
Advances on Cellular Clonotypic Immunity in Amyotrophic Lateral Sclerosis


 , ,
, , 
 and
and 
Abstract
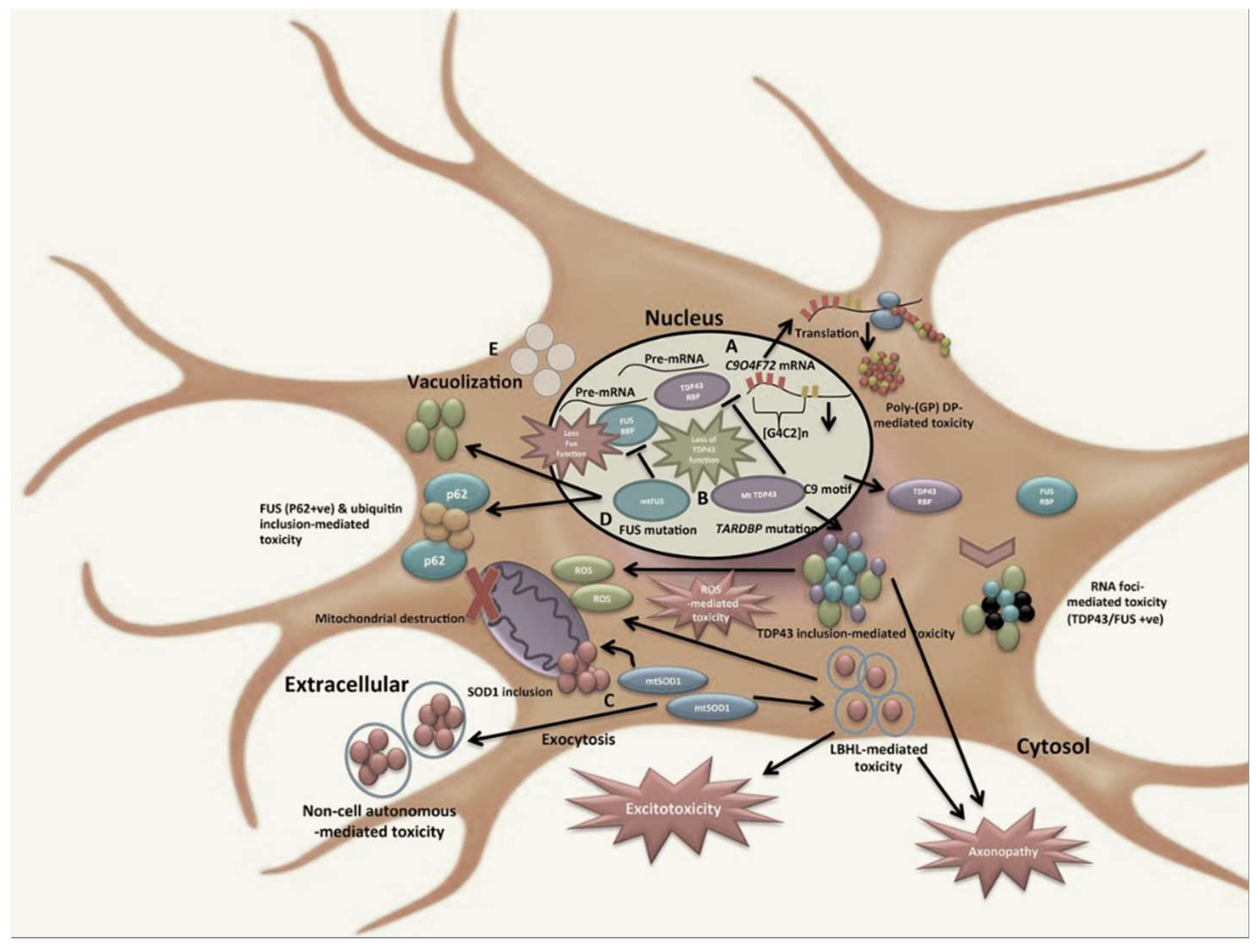
1. Introduction
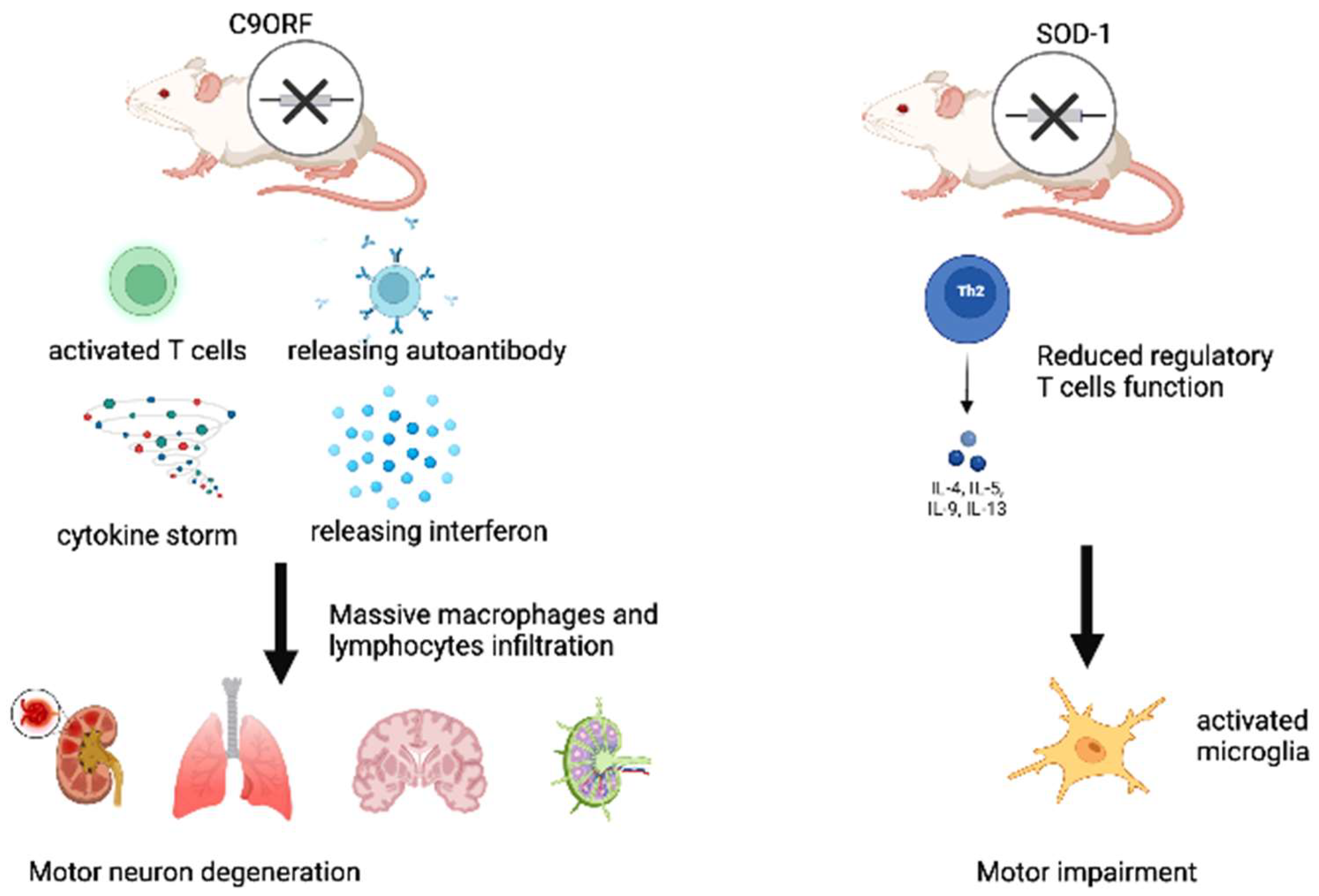
2. Recent Evidence on Clonotypic Immunity in ALS
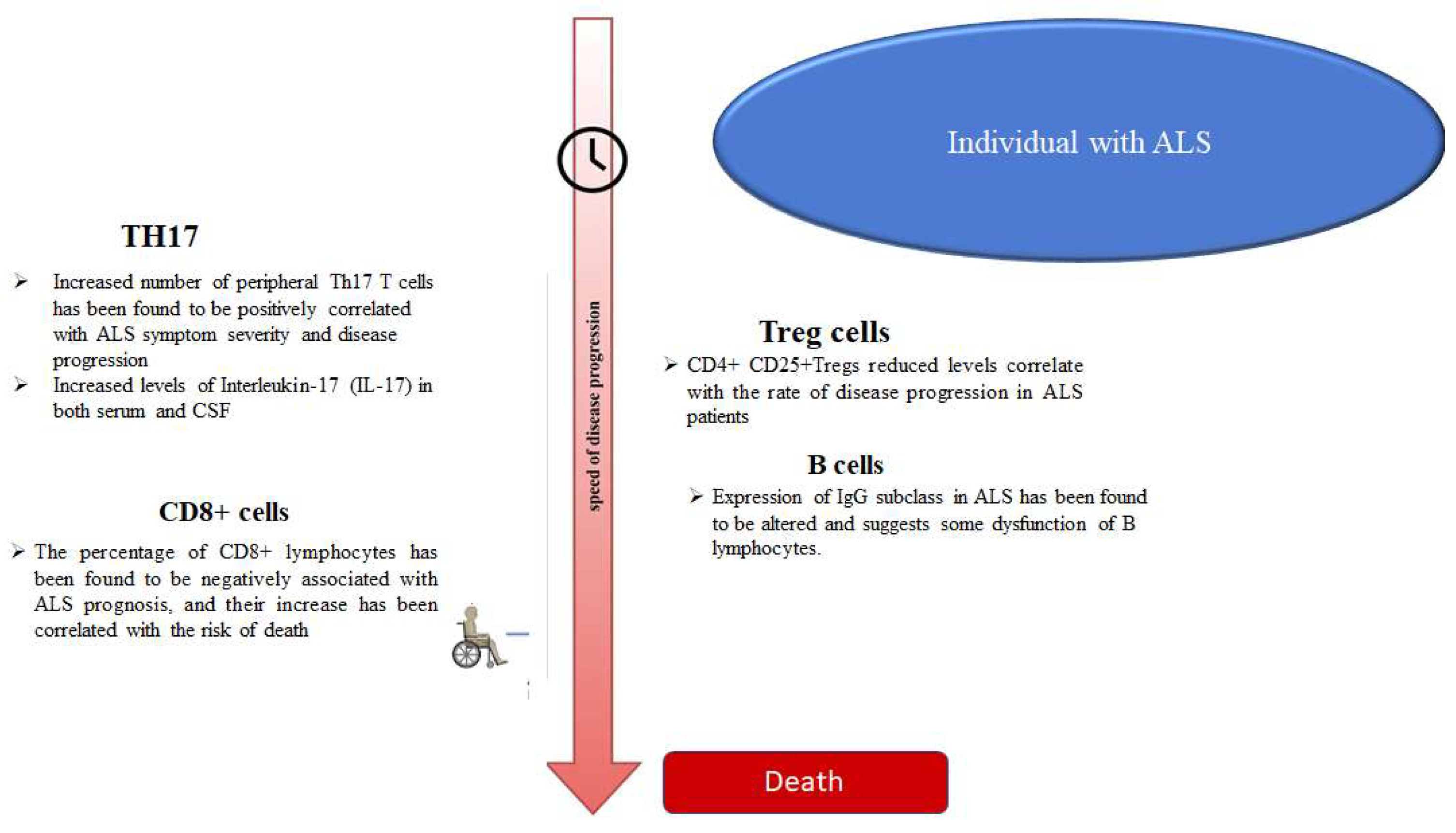
3. T Helper 17 Cells (Th17)
4. CD8+ T Cells
5. Natural Killer (NK) Cells
6. Regulatory T (Treg) Cells
7. B-Cells and Immunoglobulins
8. Immunotherapy in ALS: Promising Data
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hulisz, D. Amyotrophic Lateral Sclerosis: Disease State Overview. Am. J. Manag. Care 2018, 24, S320–S326. [Google Scholar] [PubMed]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Liu, E.; Karpf, L.; Bohl, D. Neuroinflammation in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia and the Interest of Induced Pluripotent Stem Cells to Study Immune Cells Interactions With Neurons. Front. Mol. Neurosci. 2021, 14, 767041. [Google Scholar] [CrossRef] [PubMed]
- Mayne, K.; White, J.A.; Mcmurran, C.E.; Rivera, F.J.; De La Fuente, A.G. Aging and Neurodegenerative Disease: Is the Adaptive Immune System a Friend or Foe? Front. Aging Neurosci. 2020, 12, 572090. [Google Scholar] [CrossRef]
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010, 125, S3–S23. [Google Scholar] [CrossRef]
- Ellwardt, E.; Walsh, J.T.; Kipnis, J.; Zipp, F. Understanding the Role of T Cells in CNS Homeostasis. Trends Immunol. 2016, 37, 154–165. [Google Scholar] [CrossRef]
- Radjavi, A.; Smirnov, I.; Kipnis, J. Brain antigen-reactive CD4+ T cells are sufficient to support learning behavior in mice with limited T cell repertoire. Brain Behav. Immun. 2014, 35, 58–63. [Google Scholar] [CrossRef]
- Longinetti, E.; Sveinsson, O.; Press, R.; Ye, W.; Ingre, C.; Piehl, F.; Fang, F. ALS patients with concurrent neuroinflammatory disorders; a nationwide clinical records study. Amyotroph. Lateral Scler. Front. Degener. 2022, 23, 209–219. [Google Scholar] [CrossRef]
- Yang, Y.; Pan, D.; Gong, Z.; Tang, J.; Li, Z.; Ding, F.; Liu, M.; Zhang, M. Decreased blood CD4+ T lymphocyte helps predict cognitive impairment in patients with amyotrophic lateral sclerosis. BMC Neurol. 2021, 21, 157. [Google Scholar] [CrossRef]
- Mori, K.; Weng, S.-M.; Arzberger, T.; May, S.; Rentzsch, K.; Kremmer, E.; Schmid, B.; Kretzschmar, H.A.; Cruts, M.; Van Broeckhoven, C.; et al. The C9orf72 GGGGCC Repeat Is Translated into Aggregating Dipeptide-Repeat Proteins in FTLD/ALS. Science 2013, 339, 1335–1338. [Google Scholar] [CrossRef]
- Atanasio, A.; Decman, V.; White, D.; Ramos, M.; Ikiz, B.; Lee, H.-C.; Siao, C.-J.; Brydges, S.; LaRosa, E.; Bai, Y.; et al. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production and glomerulonephropathy in mice. Sci. Rep. 2016, 6, 23204. [Google Scholar] [CrossRef] [PubMed]
- McCauley, M.E.; O’Rourke, J.G.; Yáñez, A.; Markman, J.L.; Ho, R.; Wang, X.; Chen, S.; Lall, D.; Jin, M.; Muhammad, A.K.M.G.; et al. C9orf72 in myeloid cells suppresses STING-induced inflammation. Nature 2020, 585, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Burberry, A.; Suzuki, N.; Wang, J.-Y.; Moccia, R.; Mordes, D.A.; Stewart, M.H.; Suzuki-Uematsu, S.; Ghosh, S.; Singh, A.; Merkle, F.T.; et al. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci. Transl. Med. 2016, 8, 347ra93. [Google Scholar] [CrossRef] [PubMed]
- Sudria-Lopez, E.; Koppers, M.; De Wit, M.; Van Der Meer, C.; Westeneng, H.-J.; Zundel, C.A.C.; Youssef, S.A.; Harkema, L.; De Bruin, A.; Veldink, J.H.; et al. Full ablation of C9orf72 in mice causes immune system-related pathology and neoplastic events but no motor neuron defects. Acta Neuropathol. 2016, 132, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Chiu, I.M.; Chen, A.; Zheng, Y.; Kosaras, B.; Tsiftsoglou, S.A.; Vartanian, T.K.; Brown, R.H.; Carroll, M.C. T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proc. Natl. Acad. Sci. USA 2008, 105, 17913–17918. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.-X.; Zhai, H.; Bigio, E.H.; Yan, J.; Fecto, F.; Ajroud, K.; Mishra, M.; Ajroud-Driss, S.; Heller, S.; Sufit, R.; et al. FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann. Neurol. 2010, 67, 739–748. [Google Scholar] [CrossRef]
- Bright, F.; Chan, G.; van Hummel, A.; Ittner, L.; Ke, Y. TDP-43 and Inflammation: Implications for Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Int. J. Mol. Sci. 2021, 22, 7781. [Google Scholar] [CrossRef]
- Zhao, W.; Beers, D.R.; Liao, B.; Henkel, J.S.; Appel, S.H. Regulatory T lymphocytes from ALS mice suppress microglia and effector T lymphocytes through different cytokine-mediated mechanisms. Neurobiol. Dis. 2012, 48, 418–428. [Google Scholar] [CrossRef]
- Boillée, S.; Yamanaka, K.; Lobsiger, C.S.; Copeland, N.G.; Jenkins, N.A.; Kassiotis, G.; Kollias, G.; Cleveland, D.W. Onset and Progression in Inherited ALS Determined by Motor Neurons and Microglia. Science 2006, 312, 1389–1392. [Google Scholar] [CrossRef]
- Hooten, K.G.; Beers, D.R.; Zhao, W.; Appel, S.H. Protective and Toxic Neuroinflammation in Amyotrophic Lateral Sclerosis. Neurotherapeutics 2015, 12, 364–375. [Google Scholar] [CrossRef]
- Seksenyan, A.; Ron-Harel, N.; Azoulay, D.; Cahalon, L.; Cardon, M.; Rogeri, P.; Ko, M.K.; Weil, M.; Bulvik, S.; Rechavi, G.; et al. Thymic involution, a co-morbidity factor in amyotrophic lateral sclerosis. J. Cell. Mol. Med. 2009, 14, 2470–2482. [Google Scholar] [CrossRef] [PubMed]
- Ehrhart, J.; Smith, A.J.; Kuzmin-Nichols, N.; Zesiewicz, T.A.; Jahan, I.; Shytle, R.D.; Kim, S.-H.; Sanberg, C.D.; Vu, T.H.; Gooch, C.L.; et al. Humoral factors in ALS patients during disease progression. J. Neuroinflamm. 2015, 12, 127. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, F.; Munitic, I.; Amedei, A.; Berry, J.D.; Feldman, E.L.; Aronica, E.; Nardo, G.; Van Weehaeghe, D.; Niccolai, E.; Prtenjaca, N.; et al. Interplay between immunity and amyotrophic lateral sclerosis: Clinical impact. Neurosci. Biobehav. Rev. 2021, 127, 958–978. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; He, L.; Ren, M.; Lu, Y.; Meng, H.; Yin, D.; Chen, S.; Zhou, Q. Paraneoplastic Amyotrophic Lateral Sclerosis: Case Series and Literature Review. Brain Sci. 2022, 12, 1053. [Google Scholar] [CrossRef]
- Jin, M.; Günther, R.; Akgün, K.; Hermann, A.; Ziemssen, T. Peripheral proinflammatory Th1/Th17 immune cell shift is linked to disease severity in amyotrophic lateral sclerosis. Sci. Rep. 2020, 10, 5941. [Google Scholar] [CrossRef]
- Jin, M.; Akgün, K.; Ziemssen, T.; Kipp, M.; Günther, R.; Hermann, A. Interleukin-17 and Th17 Lymphocytes Directly Impair Motoneuron Survival of Wildtype and FUS-ALS Mutant Human iPSCs. Int. J. Mol. Sci. 2021, 22, 8042. [Google Scholar] [CrossRef]
- Rentzos, M.; Rombos, A.; Nikolaou, C.; Zoga, M.; Zouvelou, V.; Dimitrakopoulos, A.; Alexakis, T.; Tsoutsou, A.; Samakovli, A.; Michalopoulou, M.; et al. Interleukin-17 and interleukin-23 are elevated in serum and cerebrospinal fluid of patients with ALS: A reflection of Th17 cells activation? Acta Neurol. Scand. 2010, 122, 425–429. [Google Scholar] [CrossRef]
- Saresella, M.; Piancone, F.; Tortorella, P.; Marventano, I.; Gatti, A.; Caputo, D.; Lunetta, C.; Corbo, M.; Rovaris, M.; Clerici, M. T helper-17 activation dominates the immunologic milieu of both amyotrophic lateral sclerosis and progressive multiple sclerosis. Clin. Immunol. 2013, 148, 79–88. [Google Scholar] [CrossRef]
- Fiala, M.; Chattopadhay, M.; La Cava, A.; Tse, E.; Liu, G.; Lourenco, E.; Eskin, A.; Liu, P.T.; Magpantay, L.; Tse, S.; et al. IL-17A is increased in the serum and in spinal cord CD8 and mast cells of ALS patients. J. Neuroinflamm. 2010, 7, 76. [Google Scholar] [CrossRef]
- Rolfes, L.; Schulte-Mecklenbeck, A.; Schreiber, S.; Vielhaber, S.; Herty, M.; Marten, A.; Pfeuffer, S.; Ruck, T.; Wiendl, H.; Gross, C.C.; et al. Amyotrophic lateral sclerosis patients show increased peripheral and intrathecal T-cell activation. Brain Commun. 2021, 3, fcab157. [Google Scholar] [CrossRef]
- Cui, C.; Ingre, C.; Yin, L.; Li, X.; Andersson, J.; Seitz, C.; Ruffin, N.; Pawitan, Y.; Piehl, F.; Fang, F. Correlation between leukocyte phenotypes and prognosis of amyotrophic lateral sclerosis. eLife 2022, 11, e74065. [Google Scholar] [CrossRef] [PubMed]
- Chiot, A.; Lobsiger, C.S.; Boillée, S. New insights on the disease contribution of neuroinflammation in amyotrophic lateral sclerosis. Curr. Opin. Neurol. 2019, 32, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Coque, E.; Salsac, C.; Espinosa-Carrasco, G.; Varga, B.; Degauque, N.; Cadoux, M.; Crabé, R.; Virenque, A.; Soulard, C.; Fierle, J.K.; et al. Cytotoxic CD8 + T lymphocytes expressing ALS-causing SOD1 mutant selectively trigger death of spinal motoneurons. Proc. Natl. Acad. Sci. USA 2019, 116, 2312–2317. [Google Scholar] [CrossRef]
- Nardo, G.; Trolese, M.C.; Verderio, M.; Mariani, A.; De Paola, M.; Riva, N.; Dina, G.; Panini, N.; Erba, E.; Quattrini, A.; et al. Counteracting roles of MHCI and CD8+ T cells in the peripheral and central nervous system of ALS SOD1G93A mice. Mol. Neurodegener. 2018, 13, 42. [Google Scholar] [CrossRef] [PubMed]
- Murdock, B.J.; Zhou, T.; Kashlan, S.R.; Little, R.J.; Goutman, S.; Feldman, E.L. Correlation of Peripheral Immunity With Rapid Amyotrophic Lateral Sclerosis Progression. JAMA Neurol. 2017, 74, 1446–1454. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, S.; Cocozza, G.; Porzia, A.; Inghilleri, M.; Raspa, M.; Scavizzi, F.; Aronica, E.; Bernardini, G.; Peng, L.; Ransohoff, R.M.; et al. Natural killer cells modulate motor neuron-immune cell cross talk in models of Amyotrophic Lateral Sclerosis. Nat. Commun. 2020, 11, 1773. [Google Scholar] [CrossRef]
- Murdock, B.J.; Famie, J.P.; Piecuch, C.E.; Raue, K.D.; Mendelson, F.E.; Pieroni, C.H.; Iniguez, S.D.; Zhao, L.; Goutman, S.A.; Feldman, E.L. Natural killer cells associate with amyotrophic lateral sclersois in a sex- and age-dependent manner. JCI Insight 2021, 6, e147129. [Google Scholar] [CrossRef] [PubMed]
- La Bella, V.; Iannitto, E.; Cuffaro, L.; Spataro, R. A rapidly progressive motor neuron disease associated to a natural killer cells leukaemia. J. Neurol. Sci. 2019, 398, 117–118. [Google Scholar] [CrossRef]
- Henkel, J.S.; Beers, D.R.; Wen, S.; Rivera, A.L.; Toennis, K.M.; Appel, J.E.; Zhao, W.; Moore, D.H.; Powell, S.Z.; Appel, S.H. Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol. Med. 2013, 5, 64–79. [Google Scholar] [CrossRef]
- Sheean, R.K.; McKay, F.C.; Cretney, E.; Bye, C.; Perera, N.D.; Tomas, D.; Weston, R.A.; Scheller, K.J.; Djouma, E.; Menon, P.; et al. Association of Regulatory T-Cell Expansion With Progression of Amyotrophic Lateral Sclerosis. JAMA Neurol. 2018, 75, 681–689. [Google Scholar] [CrossRef]
- Beers, D.R.; Zhao, W.; Wang, J.; Zhang, X.; Wen, S.; Neal, D.; Thonhoff, J.R.; Alsuliman, A.S.; Shpall, E.J.; Rezvani, K.; et al. ALS patients’ regulatory T lymphocytes are dysfunctional, and correlate with disease progression rate and severity. JCI Insight 2017, 2, e89530. [Google Scholar] [CrossRef] [PubMed]
- Westarp, M.E.; Fuchs, D.; Bartmann, P.; Hoff-Jörgensen, R.; Clausen, J.; Wachter, H.; Kornhuber, H.H. Amyotrophic lateral sclerosis an enigmatic disease with B-cellular and anti-retroviral immune responses. Eur. J. Med. 1993, 2, 327–332. [Google Scholar] [PubMed]
- Naor, S.; Keren, Z.; Bronshtein, T.; Goren, E.; Machluf, M.; Melamed, D. Development of ALS-like disease in SOD-1 mice deficient of B lymphocytes. J. Neurol. 2009, 256, 1228–1235. [Google Scholar] [CrossRef] [PubMed]
- Niebroj-Dobosz, I.; Dziewulska, D.; Janik, P. Auto-antibodies against proteins of spinal cord cells in cerebrospinal fluid of patients with amyotrophic lateral sclerosis (ALS). Folia Neuropathol. 2006, 44, 191–196. [Google Scholar] [PubMed]
- van Blitterswijk, M.; Gulati, S.; Smoot, E.; Jaffa, M.; Maher, N.; Hyman, B.T.; Ivinson, A.J.; Scherzer, C.; Schoenfeld, D.A.; Cudkowicz, M.E.; et al. Anti-superoxide dismutase antibodies are associated with survival in patients with sporadic amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2011, 12, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, J.I.; Tajti, J.; Appel, S.H. Lymphocytic infiltrates in the spinal cord in amyotrophic lateral sclerosis. Arch Neurol. 1993, 50, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Ostermeyer-Shoaib, B.; Patten, B.M. IgG subclass deficiency in amyotrophic lateral sclerosis. Acta Neurol. Scand. 2009, 87, 192–194. [Google Scholar] [CrossRef]
- Appel, S.H.; Engelhardt, J.I.; García, J.; Stefani, E. Autoimmunity and ALS: A Comparison of Animal Models of Immune-Mediated Motor Neuron Destruction and Human ALS. Adv. Neurol. 1991, 56, 405–412. [Google Scholar]
- Engelhardt, J.I.; Siklós, L.; Appel, S.H. Altered Calcium Homeostasis and Ultrastructure in Motoneurons of Mice Caused by Passively Transferred Anti-motoneuronal IgG. J. Neuropathol. Exp. Neurol. 1997, 56, 21–39. [Google Scholar] [CrossRef]
- Engelhardt, J.I.; Siklós, L.; Kőműves, L.; Smith, R.G.; Appel, S.H. Antibodies to calcium channels from ALS patients passively transferred to mice selectively increase intracellular calcium and induce ultrastructural changes in motoneurons. Synapse 1995, 20, 185–199. [Google Scholar] [CrossRef]
- Polgár, T.F.; Meszlényi, V.; Nógrádi, B.; Körmöczy, L.; Spisák, K.; Tripolszki, K.; Széll, M.; Obál, I.; Engelhardt, J.I.; Siklós, L.; et al. Passive Transfer of Blood Sera from ALS Patients with Identified Mutations Results in Elevated Motoneuronal Calcium Level and Loss of Motor Neurons in the Spinal Cord of Mice. Int. J. Mol. Sci. 2021, 22, 9994. [Google Scholar] [CrossRef] [PubMed]
- Obál, I.; Nógrádi, B.; Meszlényi, V.; Patai, R.; Ricken, G.; Kovacs, G.G.; Tripolszki, K.; Széll, M.; Siklós, L.; Engelhardt, J.I. Experimental Motor Neuron Disease Induced in Mice with Long-Term Repeated Intraperitoneal Injections of Serum from ALS Patients. Int. J. Mol. Sci. 2019, 20, 2573. [Google Scholar] [CrossRef] [PubMed]
- Thonhoff, J.R.; Beers, D.R.; Zhao, W.; Pleitez, M.; Simpson, E.P.; Berry, J.D.; Cudkowicz, M.E.; Appel, S.H. Expanded autologous regulatory T-lymphocyte infusions in ALS. Neurol. Neuroimmunol. Neuroinflamm. 2018, 5, e465. [Google Scholar] [CrossRef] [PubMed]
- Camu, W.; Mickunas, M.; Veyrune, J.-L.; Payan, C.; Garlanda, C.; Locati, M.; Juntas-Morales, R.; Pageot, N.; Malaspina, A.; Andreasson, U.; et al. Repeated 5-day cycles of low dose aldesleukin in amyotrophic lateral sclerosis (IMODALS): A phase 2a randomised, double-blind, placebo-controlled trial. eBioMedicine 2020, 59, 102844. [Google Scholar] [CrossRef]
- Vucic, S.; Henderson, R.D.; Mathers, S.; Needham, M.; Schultz, D.; Kiernan, M.C. The TEALS study group Safety and efficacy of dimethyl fumarate in ALS: Randomised controlled study. Ann. Clin. Transl. Neurol. 2021, 8, 1991–1999. [Google Scholar] [CrossRef]
- Vallarola, A.; Sironi, F.; Tortarolo, M.; Gatto, N.; De Gioia, R.; Pasetto, L.; De Paola, M.; Mariani, A.; Ghosh, S.; Watson, R.; et al. RNS60 exerts therapeutic effects in the SOD1 ALS mouse model through protective glia and peripheral nerve rescue. J. Neuroinflamm. 2018, 15, 65. [Google Scholar] [CrossRef]
- Paganoni, S.; Alshikho, M.J.; Luppino, S.; Chan, J.; Ba, L.P.; Schoenfeld, D.; Dpt, P.L.A.; Babu, S.; Zürcher, N.R.; Loggia, M.L.; et al. A pilot trial of RNS60 in amyotrophic lateral sclerosis. Muscle Nerve 2018, 59, 303–308. [Google Scholar] [CrossRef]
- Beghi, E.; Pupillo, E.; Bianchi, E.; Bonetto, V.; Luotti, S.; Pasetto, L.; Bendotti, C.; Tortarolo, M.; Sironi, F.; Camporeale, L.; et al. Effect of RNS60 in amyotrophic lateral sclerosis: A phase II multicentre, randomized, double-blind, placebo-controlled trial. Eur. J. Neurol. 2022; online ahead of print. [Google Scholar] [CrossRef]
- Berry, J.D.; Paganoni, S.; Atassi, N.; Macklin, E.A.; Goyal, N.; Rivner, M.; Simpson, E.; Appel, S.; Grasso, D.L.; Mejia, N.I.; et al. Phase IIa trial of fingolimod for amyotrophic lateral sclerosis demonstrates acceptable acute safety and tolerability. Muscle Nerve 2017, 56, 1077–1084. [Google Scholar] [CrossRef]
- Meininger, V.; Drory, V.E.; Leigh, P.N.; Ludolph, A.; Robberecht, W.; Silani, V. Glatiramer acetate has no impact on disease progression in ALS at 40 mg/day: A double- blind, randomized, multicentre, placebo-controlled trial. Amyotroph. Lateral Scler. 2009, 10, 378–383. [Google Scholar] [CrossRef]
- Mizwicki, M.T.; Milan, F.; Magpantay, L.; Aziz, N.; Sayre, J.; Liu, G.; Siani, A.; Chan, D.; Martinez-Maza, O.; Chattopadhyay, M.; et al. Tocili-zumab Attenuates Inflammation in ALS Patients through Inhibition of IL6 Receptor Signaling. Am. J. Neurodegener. Dis. 2012, 1, 305. [Google Scholar]
- Lima-Junior, J.R.; Sulzer, D.; Arlehamn, C.S.L.; Sette, A. The role of immune-mediated alterations and disorders in ALS disease. Hum. Immunol. 2021, 82, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Alexianu, M.E.; Kozovska, M.; Appel, S.H. Immune reactivity in a mouse model of familial ALS correlates with disease progression. Neurology 2001, 57, 1282–1289. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Treatment | Dose and Administration | Number of Cases or Types of Animal Model | Laboratory and/or Clinical Outcomes | References |
|---|---|---|---|---|
| Aldesleukin | Intravenous Low dose of Interleukin-2 for five days at week 1, 5, and 9. | Twenty-four patients | Increase in Treg response; no significant change in terms of a reduction in circulating lymphocytes in ALS patients when compared with the placebo group. | [54] |
| Dimethyl fumarate | Oral administration of 480 mg daily for 36 weeks. | Seventy-two patients | No significant effects in terms of a reduction in circulating lymphocytes in ALS patients compared with the placebo group. | [55] |
| RNS60 | Weekly intravenous infusion and daily nebulization. | Thirteen patients | No changes in biomarkers (i.e., FOXP3 mRNA and IL-17 levels). | [56] |
| 300 μL/mouse intraperitoneally every other day. | C57BL/6-SOD1G93A | Increase in CD4+/Foxp3+ T regulatory cells and neuroprotection. | [57] | |
| Three hundred and seventy-five mL intravenously administered for 24 weeks, once a week, and 4 mL/day administered via nebulization on the other days. | Seventy-two patients | Slower decline of forced vital capacity and bulbar dysfunctions in the patients treated with RNS60 compared with the placebo group. | [58] | |
| Infusions of autologous Treg in ALS | Intravenous Tregs 106 cells/kg, initially four doses over 2 months, and in later stages, four doses over 4 months of the disease (n. 8 total infusions). | Three patients with sALS | Increase in Treg function and a slower disease progression, measured in accordance with the Appel ALS scale for each patient. | [53] |
| Fingolimod | Oral administration, dose of 0.5 mg/day for 4 weeks. | Eighteen patients with ALS | Reduction of circulating lymphocytes in ALS patients. No effects on ALSFRS-R. | [59] |
| Glatiramer acetate | Subcutaneous injection of 40 mg/day. | Three hundred and sixty-six patients with ALS enrolled in a phase II clinical trial | No effects on ALSFRS-R. | [60] |
| Tocilizumab | Treatment of PMBCs overnight with 2 μg/mL apo-G37R + 10 μg/mL tocilizumab. | Four patients with ALS | Reduction of the secretion of cytokynes and chemiokynes from the PMBCs of patients. | [61] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schirò, G.; Di Stefano, V.; Iacono, S.; Lupica, A.; Brighina, F.; Monastero, R.; Balistreri, C.R. Advances on Cellular Clonotypic Immunity in Amyotrophic Lateral Sclerosis. Brain Sci. 2022, 12, 1412. https://doi.org/10.3390/brainsci12101412
Schirò G, Di Stefano V, Iacono S, Lupica A, Brighina F, Monastero R, Balistreri CR. Advances on Cellular Clonotypic Immunity in Amyotrophic Lateral Sclerosis. Brain Sciences. 2022; 12(10):1412. https://doi.org/10.3390/brainsci12101412
Chicago/Turabian StyleSchirò, Giuseppe, Vincenzo Di Stefano, Salvatore Iacono, Antonino Lupica, Filippo Brighina, Roberto Monastero, and Carmela Rita Balistreri. 2022. "Advances on Cellular Clonotypic Immunity in Amyotrophic Lateral Sclerosis" Brain Sciences 12, no. 10: 1412. https://doi.org/10.3390/brainsci12101412
APA StyleSchirò, G., Di Stefano, V., Iacono, S., Lupica, A., Brighina, F., Monastero, R., & Balistreri, C. R. (2022). Advances on Cellular Clonotypic Immunity in Amyotrophic Lateral Sclerosis. Brain Sciences, 12(10), 1412. https://doi.org/10.3390/brainsci12101412