord. 2022, 15, 89. [Google Scholar] [CrossRef] [PubMed]
- Morassi, M.; Palmerini, F.; Nici, S.; Magni, E.; Savelli, G.; Guerra, U.P.; Chieregato, M.; Morbelli, S.; Vogrig, A. SARS-CoV-2-related encephalitis with prominent parkinsonism: Clinical and FDG-PET correlates in two patients. J. Neurol. 2021, 268, 3980–3987. [Google Scholar] [CrossRef] [PubMed]
- Rosen, B.; Kurtishi, A.; Vazquez-Jimenez, G.R.; Møller, S.G. The Intersection of Parkinson’s Disease, Viral Infections, and COVID-19. Mol. Neurobiol. 2021, 58, 4477–4486. [Google Scholar] [CrossRef] [PubMed]
- Leta, V.; Rodríguez-Violante, M.; Abundes, A.; Rukavina, K.; Teo, J.T.; Falup-Pecurariu, C.; Irincu, L.; Rota, S.; Bhidayasiri, R.; Storch, A. Parkinson’s Disease and Post–COVID-19 Syndrome: The Parkinson’s Long-COVID Spectrum. Mov. Disord. 2021, 36, 1287. [Google Scholar] [CrossRef]
- Zheng, K.S.; Dorfman, B.J.; Christos, P.J.; Khadem, N.R.; Henchcliffe, C.; Piboolnurak, P.; Nirenberg, M.J. Clinical characteristics of exacerbations in Parkinson disease. Neurology 2012, 18, 120. [Google Scholar] [CrossRef] [Green Version]
- Bougakov, D.; Podell, K.; Goldberg, E. Multiple Neuroinvasive Pathways in COVID-19. Mol. Neurobiol. 2021, 58, 564–575. [Google Scholar] [CrossRef]
- Baig, A.M. Neurological manifestations in COVID-19 caused by SARS-CoV-2. CNS Neurosci. Ther. 2020, 26, 499. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Supports: Cause/Unmasking/Not Support | In Vitro/In Vivo/Clinical | Primary Conclusions |
|---|---|---|
| Cause | Clinical | Three published single-case reports describing patients with COVID-19 developing clinical parkinsonism within 2–5 weeks of contracting SARS-CoV-2. As seen with West Nile virus and Western Equine Encephalitis virus, α-synuclein expression increases during viral infection of CNS, suggesting that SARS-CoV-2 infection could predispose individuals to the development of PD later in life. Brundin et al. [148] |
| Cause | Clinical | A case study of an elderly diabetic woman with parkinsonism with akinetic mutism following non-dyselectrolytemic osmotic demyelination syndrome, which was precipitated by COVID-19 infection induced hyperglycemic hyper-osmolar state. She was placed on levodopa/carbidopa and pramipexole, and after two months of follow-up her features of parkinsonism improved significantly, but with only mild improvement of the features associated with akinetic mutism. Ghosh et al. [205] |
| Cause | Clinical | A previously healthy 31-year-old man tested positive for COVID-19 and developed acute necrotizing encephalopathy (ANEC), with presenting features of parkinsonism and myorhythmia. Myorhythmia can also occur alongside other movement disorders, such as dystonia and parkinsonism, due to disrupted basal gangliathalamo-frontal cortical circuits. The exact pathogenesis of ANEC is not entirely clear, but systemic inflammatory insult and hypercytokinemia have been postulated to trigger necrotic brain lesions in patients with ANEC. Ong et al. [206] |
| Cause | In vitro | Identification that the SARS-CoV-2 nucleocapsid protein (N-protein) induces the aggregation of αSYN in a test tube. In the presence of N-protein, the onset of α-synuclein aggregation into amyloid fibrils is strongly accelerated, indicating that N-protein facilitates the formation of a critical nucleus for aggregation. These experiments suggest that SARS-CoV-2 infection and PD might originate from a molecular interaction between virus protein and α-synuclein. Semerdzhiev et al. [78] |
| Cause | Clinical | Three patients developed parkinsonism while infected with COVID-19, all of whom required levodopa-carbidopa therapy for recovery despite having no prodromal PD symptoms prior to COVID-19 infection. The authors concluded that parkinsonism could be a post-COVID-19 sequelae. Rao et al. [182] |
| Cause | In vivo | Authors infected macaques with SARS-CoV-2 and demonstrated brain inflammation and post-mortem studies uncovered Lewy bodies were not present in controls. They conclude that this data is a serious warning for potential COVID-19-related neurodegeneration (particularly PD given the presence of hallmark Lewy bodies) even after asymptomatic or mild infection. Philippens et al. [151] |
| Cause | Clinical | Two cases of patients with COVID-19 encephalopathy who developed parkinsonism without a history of prodromal PD symptoms. FDG-PT/CT imaging showed distinct areas of hypo- and hyper-metabolism in comparison to 48 healthy controls. Authors state that while they cannot dismiss symptom development due to unmasking, given the patients’ lack of prior PD prodromal symptoms and motor features prior to infection, rapid onset of parkinsonism after encephalitis, and lack of improvement after discontinuing neuroleptics and initiating levodopa, that is unlikely. Morassi et al. [207] |
| Unmasking | Clinical | Single case report of patient who developed parkinsonism within days of COVID-19 symptom onset. Makhoul and Jankovic [144] |
| Not Support | Clinical | CSF PCR for SARS-CoV-2 was negative for 100% (76/76) in samples analyzed that were assessed previously and were positive for SARS-CoV-2. Jarius et al. [73] |
| Not Support/Cause | In vivo | RNA viruses upregulate αSYN in neurons, which subsequently can activate the interferon-mediated-anti-viral defense mechanism in innate immunity. However, long term consequences could lead to chronic inflammation with the development or progression of PD. Rosen et al. [208] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morowitz, J.M.; Pogson, K.B.; Roque, D.A.; Church, F.C. Role of SARS-CoV-2 in Modifying Neurodegenerative Processes in Parkinson’s Disease: A Narrative Review. Brain Sci. 2022, 12, 536. https://doi.org/10.3390/brainsci12050536
Morowitz JM, Pogson KB, Roque DA, Church FC. Role of SARS-CoV-2 in Modifying Neurodegenerative Processes in Parkinson’s Disease: A Narrative Review. Brain Sciences. 2022; 12(5):536. https://doi.org/10.3390/brainsci12050536
Chicago/Turabian StyleMorowitz, Jeremy M., Kaylyn B. Pogson, Daniel A. Roque, and Frank C. Church. 2022. "Role of SARS-CoV-2 in Modifying Neurodegenerative Processes in Parkinson’s Disease: A Narrative Review" Brain Sciences 12, no. 5: 536. https://doi.org/10.3390/brainsci12050536
APA StyleMorowitz, J. M., Pogson, K. B., Roque, D. A., & Church, F. C. (2022). Role of SARS-CoV-2 in Modifying Neurodegenerative Processes in Parkinson’s Disease: A Narrative Review. Brain Sciences, 12(5), 536. https://doi.org/10.3390/brainsci12050536