
Understanding the Modulatory Effects of Cannabidiol on Alzheimer’s Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
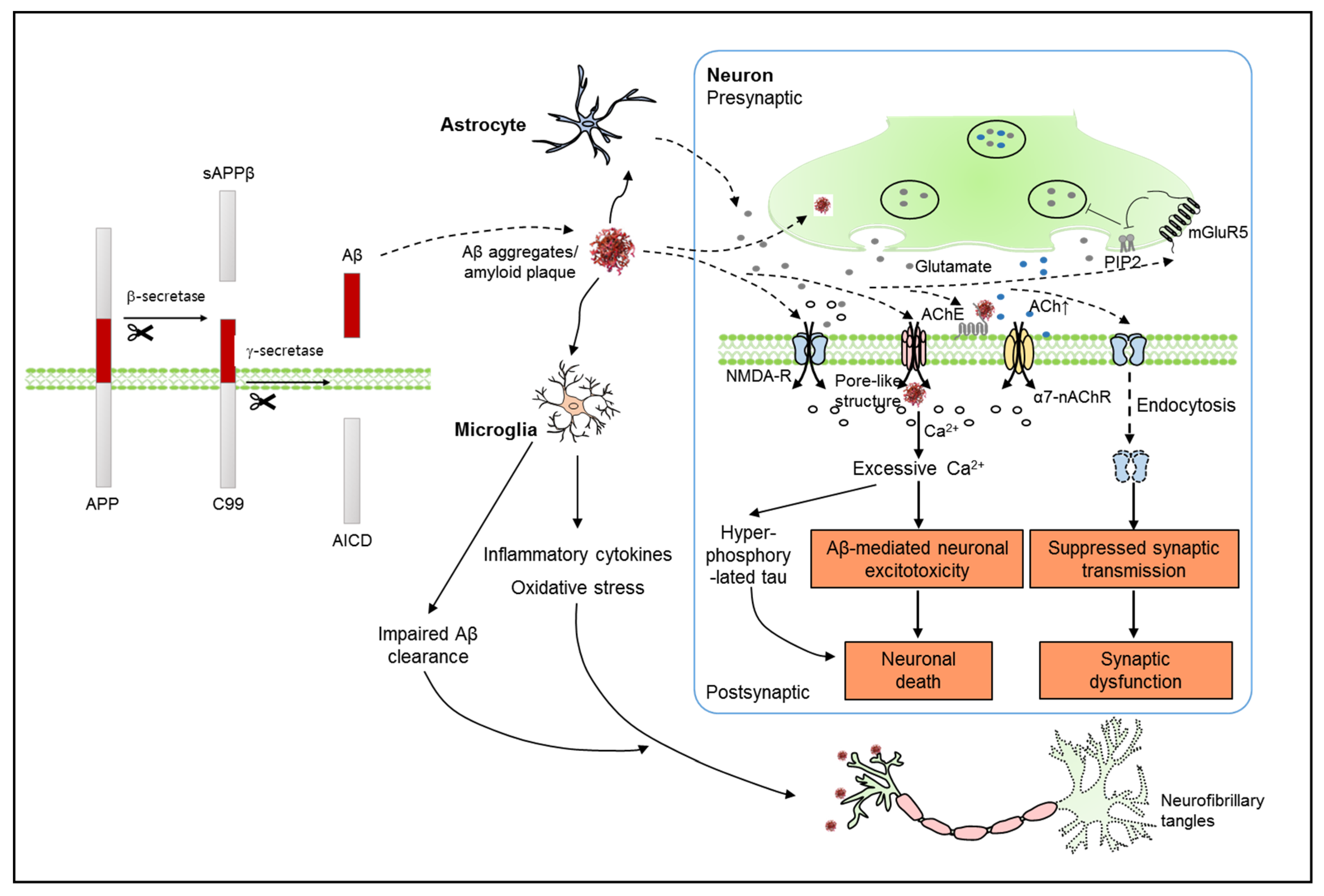
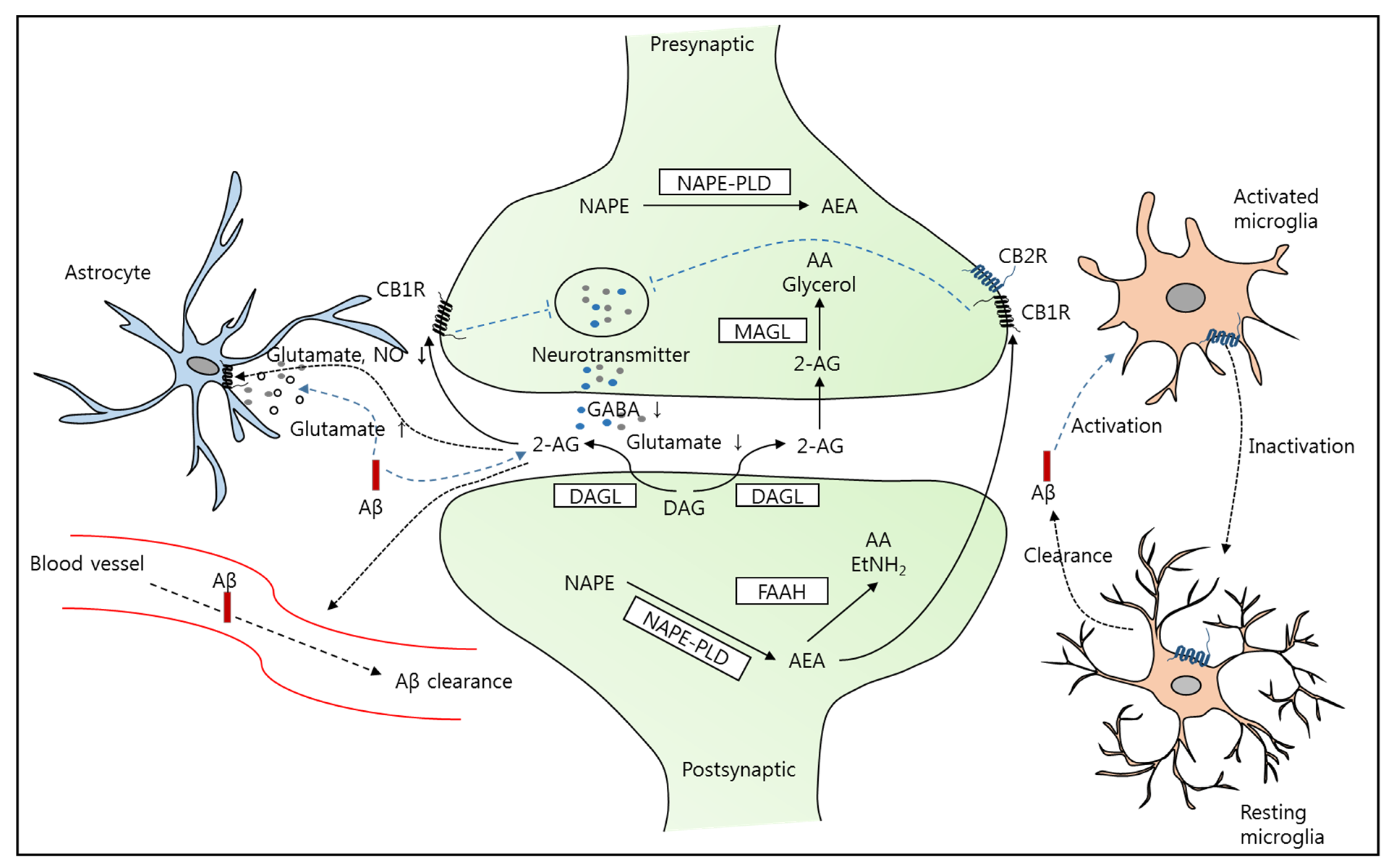
2. Alzheimer’s Disease and the Endocannabinoid System (ECS)
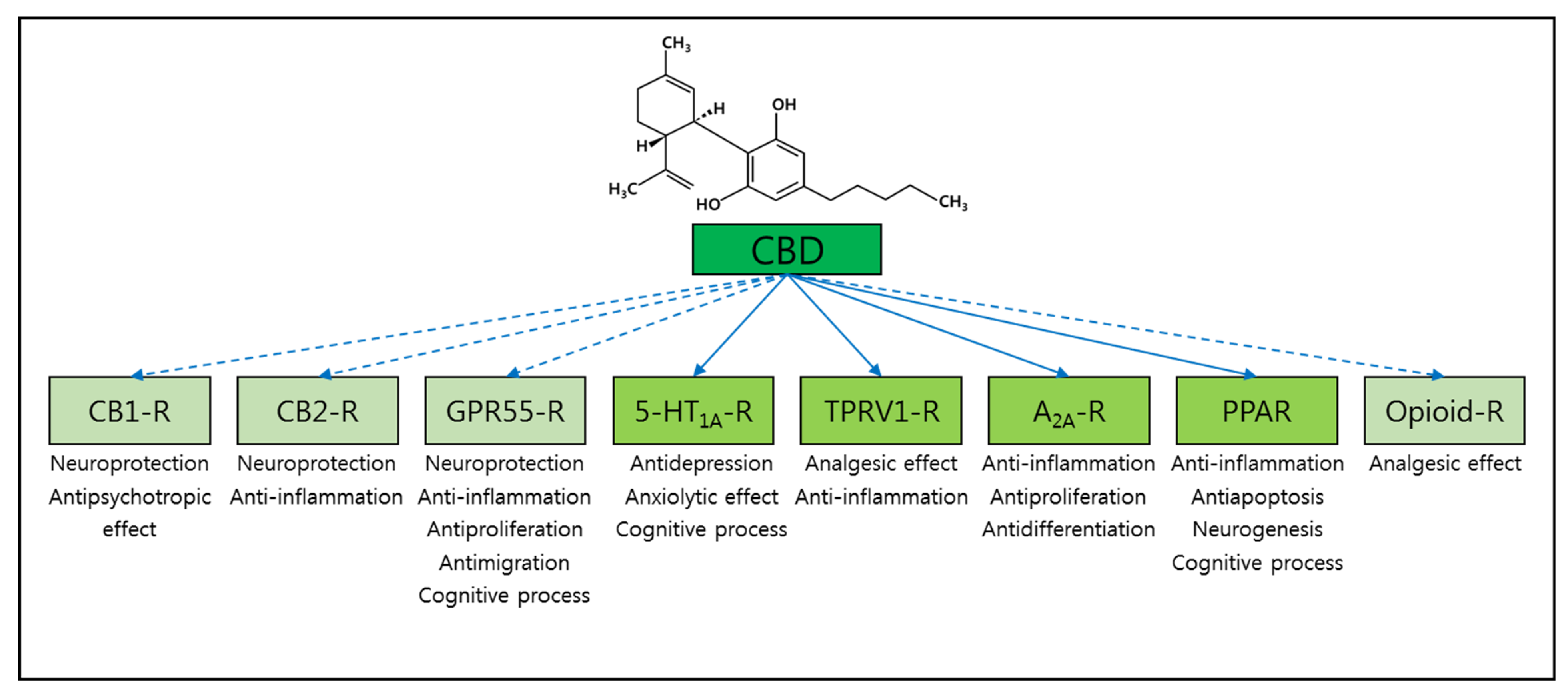
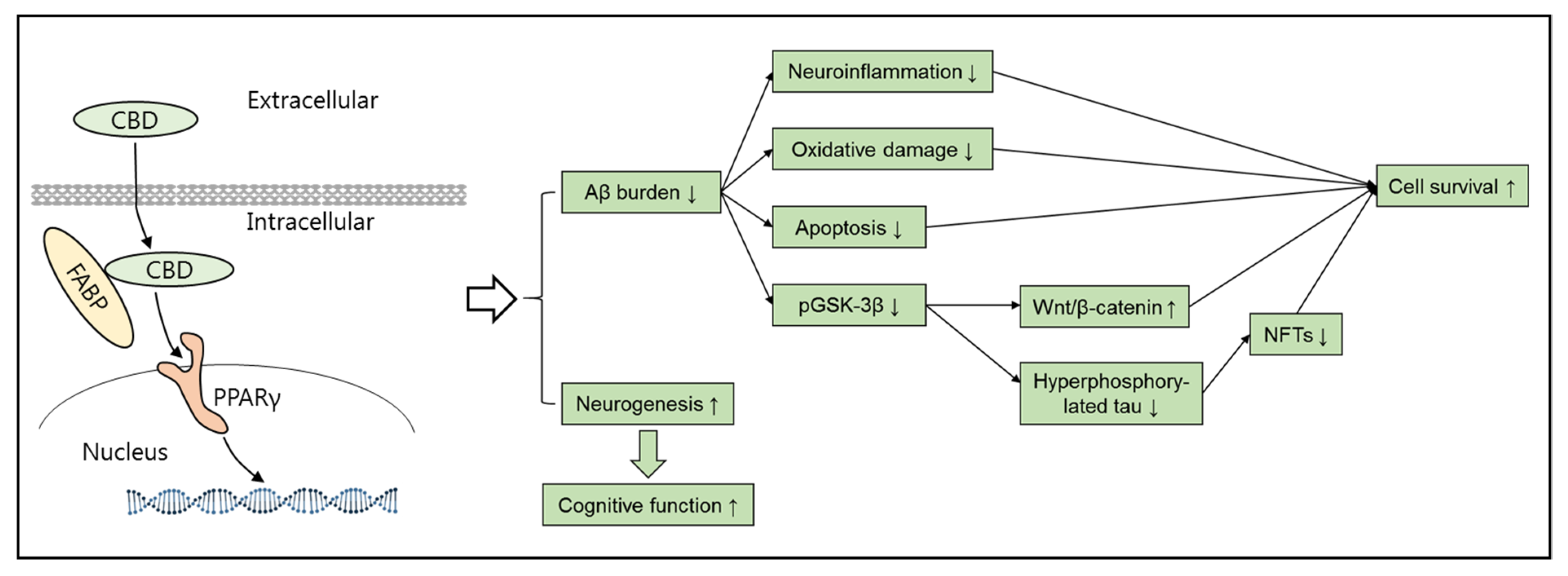
3. Alzheimer’s Disease and Cannabidiol (CBD)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviation List
| 2-AG | 2-Arachidonyl Glycerol |
| 5-HT | 5-Hydroxytryptamine |
| AChE | Acetylcholinesterase |
| AD | Alzheimer’s Disease |
| AEA | Anandamide |
| APP | Amyloid Precursor Protein |
| ATP | Adenosine Triphosphate |
| Aβ | Amyloid β |
| CB1R | Cannabinoid Receptor Type 1 |
| CB2R | Cannabinoid Receptor Type 2 |
| CBD | Cannabidiol |
| DAGL | Diacylglycerol Lipase |
| DCX | Doublecortin |
| ECS | Endocannabinoid System |
| FAAH | Fatty Acid Amide Hydrolase |
| GABA | γ-Aminobutyric Acid |
| GSK-3β | Glycogen Synthase Kinase-3β |
| LTP | Long-term Potentiation |
| MAGL | Monoglyceride Lipase |
| MAPK | Mitogen-Activated Protein Kinase |
| mGluR5 | Metabotropic Glutamate Receptor 5 |
| NAPE-PLD | N-Acyl Phosphatidyl Ethanolamine Specific Phospholipase D |
| NMDA | N-Methyl-D-Aspartate |
| NO | Nitric Oxide |
| PIP2 | Phosphatidylinositol-4,5-Bisphosphate |
| PPARγ | Peroxisome Proliferator-Activated Receptor Gamma |
| PS1 | Presenilin 1 |
| ROS | Reactive Oxygen Species |
| THC | Δ9-Tetrahydro Cannabinol |
| TRPV1 | Transient Receptor Potential Vanilloid 1 |
References
- Gonzalez, C.; Armijo, E.; Bravo-Alegria, J.; Becerra-Calixto, A.; Mays, C.E.; Soto, C. Modeling amyloid beta and tau pathology in human cerebral organoids. Mol. Psychiatry 2018, 23, 2363–2374. [Google Scholar] [CrossRef]
- Moore, S.; Evans, L.D.; Andersson, T.; Portelius, E.; Smith, J.; Dias, T.B.; Saurat, N.; McGlade, A.; Kirwan, P.; Blennow, K.; et al. APP metabolism regulates tau proteostasis in human cerebral cortex neurons. Cell Rep. 2015, 11, 689–696. [Google Scholar] [CrossRef] [Green Version]
- Hurtado, D.E.; Molina-Porcel, L.; Iba, M.; Aboagye, A.K.; Paul, S.M.; Trojanowski, J.Q.; Lee, V.M. Aβ accelerates the spatiotemporal progression of tau pathology and augments tau amyloidosis in an Alzheimer mouse model. Am. J. Pathol. 2010, 177, 1977–1988. [Google Scholar] [CrossRef]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef] [Green Version]
- Leroy, K.; Ando, K.; Laporte, V.; Dedecker, R.; Suain, V.; Authelet, M.; Heraud, C.; Pierrot, N.; Yilmaz, Z.; Octave, J.N.; et al. Lack of tau proteins rescues neuronal cell death and decreases amyloidogenic processing of APP in APP/PS1 mice. Am. J. Pathol. 2012, 181, 1928–1940. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, P.; Feng, J.; Wu, M. Dysfunction of NMDA receptors in Alzheimer’s disease. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2016, 37, 1039–1047. [Google Scholar] [CrossRef] [Green Version]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef] [Green Version]
- Demuro, A.; Mina, E.; Kayed, R.; Milton, S.C.; Parker, I.; Glabe, C.G. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J. Biol. Chem. 2005, 280, 17294–17300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demuro, A.; Smith, M.; Parker, I. Single-channel Ca2+ imaging implicates Aβ1-42 amyloid pores in Alzheimer’s disease pathology. J. Cell Biol. 2011, 195, 515–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, E.M.; Nong, Y.; Almeida, C.G.; Paul, S.; Moran, T.; Choi, E.Y.; Nairn, A.C.; Salter, M.W.; Lombroso, P.J.; Gouras, G.K.; et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat. Neurosci. 2005, 8, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.K.; Jacobi, E.; Sakimura, K.; Malinow, R.; von Engelhardt, J. NMDA receptors mediate synaptic depression, but not spine loss in the dentate gyrus of adult amyloid Beta (Abeta) overexpressing mice. Acta Neuropathol. Commun. 2018, 6, 110. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wei, M.; Wu, Y.; Qin, H.; Li, W.; Ma, X.; Cheng, J.; Ren, J.; Shen, Y.; Chen, Z.; et al. Amyloid beta oligomers suppress excitatory transmitter release via presynaptic depletion of phosphatidylinositol-4,5-bisphosphate. Nat. Commun. 2019, 10, 1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.; Hussain, M.D.; Yan, L.J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int. J. Neurosci. 2014, 124, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, P.; Loganathan, K.; Sekiguchi, M.; Matsuba, Y.; Hui, K.; Tsubuki, S.; Tanaka, M.; Iwata, N.; Saito, T.; Saido, T.C. Abeta secretion and plaque formation depend on autophagy. Cell Rep. 2013, 5, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Lleo, A.; Greenberg, S.M.; Growdon, J.H. Current pharmacotherapy for Alzheimer’s disease. Annu. Rev. Med. 2006, 57, 513–533. [Google Scholar] [CrossRef] [PubMed]
- Husna Ibrahim, N.; Yahaya, M.F.; Mohamed, W.; Teoh, S.L.; Hui, C.K.; Kumar, J. Pharmacotherapy of Alzheimer’s Disease: Seeking clarity in a time of uncertainty. Front. Pharmacol. 2020, 11, 261. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Wang, X. Antioxidant therapies for Alzheimer’s disease. Oxid. Med. Cell. Longev. 2012, 2012, 472932. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Wang, Y.; Wang, D.; Zhang, J.; Zhang, F. NSAID Exposure and risk of Alzheimer’s disease: An updated meta-analysis from cohort studies. Front. Aging Neurosci. 2018, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Sogorb-Esteve, A.; Garcia-Ayllon, M.S.; Llansola, M.; Felipo, V.; Blennow, K.; Saez-Valero, J. Inhibition of gamma-secretase leads to an increase in presenilin-1. Mol. Neurobiol. 2018, 55, 5047–5058. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Li, Y.; Xu, H.; Zhang, Y.W. The gamma-secretase complex: From structure to function. Front. Cell. Neurosci. 2014, 8, 427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imbimbo, B.P.; Watling, M. Investigational BACE inhibitors for the treatment of Alzheimer’s disease. Exp. Opin. Investig. Drugs 2019, 28, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Orgogozo, J.M.; Gilman, S.; Dartigues, J.F.; Laurent, B.; Puel, M.; Kirby, L.C.; Jouanny, P.; Dubois, B.; Eisner, L.; Flitman, S.; et al. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology 2003, 61, 46–54. [Google Scholar] [CrossRef]
- Van Dyck, C.H. Anti-amyloid-β monoclonal antibodies for Alzheimer’s disease: Pitfalls and promise. Biol. Psychiatry 2018, 83, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Hock, C.; Konietzko, U.; Streffer, J.R.; Tracy, J.; Signorell, A.; Muller-Tillmanns, B.; Lemke, U.; Henke, K.; Moritz, E.; Garcia, E.; et al. Antibodies against beta-amyloid slow cognitive decline in Alzheimer’s disease. Neuron 2003, 38, 547–554. [Google Scholar] [CrossRef] [Green Version]
- Sevigny, J.; Chiao, P.; Bussiere, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Watt, G.; Karl, T. In vivo evidence for therapeutic properties of cannabidiol (CBD) for Alzheimer’s disease. Front. Pharmacol. 2017, 8, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fride, E. The endocannabinoid-CB(1) receptor system in pre- and postnatal life. Eur. J. Pharmacol. 2004, 500, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V.; Goparaju, S.K.; Wang, L.; Liu, J.; Bátkai, S.; Járai, Z.; Fezza, F.; Miura, G.I.; Palmiter, R.D.; Sugiura, T.; et al. Leptin-regulated endocannabinoids are involved in maintaining food intake. Nature 2001, 410, 822–825. [Google Scholar] [CrossRef] [PubMed]
- Donvito, G.; Nass, S.R.; Wilkerson, J.L.; Curry, Z.A.; Schurman, L.D.; Kinsey, S.G.; Lichtman, A.H. The endogenous cannabinoid system: A budding source of targets for treating inflammatory and neuropathic pain. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2018, 43, 52–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campolongo, P.; Trezza, V. The endocannabinoid system: A key modulator of emotions and cognition. Front. Behav. Neurosci. 2012, 6, 73. [Google Scholar] [CrossRef] [Green Version]
- Marsicano, G.; Lafenetre, P. Roles of the endocannabinoid system in learning and memory. Curr. Top. Behav. Neurosci. 2009, 1, 201–230. [Google Scholar] [CrossRef] [PubMed]
- Wu, J. Cannabis, cannabinoid receptors, and endocannabinoid system: Yesterday, today, and tomorrow. Acta Pharmacol. Sin. 2019, 40, 297–299. [Google Scholar] [CrossRef]
- Zou, S.; Kumar, U. Cannabinoid receptors and the endocannabinoid system: Signaling and function in the central nervous system. Int. J. Mol. Sci. 2018, 19, 833. [Google Scholar] [CrossRef] [Green Version]
- Benito, C.; Nunez, E.; Tolon, R.M.; Carrier, E.J.; Rabano, A.; Hillard, C.J.; Romero, J. Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaque-associated glia in Alzheimer’s disease brains. J. Neurosci. Off. J. Soci. Neurosci. 2003, 23, 11136–11141. [Google Scholar] [CrossRef] [Green Version]
- Solas, M.; Francis, P.T.; Franco, R.; Ramirez, M.J. CB2 receptor and amyloid pathology in frontal cortex of Alzheimer’s disease patients. Neurobiol. Aging 2013, 34, 805–808. [Google Scholar] [CrossRef]
- Manuel, I.; Gonzalez de San Roman, E.; Giralt, M.T.; Ferrer, I.; Rodriguez-Puertas, R. Type-1 cannabinoid receptor activity during Alzheimer’s disease progression. J. Alzheimer’s Dis. JAD 2014, 42, 761–766. [Google Scholar] [CrossRef] [Green Version]
- Galán-Ganga, M.; Rodríguez-Cueto, C.; Merchán-Rubira, J.; Hernández, F.; Ávila, J.; Posada-Ayala, M.; Lanciego, J.L.; Luengo, E.; Lopez, M.G.; Rábano, A.; et al. Cannabinoid receptor CB2 ablation protects against TAU induced neurodegeneration. Acta Neuropathol. Commun. 2021, 9, 90. [Google Scholar] [CrossRef] [PubMed]
- Mulder, J.; Zilberter, M.; Pasquare, S.J.; Alpar, A.; Schulte, G.; Ferreira, S.G.; Kofalvi, A.; Martin-Moreno, A.M.; Keimpema, E.; Tanila, H.; et al. Molecular reorganization of endocannabinoid signalling in Alzheimer’s disease. Brain J. Neurol. 2011, 134, 1041–1060. [Google Scholar] [CrossRef]
- Basavarajappa, B.S.; Shivakumar, M.; Joshi, V.; Subbanna, S. Endocannabinoid system in neurodegenerative disorders. J. Neurochem. 2017, 142, 624–648. [Google Scholar] [CrossRef] [PubMed]
- Walther, S.; Mahlberg, R.; Eichmann, U.; Kunz, D. Delta-9-tetrahydrocannabinol for nighttime agitation in severe dementia. Psychopharmacology 2006, 185, 524–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volicer, L.; Stelly, M.; Morris, J.; McLaughlin, J.; Volicer, B.J. Effects of dronabinol on anorexia and disturbed behavior in patients with Alzheimer’s disease. Int. J. Geriatr. Psychiatry 1997, 12, 913–919. [Google Scholar] [CrossRef]
- Cooray, R.; Gupta, V.; Suphioglu, C. Current aspects of the endocannabinoid system and targeted THC and CBD phytocannabinoids as potential therapeutics for Parkinson’s and Alzheimer’s diseases: A review. Mol. Neurobiol. 2020, 57, 4878–4890. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Mechoulam, R. Is lipid signaling through cannabinoid 2 receptors part of a protective system? Prog. Lipid Res. 2011, 50, 193–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katona, I.; Freund, T.F. Multiple functions of endocannabinoid signaling in the brain. Annu. Rev. Neurosci. 2012, 35, 529–558. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Wang, M.; Liu, W.; Ma, Z.; Wu, J. Activation of cannabinoid receptor 2 protects rat hippocampal neurons against Aβ-induced neuronal toxicity. Neurosci. Lett. 2020, 735, 135207. [Google Scholar] [CrossRef]
- Van der Stelt, M.; Mazzola, C.; Esposito, G.; Matias, I.; Petrosino, S.; De Filippis, D.; Micale, V.; Steardo, L.; Drago, F.; Iuvone, T.; et al. Endocannabinoids and beta-amyloid-induced neurotoxicity in vivo: Effect of pharmacological elevation of endocannabinoid levels. Cell. Mol. Life Sci. CMLS 2006, 63, 1410–1424. [Google Scholar] [CrossRef]
- Gajardo-Gomez, R.; Labra, V.C.; Maturana, C.J.; Shoji, K.F.; Santibanez, C.A.; Saez, J.C.; Giaume, C.; Orellana, J.A. Cannabinoids prevent the amyloid beta-induced activation of astroglial hemichannels: A neuroprotective mechanism. Glia 2017, 65, 122–137. [Google Scholar] [CrossRef]
- Tanveer, R.; Gowran, A.; Noonan, J.; Keating, S.E.; Bowie, A.G.; Campbell, V.A. The endocannabinoid, anandamide, augments Notch-1 signaling in cultured cortical neurons exposed to amyloid-beta and in the cortex of aged rats. J. Biol. Chem. 2012, 287, 34709–34721. [Google Scholar] [CrossRef] [Green Version]
- Bachmeier, C.; Beaulieu-Abdelahad, D.; Mullan, M.; Paris, D. Role of the cannabinoid system in the transit of beta-amyloid across the blood-brain barrier. Mol. Cell. Neurosci. 2013, 56, 255–262. [Google Scholar] [CrossRef]
- Milton, N.G. Anandamide and noladin ether prevent neurotoxicity of the human amyloid-beta peptide. Neurosci. Lett. 2002, 332, 127–130. [Google Scholar] [CrossRef]
- Esposito, G.; De Filippis, D.; Steardo, L.; Scuderi, C.; Savani, C.; Cuomo, V.; Iuvone, T. CB1 receptor selective activation inhibits beta-amyloid-induced iNOS protein expression in C6 cells and subsequently blunts tau protein hyperphosphorylation in co-cultured neurons. Neurosci. Lett. 2006, 404, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Patricio-Martinez, A.; Sanchez-Zavaleta, R.; Angulo-Cruz, I.; Gutierrez-Praxedis, L.; Ramirez, E.; Martinez-Garcia, I.; Limon, I.D. The acute activation of the CB1 receptor in the hippocampus decreases neurotoxicity and prevents spatial memory impairment in rats lesioned with beta-amyloid 25-35. Neuroscience 2019, 416, 239–254. [Google Scholar] [CrossRef]
- Haghani, M.; Janahmadi, M.; Shabani, M. Protective effect of cannabinoid CB1 receptor activation against altered intrinsic repetitive firing properties induced by Abeta neurotoxicity. Neurosci. Lett. 2012, 507, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Stumm, C.; Hiebel, C.; Hanstein, R.; Purrio, M.; Nagel, H.; Conrad, A.; Lutz, B.; Behl, C.; Clement, A.B. Cannabinoid receptor 1 deficiency in a mouse model of Alzheimer’s disease leads to enhanced cognitive impairment despite of a reduction in amyloid deposition. Neurobiol. Aging 2013, 34, 2574–2584. [Google Scholar] [CrossRef] [PubMed]
- Belue, R.C.; Howlett, A.C.; Westlake, T.M.; Hutchings, D.E. The ontogeny of cannabinoid receptors in the brain of postnatal and aging rats. Neurotoxicol. Teratol. 1995, 17, 25–30. [Google Scholar] [CrossRef]
- Aso, E.; Ferrer, I. CB2 cannabinoid receptor as potential target against Alzheimer’s disease. Front. Neurosci. 2016, 10, 243. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, B.G.; Blazquez, C.; Gomez del Pulgar, T.; Guzman, M.; de Ceballos, M.L. Prevention of Alzheimer’s disease pathology by cannabinoids: Neuroprotection mediated by blockade of microglial activation. J. Neurosci. Off. J. Soci. Neurosci. 2005, 25, 1904–1913. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Shi, J.; Wang, B.; Li, J.; Jia, H. CB2 cannabinoid receptor agonist ameliorates novel object recognition but not spatial memory in transgenic APP/PS1 mice. Neurosci. Lett. 2019, 707, 134286. [Google Scholar] [CrossRef]
- Aso, E.; Andres-Benito, P.; Carmona, M.; Maldonado, R.; Ferrer, I. Cannabinoid receptor 2 participates in amyloid-beta processing in a mouse model of Alzheimer’s disease but plays a minor role in the therapeutic properties of a aannabis-based medicine. J. Alzheimer’s Dis. JAD 2016, 51, 489–500. [Google Scholar] [CrossRef] [Green Version]
- Schmole, A.C.; Lundt, R.; Toporowski, G.; Hansen, J.N.; Beins, E.; Halle, A.; Zimmer, A. Cannabinoid receptor 2-deficiency ameliorates disease symptoms in a mouse model with Alzheimer’s disease-like pathology. J. Alzheimer’s Dis. JAD 2018, 64, 379–392. [Google Scholar] [CrossRef]
- Lopez, A.; Aparicio, N.; Pazos, M.R.; Grande, M.T.; Barreda-Manso, M.A.; Benito-Cuesta, I.; Vazquez, C.; Amores, M.; Ruiz-Perez, G.; Garcia-Garcia, E.; et al. Cannabinoid CB2 receptors in the mouse brain: Relevance for Alzheimer’s disease. J. Neuroinflam. 2018, 15, 158. [Google Scholar] [CrossRef] [Green Version]
- Tolon, R.M.; Nunez, E.; Pazos, M.R.; Benito, C.; Castillo, A.I.; Martinez-Orgado, J.A.; Romero, J. The activation of cannabinoid CB2 receptors stimulates in situ and in vitro beta-amyloid removal by human macrophages. Brain Res. 2009, 1283, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Talarico, G.; Trebbastoni, A.; Bruno, G.; de Lena, C. Modulation of the cannabinoid system: A new perspective for the treatment of the Alzheimer’s disease. Cur. Neuropharmacol. 2019, 17, 176–183. [Google Scholar] [CrossRef]
- Aso, E.; Sanchez-Pla, A.; Vegas-Lozano, E.; Maldonado, R.; Ferrer, I. Cannabis-based medicine reduces multiple pathological processes in AbetaPP/PS1 mice. J. Alzheimer’s Dis. JAD 2015, 43, 977–991. [Google Scholar] [CrossRef] [Green Version]
- Casarejos, M.J.; Perucho, J.; Gomez, A.; Munoz, M.P.; Fernandez-Estevez, M.; Sagredo, O.; Fernandez Ruiz, J.; Guzman, M.; de Yebenes, J.G.; Mena, M.A. Natural cannabinoids improve dopamine neurotransmission and tau and amyloid pathology in a mouse model of tauopathy. J. Alzheimer’s Dis. JAD 2013, 35, 525–539. [Google Scholar] [CrossRef] [Green Version]
- Eubanks, L.M.; Rogers, C.J.; Beuscher, A.E.t.; Koob, G.F.; Olson, A.J.; Dickerson, T.J.; Janda, K.D. A molecular link between the active component of marijuana and Alzheimer’s disease pathology. Mol. Pharm. 2006, 3, 773–777. [Google Scholar] [CrossRef] [Green Version]
- Pertwee, R.G. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: Delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br. J. Pharmacol. 2008, 153, 199–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dos Santos, R.G.; Hallak, J.E.C.; Crippa, J.A.S. Neuropharmacological effects of the main phytocannabinoids: A narrative review. Adv. Exp. Med. Biol. 2021, 1264, 29–45. [Google Scholar] [CrossRef]
- Dash, R.; Ali, M.C.; Jahan, I.; Munni, Y.A.; Mitra, S.; Hannan, M.A.; Timalsina, B.; Oktaviani, D.F.; Choi, H.J.; Moon, I.S. Emerging potential of cannabidiol in reversing proteinopathies. Aging Res. Rev. 2021, 65, 101209. [Google Scholar] [CrossRef] [PubMed]
- Petitet, F.; Jeantaud, B.; Reibaud, M.; Imperato, A.; Dubroeucq, M.C. Complex pharmacology of natural cannabinoids: Evidence for partial agonist activity of delta9-tetrahydrocannabinol and antagonist activity of cannabidiol on rat brain cannabinoid receptors. Life Sci. 1998, 63, PL1–PL6. [Google Scholar] [CrossRef]
- Thomas, A.; Baillie, G.L.; Phillips, A.M.; Razdan, R.K.; Ross, R.A.; Pertwee, R.G. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br. J. Pharmacol. 2007, 150, 613–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunn, C.A.; Fine, J.S.; Rojas-Triana, A.; Jackson, J.V.; Fan, X.; Kung, T.T.; Gonsiorek, W.; Schwarz, M.A.; Lavey, B.; Kozlowski, J.A.; et al. A novel cannabinoid peripheral cannabinoid receptor-selective inverse agonist blocks leukocyte recruitment in vivo. J. Pharmacol. Exp. Ther. 2006, 316, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, Y.; Tian, D.; Tian, L.; Ju, X.; Qi, L.; Wang, Y.; Liang, C. Overview of cannabidiol (CBD) and its analogues: Structures, biological activities, and neuroprotective mechanisms in epilepsy and Alzheimer’s disease. Eur. J. Med. Chem. 2020, 192, 112163. [Google Scholar] [CrossRef]
- Ryberg, E.; Larsson, N.; Sjogren, S.; Hjorth, S.; Hermansson, N.O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar] [CrossRef]
- Lauckner, J.E.; Jensen, J.B.; Chen, H.Y.; Lu, H.C.; Hille, B.; Mackie, K. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc. Natl. Acad. Sci. USA 2008, 105, 2699–2704. [Google Scholar] [CrossRef] [Green Version]
- Marichal-Cancino, B.A.; Fajardo-Valdez, A.; Ruiz-Contreras, A.E.; Mendez-Diaz, M.; Prospero-Garcia, O. Advances in the physiology of GPR55 in the central nervous system. Cur. Neuropharmacol. 2017, 15, 771–778. [Google Scholar] [CrossRef] [Green Version]
- De Gregorio, D.; McLaughlin, R.J.; Posa, L.; Ochoa-Sanchez, R.; Enns, J.; Lopez-Canul, M.; Aboud, M.; Maione, S.; Comai, S.; Gobbi, G. Cannabidiol modulates serotonergic transmission and reverses both allodynia and anxiety-like behavior in a model of neuropathic pain. Pain 2019, 160, 136–150. [Google Scholar] [CrossRef] [PubMed]
- Costa, B.; Giagnoni, G.; Franke, C.; Trovato, A.E.; Colleoni, M. Vanilloid TRPV1 receptor mediates the antihyperalgesic effect of the nonpsychoactive cannabinoid, cannabidiol, in a rat model of acute inflammation. Br. J. Pharmacol. 2004, 143, 247–250. [Google Scholar] [CrossRef] [Green Version]
- Svob Strac, D.; Pivac, N.; Muck-Seler, D. The serotonergic system and cognitive function. Transl. Neurosci. 2016, 7, 35–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magen, I.; Avraham, Y.; Ackerman, Z.; Vorobiev, L.; Mechoulam, R.; Berry, E.M. Cannabidiol ameliorates cognitive and motor impairments in bile-duct ligated mice via 5-HT1A receptor activation. Br. J. Pharmacol. 2010, 159, 950–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuji, F.; Murai, M.; Oki, K.; Seki, I.; Ueda, K.; Inoue, H.; Nagelkerken, L.; Sasano, M.; Aono, H. Transient receptor potential vanilloid 1 agonists as candidates for anti-inflammatory and immunomodulatory agents. Eur. J. Pharmacol. 2010, 627, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Stampanoni Bassi, M.; Gentile, A.; Iezzi, E.; Zagaglia, S.; Musella, A.; Simonelli, I.; Gilio, L.; Furlan, R.; Finardi, A.; Marfia, G.A.; et al. Transient Receptor Potential Vanilloid 1 Modulates Central Inflammation in Multiple Sclerosis. Front. Neurol. 2019, 10, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mecha, M.; Feliu, A.; Inigo, P.M.; Mestre, L.; Carrillo-Salinas, F.J.; Guaza, C. Cannabidiol provides long-lasting protection against the deleterious effects of inflammation in a viral model of multiple sclerosis: A role for A2A receptors. Neurobiol. Dis. 2013, 59, 141–150. [Google Scholar] [CrossRef]
- O’Sullivan, S.E. An update on PPAR activation by cannabinoids. Br. J. Pharmacol 2016, 173, 1899–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, R.; Reyes-Resina, I.; Aguinaga, D.; Lillo, A.; Jimenez, J.; Raich, I.; Borroto-Escuela, D.O.; Ferreiro-Vera, C.; Canela, E.I.; Sanchez de Medina, V.; et al. Potentiation of cannabinoid signaling in microglia by adenosine A2A receptor antagonists. Glia 2019, 67, 2410–2423. [Google Scholar] [CrossRef]
- Peters, J.M.; Shah, Y.M.; Gonzalez, F.J. The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention. Nat. Rev. Cancer 2012, 12, 181–195. [Google Scholar] [CrossRef]
- Elmes, M.W.; Kaczocha, M.; Berger, W.T.; Leung, K.; Ralph, B.P.; Wang, L.; Sweeney, J.M.; Miyauchi, J.T.; Tsirka, S.E.; Ojima, I.; et al. Fatty acid-binding proteins (FABPs) are intracellular carriers for Delta9-tetrahydrocannabinol (THC) and cannabidiol (CBD). J. Biol. Chem. 2015, 290, 8711–8721. [Google Scholar] [CrossRef] [Green Version]
- Silvestro, S.; Schepici, G.; Bramanti, P.; Mazzon, E. Molecular targets of cannabidiol in eExperimental models of neurological disease. Molecules 2020, 25, 5186. [Google Scholar] [CrossRef]
- Kathmann, M.; Flau, K.; Redmer, A.; Trankle, C.; Schlicker, E. Cannabidiol is an allosteric modulator at mu- and delta-opioid receptors. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2006, 372, 354–361. [Google Scholar] [CrossRef]
- Teng, L.; Zhao, J.; Wang, F.; Ma, L.; Pei, G. A GPCR/secretase complex regulates beta- and gamma-secretase specificity for Abeta production and contributes to AD pathogenesis. Cell Res. 2010, 20, 138–153. [Google Scholar] [CrossRef]
- Hughes, B.; Herron, C.E. Cannabidiol reverses deficits in hippocampal LTP in a model of Alzheimer’s disease. Neurochem. Res. 2019, 44, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Martin-Moreno, A.M.; Reigada, D.; Ramirez, B.G.; Mechoulam, R.; Innamorato, N.; Cuadrado, A.; de Ceballos, M.L. Cannabidiol and other cannabinoids reduce microglial activation in vitro and in vivo: Relevance to Alzheimer’s disease. Mol. Pharmacol. 2011, 79, 964–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, D.; Spiro, A.S.; Jenner, A.M.; Garner, B.; Karl, T. Long-term cannabidiol treatment prevents the development of social recognition memory deficits in Alzheimer’s disease transgenic mice. J. Alzheimer’s Dis. JAD 2014, 42, 1383–1396. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Low, J.K.; Logge, W.; Garner, B.; Karl, T. Chronic cannabidiol treatment improves social and object recognition in double transgenic APPswe/PS1E9 mice. Psychopharmacology 2014, 231, 3009–3017. [Google Scholar] [CrossRef]
- Watt, G.; Chesworth, R.; Przybyla, M.; Ittner, A.; Garner, B.; Ittner, L.M.; Karl, T. Chronic cannabidiol (CBD) treatment did not exhibit beneficial effects in 4-month-old male TAU58/2 transgenic mice. Pharmacol. Biochem. Behav. 2020, 196, 172970. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, C.; Steardo, L.; Esposito, G. Cannabidiol promotes amyloid precursor protein ubiquitination and reduction of beta amyloid expression in SHSY5YAPP+ cells through PPARgamma involvement. Phytother. Res. PTR 2014, 28, 1007–1013. [Google Scholar] [CrossRef]
- Hao, F.; Feng, Y. Cannabidiol (CBD) enhanced the hippocampal immune response and autophagy of APP/PS1 Alzheimer’s mice uncovered by RNA-seq. Life Sci. 2021, 264, 118624. [Google Scholar] [CrossRef]
- Esposito, G.; De Filippis, D.; Carnuccio, R.; Izzo, A.A.; Iuvone, T. The marijuana component cannabidiol inhibits beta-amyloid-induced tau protein hyperphosphorylation through Wnt/beta-catenin pathway rescue in PC12 cells. J. Mol. Med. (Berl. Ger.) 2006, 84, 253–258. [Google Scholar] [CrossRef]
- Hernandez, F.; Lucas, J.J.; Avila, J. GSK3 and tau: Two convergence points in Alzheimer’s disease. J. Alzheimer’s Dis. JAD 2013, 33 (Suppl. 1), S141–S144. [Google Scholar] [CrossRef] [Green Version]
- Iuvone, T.; Esposito, G.; Esposito, R.; Santamaria, R.; Di Rosa, M.; Izzo, A.A. Neuroprotective effect of cannabidiol, a non-psychoactive component from Cannabis sativa, on beta-amyloid-induced toxicity in PC12 cells. J. Neurochem. 2004, 89, 134–141. [Google Scholar] [CrossRef]
- Esposito, G.; De Filippis, D.; Maiuri, M.C.; De Stefano, D.; Carnuccio, R.; Iuvone, T. Cannabidiol inhibits inducible nitric oxide synthase protein expression and nitric oxide production in beta-amyloid stimulated PC12 neurons through p38 MAP kinase and NF-kappaB involvement. Neurosci. Lett. 2006, 399, 91–95. [Google Scholar] [CrossRef]
- Esposito, G.; Scuderi, C.; Valenza, M.; Togna, G.I.; Latina, V.; De Filippis, D.; Cipriano, M.; Carratu, M.R.; Iuvone, T.; Steardo, L. Cannabidiol reduces Abeta-induced neuroinflammation and promotes hippocampal neurogenesis through PPARgamma involvement. PLoS ONE 2011, 6, e28668. [Google Scholar] [CrossRef] [PubMed]
- Pecina-Slaus, N. Wnt signal transduction pathway and apoptosis: A review. Cancer Cell Int. 2010, 10, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallee, A.; Lecarpentier, Y.; Guillevin, R.; Vallee, J.N. Effects of cannabidiol interactions with Wnt/beta-catenin pathway and PPARgamma on oxidative stress and neuroinflammation in Alzheimer’s disease. Acta Biochim. Biophys. Sin. 2017, 49, 853–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libro, R.; Diomede, F.; Scionti, D.; Piattelli, A.; Grassi, G.; Pollastro, F.; Bramanti, P.; Mazzon, E.; Trubiani, O. Cannabidiol modulates the expression of Alzheimer’s disease-related genes in mesenchymal stem cells. Int. J. Mol. Sci. 2016, 18, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiong, Y.; Lim, C.-S. Understanding the Modulatory Effects of Cannabidiol on Alzheimer’s Disease. Brain Sci. 2021, 11, 1211. https://doi.org/10.3390/brainsci11091211
Xiong Y, Lim C-S. Understanding the Modulatory Effects of Cannabidiol on Alzheimer’s Disease. Brain Sciences. 2021; 11(9):1211. https://doi.org/10.3390/brainsci11091211
Chicago/Turabian StyleXiong, Yinyi, and Chae-Seok Lim. 2021. "Understanding the Modulatory Effects of Cannabidiol on Alzheimer’s Disease" Brain Sciences 11, no. 9: 1211. https://doi.org/10.3390/brainsci11091211
APA StyleXiong, Y., & Lim, C.-S. (2021). Understanding the Modulatory Effects of Cannabidiol on Alzheimer’s Disease. Brain Sciences, 11(9), 1211. https://doi.org/10.3390/brainsci11091211