
CSF Secretion Is Not Altered by NKCC1 Nor TRPV4 Antagonism in Healthy Rats

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
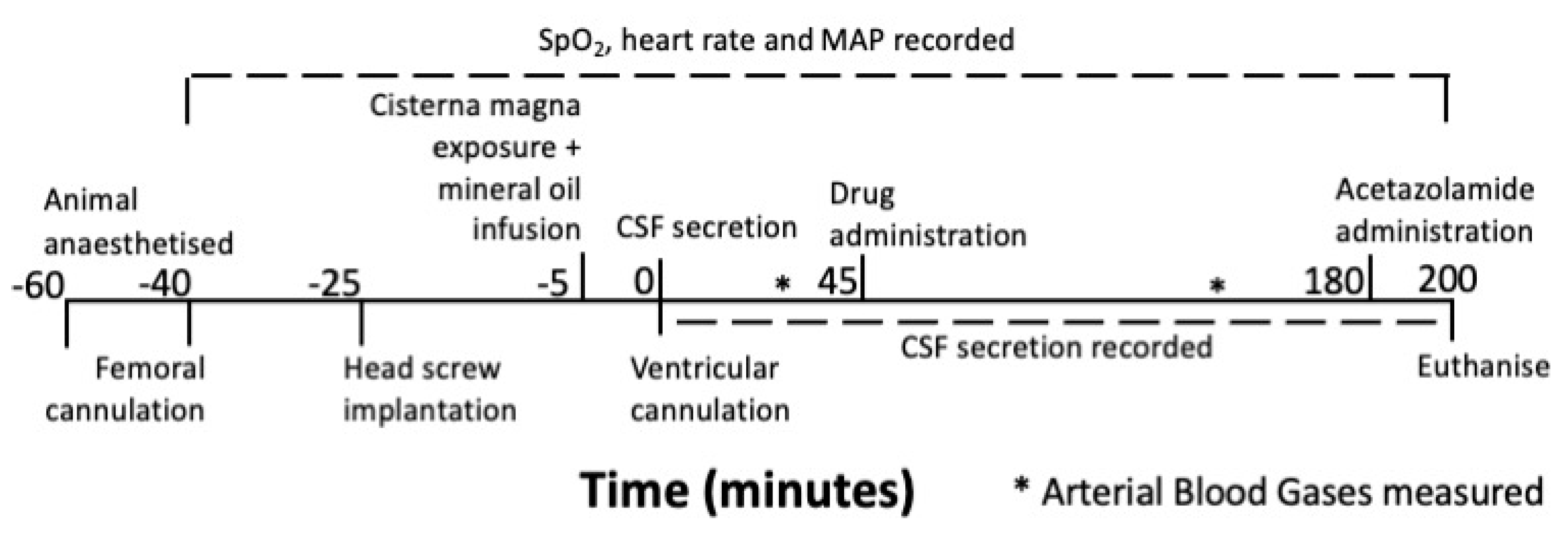
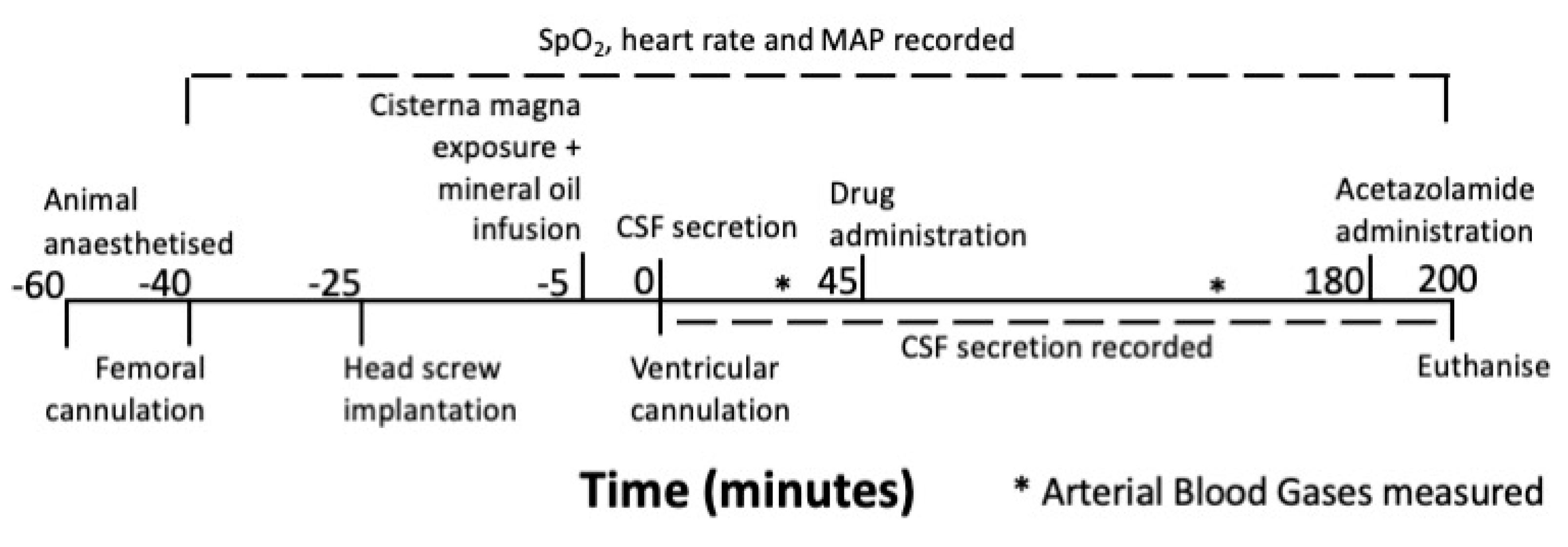
2.1. Experimental Design
2.2. Animals
2.3. Anaesthesia and Monitoring
2.4. Measuring CSF secretion
2.5. Drug Administration
2.6. Statistics
3. Results
3.1. Exclusions
3.2. Physiological Parameters
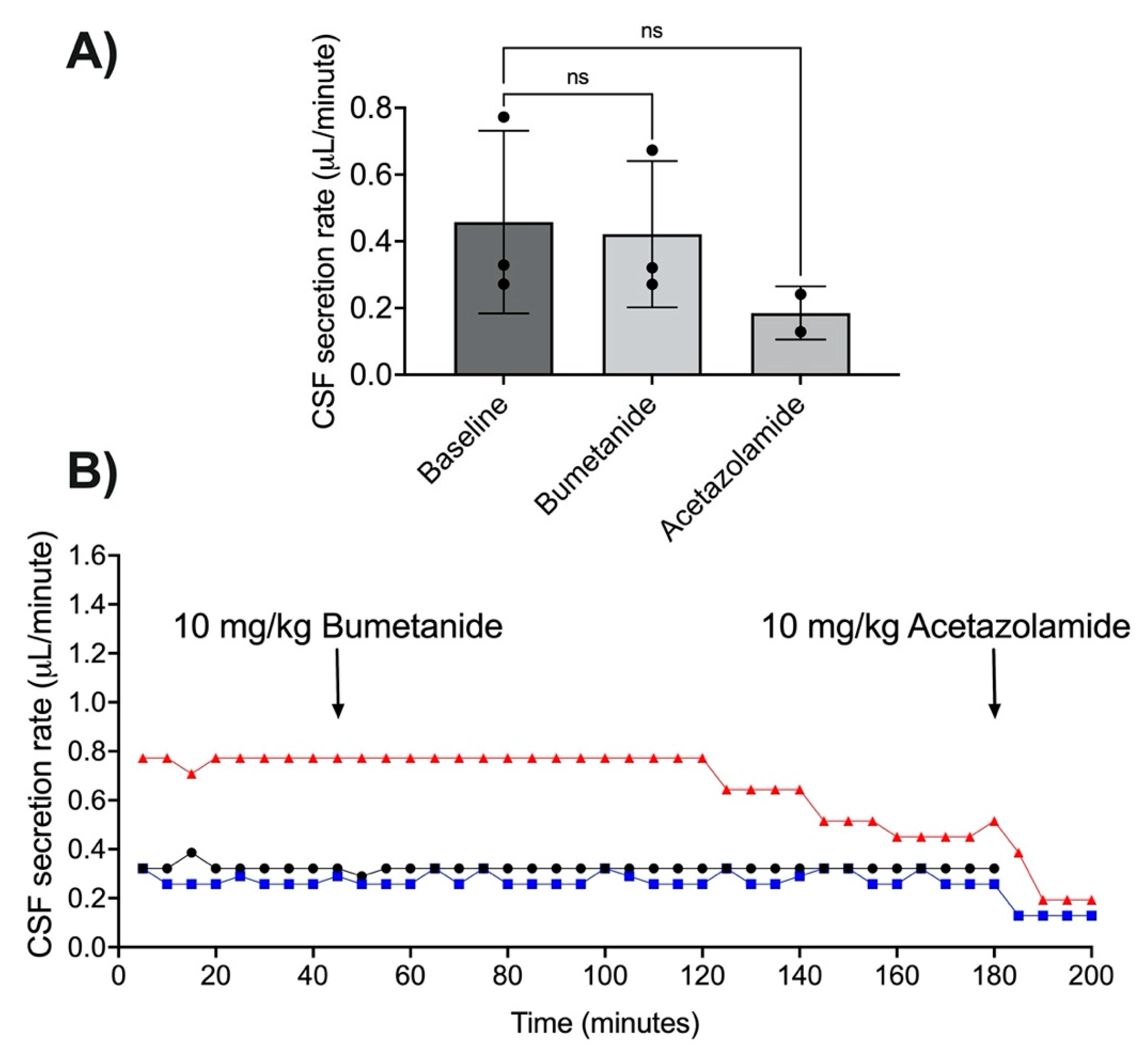
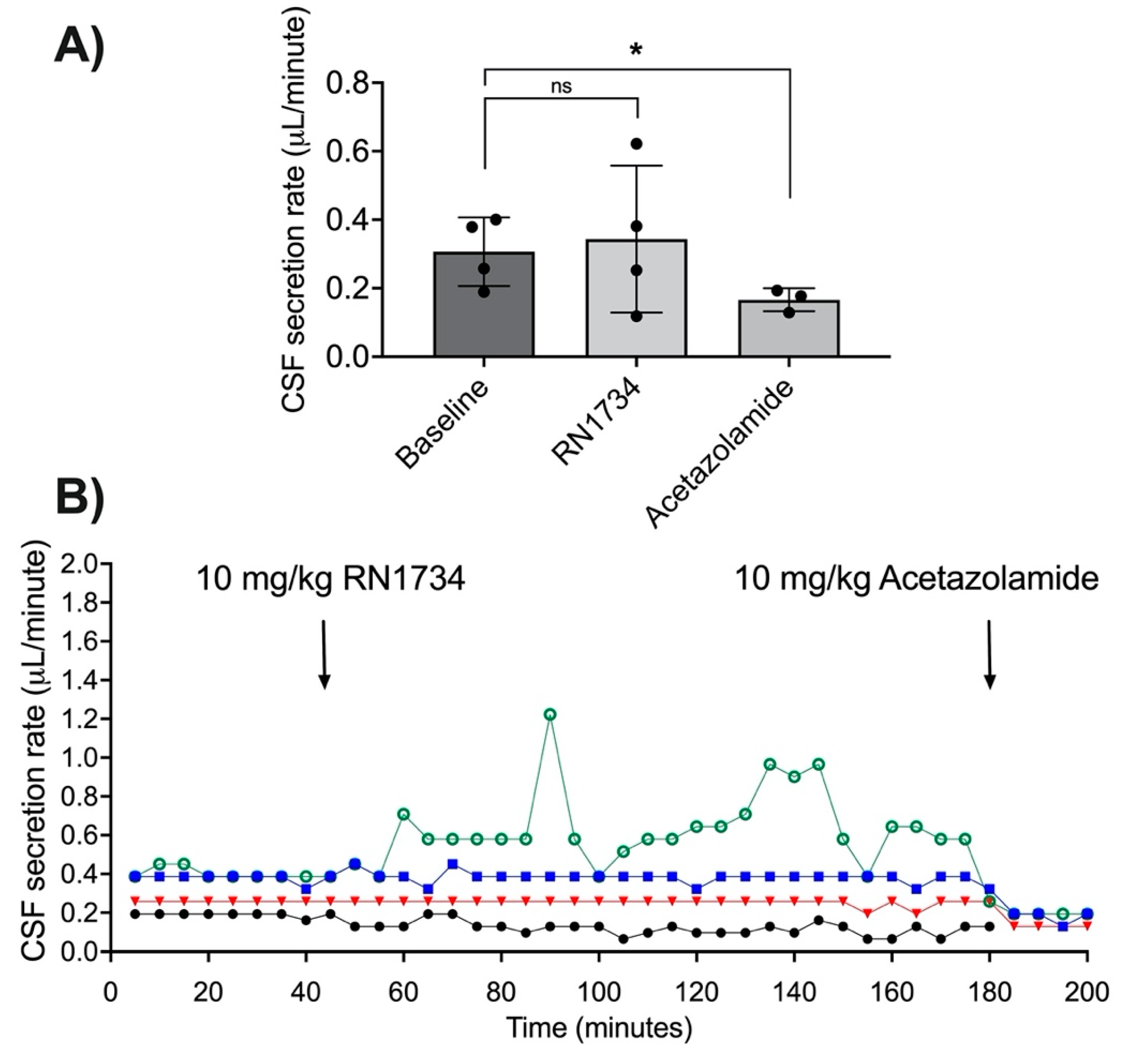
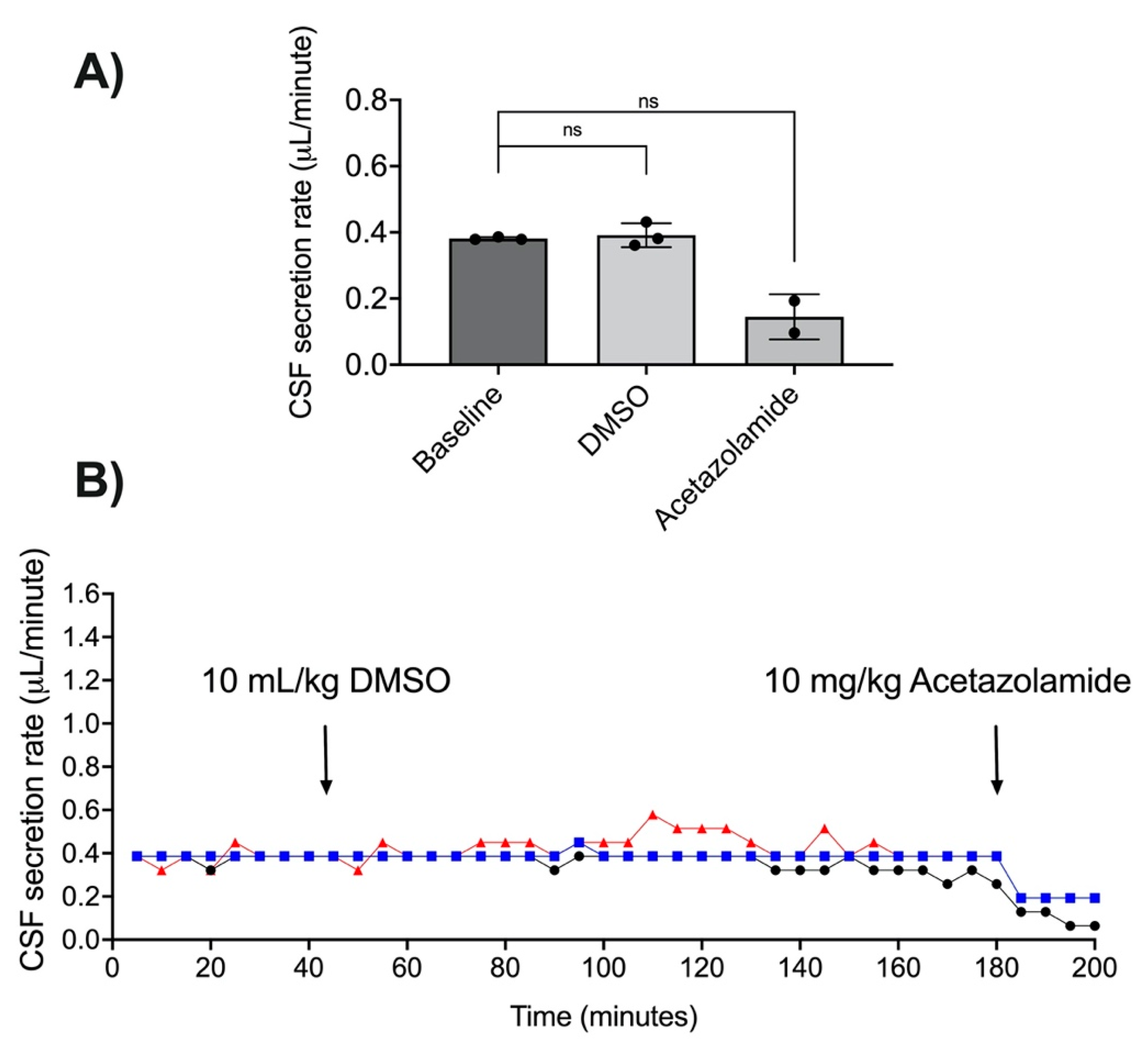
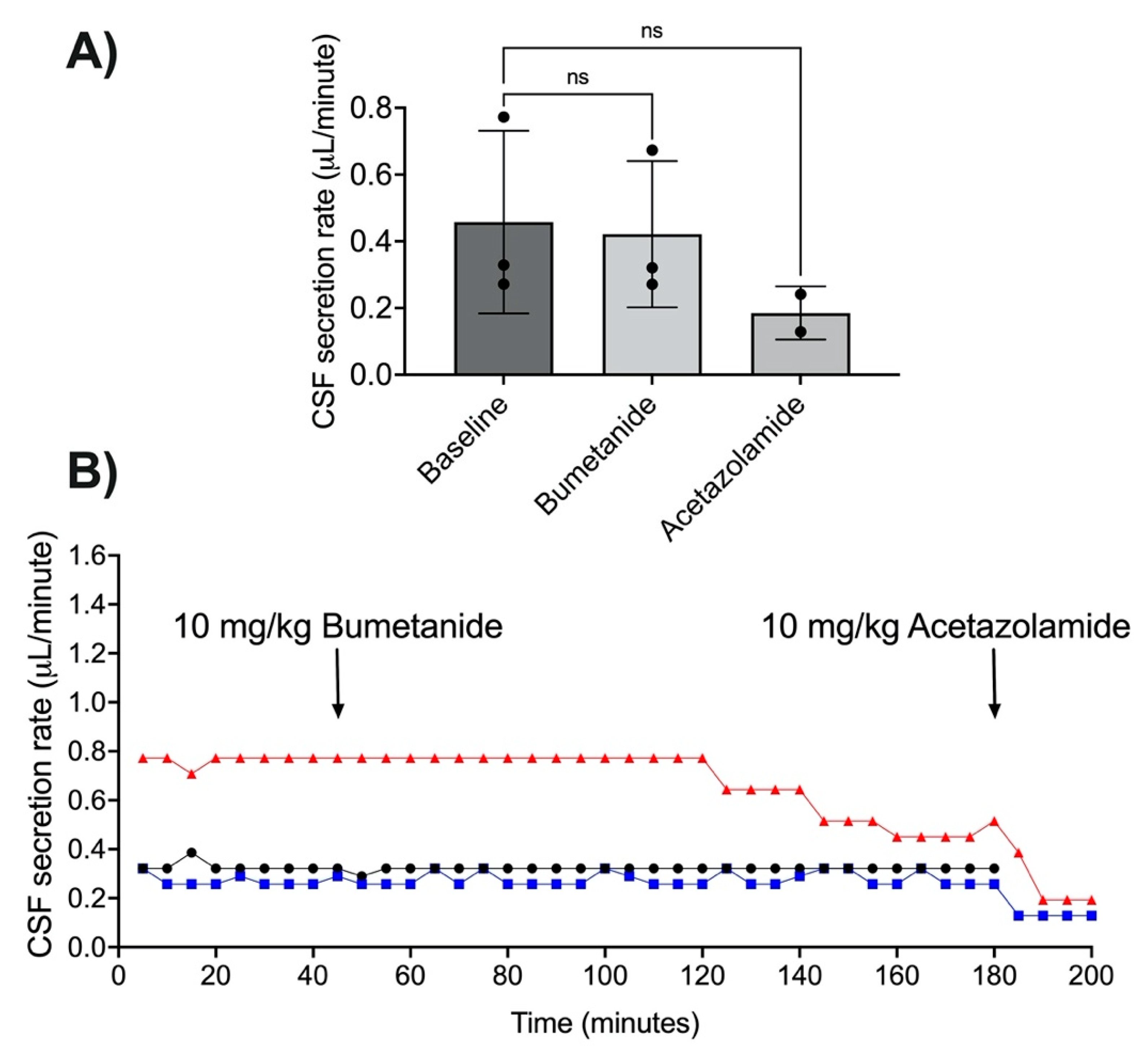
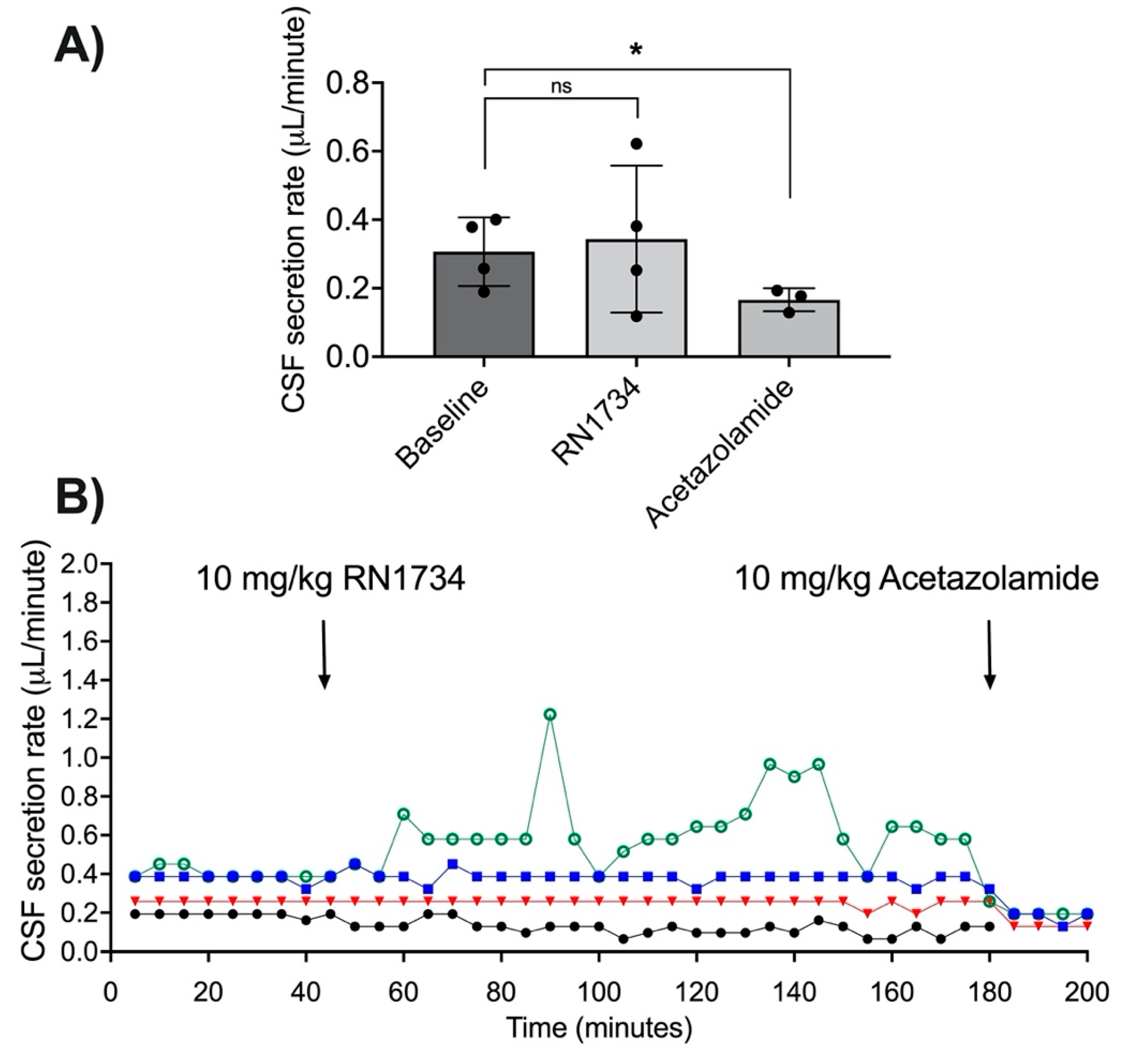
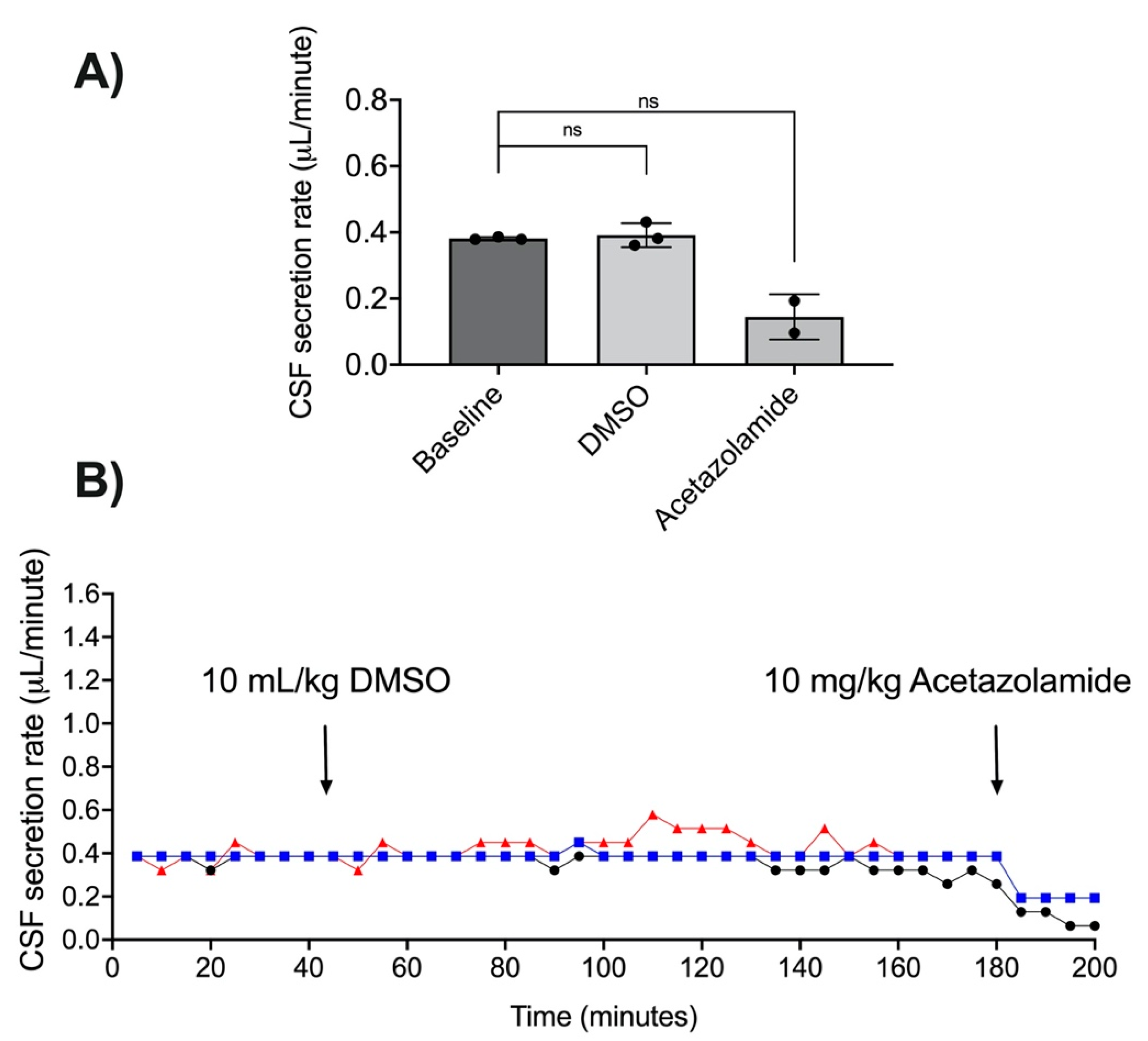
3.3. Acetazolamide and CSF Secretion
3.4. NKCC1 Antagonism with Bumetanide
3.5. TRPV4 Antagonism with RN1734
3.6. DMSO as a Vehicle for RN1734 Administration
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leinonen, V.; Vanninen, R.; Rauramaa, T. Cerebrospinal fluid circulation and hydrocephalus. Handb. Clin. Neurol. 2017, 145, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Jergović, I.; Budinčević, H.; Planjar-Prvan, M.; Bielen, I. Transient Obstructive Hydrocephalus in Patients with Intracerebral Hemorrhage: Report of Two Cases. Acta Clin. Croat. 2016, 55, 497–500. [Google Scholar] [CrossRef] [Green Version]
- Goudie, C.; Burr, J.; Blaikie, A. Incidence of idiopathic intracranial hypertension in Fife. Scott. Med. J. 2019, 64, 62–66. [Google Scholar] [CrossRef] [Green Version]
- Unterberg, A.W.; Stover, J.; Kress, B.; Kiening, K.L. Edema and brain trauma. Neuroscience 2004, 129, 1021–1029. [Google Scholar] [CrossRef]
- Sorby-Adams, A.J.; Leonard, A.V.; Hoving, J.W.; Yassi, N.; Vink, R.; Wells, A.J.; Turner, R.J. NK1-r Antagonist Treatment Comparable to Decompressive Craniectomy in Reducing Intracranial Pressure Following Stroke. Front. Neurosci. 2019, 13, 681. [Google Scholar] [CrossRef]
- Sorby-Adams, A.J.; Leonard, A.V.; Elms, L.E.; Marian, O.C.; Hoving, J.W.; Yassi, N.; Vink, R.; Thornton, E.; Turner, R.J. Determining the Temporal Profile of Intracranial Pressure Changes Following Transient Stroke in an Ovine Model. Front. Neurosci. 2019, 13, 587. [Google Scholar] [CrossRef]
- Schubert, G.A.; Seiz, M.; Hegewald, A.A.; Manville, J.; Thomé, C. Hypoperfusion in the acute phase of subarachnoid hemorrhage. Acta Neurochir. Suppl. 2011, 110, 35–38. [Google Scholar] [CrossRef]
- van Asch, C.J.; van der Schaaf, I.C.; Rinkel, G.J. Acute hydrocephalus and cerebral perfusion after aneurysmal subarachnoid hemorrhage. AJNR Am. J. Neuroradiol. 2010, 31, 67–70. [Google Scholar] [CrossRef] [Green Version]
- Griebel, R.W.; Black, P.M.; Pile-Spellman, J.; Strauss, W.H. The Importance of “Accessory” Outflow Pathways in Hydrocephalus after Experimental Subarachnoid Hemorrhage. Neurosurgery 1989, 24, 187–192. [Google Scholar] [CrossRef]
- Ropper, A.H.; Shafran, B. Brain edema after stroke. Clinical syndrome and intracranial pressure. Arch. Neurol. 1984, 41, 26–29. [Google Scholar] [CrossRef]
- Beard, D.J.; Logan, C.L.; McLeod, D.D.; Hood, R.J.; Pepperall, D.; Murtha, L.A.; Spratt, N.J. Ischemic penumbra as a trigger for intracranial pressure rise—A potential cause for collateral failure and infarct progression? J. Cereb. Blood Flow Metab. 2016, 36, 917–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murtha, L.A.; McLeod, D.D.; McCann, S.K.; Pepperall, D.; Chung, S.; Levi, C.R.; Calford, M.B.; Spratt, N.J. Short-duration hypothermia after ischemic stroke prevents delayed intracranial pressure rise. Int. J. Stroke 2014, 9, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Murtha, L.A.; McLeod, D.D.; Pepperall, D.; McCann, S.K.; Beard, D.J.; Tomkins, A.J.; Holmes, W.M.; McCabe, C.; Macrae, I.M.; Spratt, N.J. Intracranial pressure elevation after ischemic stroke in rats: Cerebral edema is not the only cause, and short-duration mild hypothermia is a highly effective preventive therapy. J. Cereb. Blood Flow Metab. 2015, 35, 592–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alshuhri, M.S.; Gallagher, L.; McCabe, C.; Holmes, W.M. Change in CSF Dynamics Responsible for ICP Elevation After Ischemic Stroke in Rats: A New Mechanism for Unexplained END? Transl. Stroke Res. 2019, 11, 310–318. [Google Scholar] [CrossRef]
- Kotwica, Z.; Hårdemark, H.G.; Persson, L. Intracranial pressure changes following middle cerebral artery occlusion in rats. Res. Exp. Med. 1991, 191, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Robba, C.; Citerio, G. How I manage intracranial hypertension. Crit. Care 2019, 23, 243. [Google Scholar] [CrossRef] [Green Version]
- Montemurro, N.; Santoro, G.; Marani, W.; Petrella, G. Posttraumatic synchronous double acute epidural hematomas: Two craniotomies, single skin incision. Surg. Neurol. Int. 2020, 11, 435. [Google Scholar] [CrossRef]
- Pinto, V.L.; Tadi, P.; Adeyinka, A. Increased Intracranial Pressure. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Vogh, B.P.; Langham, M.R., Jr. The effect of furosemide and bumetanide on cerebrospinal fluid formation. Brain Res. 1981, 221, 171–183. [Google Scholar] [CrossRef]
- Uldall, M.; Botfield, H.; Jansen-Olesen, I.; Sinclair, A.; Jensen, R. Acetazolamide lowers intracranial pressure and modulates the cerebrospinal fluid secretion pathway in healthy rats. Neurosci. Lett. 2017, 645, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Williamson, M.R.; Wilkinson, C.M.; Dietrich, K.; Colbourne, F. Acetazolamide Mitigates Intracranial Pressure Spikes Without Affecting Functional Outcome After Experimental Hemorrhagic Stroke. Transl. Stroke Res. 2019, 10, 428–439. [Google Scholar] [CrossRef]
- Supuran, C.T. Acetazolamide for the treatment of idiopathic intracranial hypertension. Expert Rev. Neurother. 2015, 15, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.; Chan, H.; Thorne, L.; Watkins, L.; Toma, A. TM3-4 The effect of acetazolamide on intracranial pressure: Primary study with prolonged continuous intracranial pressure monitoring. J. Neurol. Neurosurg. Psychiatry 2019, 90, e16. [Google Scholar] [CrossRef]
- Woster, P.S.; LeBlanc, K.L. Management of elevated intracranial pressure. Clin. Pharm. 1990, 9, 762–772. [Google Scholar]
- Libenson, M.H.; Kaye, E.M.; Rosman, N.P.; Gilmore, H.E. Acetazolamide and furosemide for posthemorrhagic hydrocephalus of the newborn. Pediatric Neurol. 1999, 20, 185–191. [Google Scholar] [CrossRef]
- Oernbo, E.K.; Lykke, K.; Steffensen, A.B.; Töllner, K.; Kruuse, C.; Rath, M.F.; Löscher, W.; MacAulay, N. Cerebral influx of Na+ and Cl− as the osmotherapy-mediated rebound response in rats. Fluids Barriers CNS 2018, 15, 27. [Google Scholar] [CrossRef]
- Brinker, T.; Stopa, E.; Morrison, J.; Klinge, P. A new look at cerebrospinal fluid circulation. Fluids Barriers CNS 2014, 11, 10. [Google Scholar] [CrossRef] [Green Version]
- Kimelberg, H.K. Water homeostasis in the brain: Basic concepts. Neuroscience 2004, 129, 851–860. [Google Scholar] [CrossRef]
- Redzic, Z. Molecular biology of the blood-brain and the blood-cerebrospinal fluid barriers: Similarities and differences. Fluids Barriers CNS 2011, 8, 3. [Google Scholar] [CrossRef] [Green Version]
- Oresković, D.; Klarica, M. The formation of cerebrospinal fluid: Nearly a hundred years of interpretations and misinterpretations. Brain Res. Rev. 2010, 64, 241–262. [Google Scholar] [CrossRef] [Green Version]
- Javaheri, S.; Wagner, K.R. Bumetanide decreases canine cerebrospinal fluid production. In vivo evidence for NaCl cotransport in the central nervous system. J. Clin. Investig. 1993, 92, 2257–2261. [Google Scholar] [CrossRef] [Green Version]
- Bairamian, D.; Johanson, C.E.; Parmelee, J.T.; Epstein, M.H. Potassium cotransport with sodium and chloride in the choroid plexus. J. Neurochem. 1991, 56, 1623–1629. [Google Scholar] [CrossRef]
- Verkman, A.S. Aquaporins. Curr. Biol. 2013, 23, R52–R55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speake, T.; Freeman, L.J.; Brown, P.D. Expression of aquaporin 1 and aquaporin 4 water channels in rat choroid plexus. Biochim. Biophys. Acta 2003, 1609, 80–86. [Google Scholar] [CrossRef]
- Johnson, D.C.; Singer, S.; Hoop, B.; Kazemi, H. Chloride flux from blood to CSF: Inhibition by furosemide and bumetanide. J. Appl. Physiol. 1987, 63, 1591–1600. [Google Scholar] [CrossRef] [PubMed]
- Pollay, M.; Fullenwider, C.; Roberts, P.A.; Stevens, F.A. Effect of mannitol and furosemide on blood-brain osmotic gradient and intracranial pressure. J. Neurosurg. 1983, 59, 945–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, P.A.; Pollay, M.; Engles, C.; Pendleton, B.; Reynolds, E.; Stevens, F.A. Effect on intracranial pressure of furosemide combined with varying doses and administration rates of mannitol. J. Neurosurg. 1987, 66, 440–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoeman, J.F. Childhood Pseudotumor Cerebri: Clinical and Intracranial Pressure Response to Acetazolamide and Furosemide Treatment in a Case Series. J. Child Neurol. 1994, 9, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Schoeman, J.; Donald, P.; van Zyl, L.; Keet, M.; Wait, J. Tuberculous hydrocephalus: Comparison of different treatments with regard to ICP, ventricular size and clinical outcome. Dev. Med. Child Neurol. 1991, 33, 396–405. [Google Scholar] [CrossRef]
- Steffensen, A.B.; Oernbo, E.K.; Stoica, A.; Gerkau, N.J.; Barbuskaite, D.; Tritsaris, K.; Rose, C.R.; MacAulay, N. Cotransporter-mediated water transport underlying cerebrospinal fluid formation. Nat. Commun. 2018, 9, 2167. [Google Scholar] [CrossRef]
- Karimy, J.K.; Kahle, K.T.; Kurland, D.B.; Yu, E.; Gerzanich, V.; Simard, J.M. A novel method to study cerebrospinal fluid dynamics in rats. J. Neurosci. Methods 2015, 241, 78–84. [Google Scholar] [CrossRef] [Green Version]
- NSW Ministry of Health. Initial Management of Closed Head Injury in Adults, 2nd ed.; NSW Ministry of Health: St Leonards, NSW, Australia, 2011. [Google Scholar]
- Stroke Foundation. Clinical Guidelines for Stroke Management; Stroke Foundation: Melbourne, VIC, Australia, 2019. [Google Scholar]
- Narita, K.; Sasamoto, S.; Koizumi, S.; Okazaki, S.; Nakamura, H.; Inoue, T.; Takeda, S. TRPV4 regulates the integrity of the blood-cerebrospinal fluid barrier and modulates transepithelial protein transport. Faseb J. 2015, 29, 2247–2259. [Google Scholar] [CrossRef]
- Pochynyuk, O.; Zaika, O.; O’Neil, R.G.; Mamenko, M. Novel insights into TRPV4 function in the kidney. Pflugers Arch. 2013, 465, 177–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takayama, Y.; Shibasaki, K.; Suzuki, Y.; Yamanaka, A.; Tominaga, M. Modulation of water efflux through functional interaction between TRPV4 and TMEM16A/anoctamin 1. Faseb J. 2014, 28, 2238–2248. [Google Scholar] [CrossRef]
- Preston, D.; Simpson, S.; Halm, D.; Hochstetler, A.; Schwerk, C.; Schroten, H.; Blazer-Yost, B.L. Activation of TRPV4 stimulates transepithelial ion flux in a porcine choroid plexus cell line. Am. J. Physiol. Cell Physiol. 2018, 315, C357–C366. [Google Scholar] [CrossRef]
- Liedtke, W.; Choe, Y.; Martí-Renom, M.A.; Bell, A.M.; Denis, C.S.; Sali, A.; Hudspeth, A.J.; Friedman, J.M.; Heller, S. Vanilloid receptor-related osmotically activated channel (VR-OAC), a candidate vertebrate osmoreceptor. Cell 2000, 103, 525–535. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, H.; Vriens, J.; Suh, S.H.; Benham, C.D.; Droogmans, G.; Nilius, B. Heat-evoked activation of TRPV4 channels in a HEK293 cell expression system and in native mouse aorta endothelial cells. J. Biol. Chem. 2002, 277, 47044–47051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohler, R.; Heyken, W.T.; Heinau, P.; Schubert, R.; Si, H.; Kacik, M.; Busch, C.; Grgic, I.; Maier, T.; Hoyer, J. Evidence for a functional role of endothelial transient receptor potential V4 in shear stress-induced vasodilatation. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1495–1502. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, H.; Davis, J.B.; Smart, D.; Jerman, J.C.; Smith, G.D.; Hayes, P.; Vriens, J.; Cairns, W.; Wissenbach, U.; Prenen, J.; et al. Activation of TRPV4 channels (hVRL-2/mTRP12) by phorbol derivatives. J. Biol. Chem. 2002, 277, 13569–13577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jie, P.; Lu, Z.; Hong, Z.; Li, L.; Zhou, L.; Li, Y.; Zhou, R.; Zhou, Y.; Du, Y.; Chen, L.; et al. Activation of Transient Receptor Potential Vanilloid 4 is Involved in Neuronal Injury in Middle Cerebral Artery Occlusion in Mice. Mol. Neurobiol. 2016, 53, 8–17. [Google Scholar] [CrossRef]
- Hoshi, Y.; Okabe, K.; Shibasaki, K.; Funatsu, T.; Matsuki, N.; Ikegaya, Y.; Koyama, R. Ischemic Brain Injury Leads to Brain Edema via Hyperthermia-Induced TRPV4 Activation. J. Neurosci. 2018, 38, 5700. [Google Scholar] [CrossRef]
- Lu, K.T.; Huang, T.C.; Tsai, Y.H.; Yang, Y.L. Transient receptor potential vanilloid type 4 channels mediate Na-K-Cl-co-transporter-induced brain edema after traumatic brain injury. J. Neurochem. 2017, 140, 718–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hochstetler, A.E.; Smith, H.M.; Preston, D.C.; Reed, M.M.; Territo, P.R.; Shim, J.W.; Fulkerson, D.; Blazer-Yost, B.L. TRPV4 antagonists ameliorate ventriculomegaly in a rat model of hydrocephalus. JCI Insight 2020, 5, e137646. [Google Scholar] [CrossRef]
- Kumar, H.; Lim, C.S.; Choi, H.; Joshi, H.P.; Kim, K.T.; Kim, Y.H.; Park, C.K.; Kim, H.M.; Han, I.B. Elevated TRPV4 Levels Contribute to Endothelial Damage and Scarring in Experimental Spinal Cord Injury. J. Neurosci. 2020, 40, 1943–1955. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Mu, X.; Wang, H.; Song, C.; Ma, W.; Jolkkonen, J.; Zhao, C. Chloride Co-transporter NKCC1 Inhibitor Bumetanide Enhances Neurogenesis and Behavioral Recovery in Rats After Experimental Stroke. Mol. Neurobiol. 2017, 54, 2406–2414. [Google Scholar] [CrossRef] [PubMed]
- Töpfer, M.; Töllner, K.; Brandt, C.; Twele, F.; Bröer, S.; Löscher, W. Consequences of inhibition of bumetanide metabolism in rodents on brain penetration and effects of bumetanide in chronic models of epilepsy. Eur. J. Neurosci. 2014, 39, 673–687. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Shangguan, Y.; Barks, J.D.E.; Silverstein, F.S. Bumetanide augments the neuroprotective efficacy of phenobarbital plus hypothermia in a neonatal hypoxia–ischemia model. Pediatric Res. 2012, 71, 559–565. [Google Scholar] [CrossRef] [Green Version]
- Martins, A.N.; Newby, N.; Doyle, T.F. Sources of error in measuring cerebrospinal fluid formation by ventriculocisternal perfusion. J. Neurol. Neurosurg. Psychiatry 1977, 40, 645–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrini, P.; Montemurro, N.; Iannelli, A. The Contribution of Carlo Giacomini (1840–1898): The Limbus Giacomini and Beyond. Neurosurgery 2012, 72, 475–482. [Google Scholar] [CrossRef]
- Wilkinson, C.M.; Fedor, B.A.; Aziz, J.R.; Nadeau, C.A.; Brar, P.S.; Clark, J.J.A.; Colbourne, F. Failure of bumetanide to improve outcome after intracerebral hemorrhage in rat. PLoS ONE 2019, 14, e0210660. [Google Scholar] [CrossRef] [Green Version]
- Damkier, H.H.; Brown, P.D.; Praetorius, J. Epithelial pathways in choroid plexus electrolyte transport. Physiology 2010, 25, 239–249. [Google Scholar] [CrossRef] [Green Version]
- Artru, A.A. Isoflurane Does Not Increase the Rate of CSF Production in the Dog. Anesthesiology 1984, 60, 193–197. [Google Scholar] [CrossRef]
- Liu, G.; Mestre, H.; Sweeney, A.M.; Sun, Q.; Weikop, P.; Du, T.; Nedergaard, M. Direct Measurement of Cerebrospinal Fluid Production in Mice. Cell Rep. 2020, 33, 108524. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RN1734 (n = 3) | DMSO (n = 4) | Bumetanide (n = 3) | ||||
|---|---|---|---|---|---|---|
| Baseline | Post-treatment | Baseline | Post-treatment | Baseline | Post-treatment | |
| SpO2 (%) | 97.5 ± 2.1 | 97.3 ± 1.5 | 97.7 ± 2.3 | 97.3 ± 2.08 | 98.0 ± 1.7 | 98.7 ± 0.6 |
| Heart rate (BPM) | 391 ± 54 | 403 ± 45 | 388 ± 13 | 391 ± 30 | 406 ± 30 | 411 ± 43 |
| Mean arterial pressure (mmHg) | 104.8 ± 3 | 102.5 ± 9.9 | 100 ± 11 | 100 ± 13.7 | 89.3 ± 7.2 | 84.0 ± 7 |
| Respiratory rate (per minute) | 66 ± 8 | 71 ± 4 | 69 ± 2 | 72 ± 4 | 70 ± 2 | 73 ± 2 |
| paO2 (mmHg) | 139 ± 47.6 | 136 ± 42.2 | 137 ± 30.9 | 158 ± 16.5 | 151 ± 49.3 | 238 ± 54.3 |
| paCO2 (mmHg) | 57.5 ± 4.9 | 57.7 ± 8.6 | 65.5 ± 6.6 | 62.9 ± 15.9 | 73.6 ± 22.6 | 64.4 ± 3.9 |
| pH | 7.31 ± 0.01 | 7.30 ± 0.03 | 7.23 ± 0.04 | 7.28 ± 0.09 | 7.21 ± 0.08 | 7.25 ± 0.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bothwell, S.W.; Omileke, D.; Patabendige, A.; Spratt, N.J. CSF Secretion Is Not Altered by NKCC1 Nor TRPV4 Antagonism in Healthy Rats. Brain Sci. 2021, 11, 1117. https://doi.org/10.3390/brainsci11091117
Bothwell SW, Omileke D, Patabendige A, Spratt NJ. CSF Secretion Is Not Altered by NKCC1 Nor TRPV4 Antagonism in Healthy Rats. Brain Sciences. 2021; 11(9):1117. https://doi.org/10.3390/brainsci11091117
Chicago/Turabian StyleBothwell, Steven W., Daniel Omileke, Adjanie Patabendige, and Neil J. Spratt. 2021. "CSF Secretion Is Not Altered by NKCC1 Nor TRPV4 Antagonism in Healthy Rats" Brain Sciences 11, no. 9: 1117. https://doi.org/10.3390/brainsci11091117
APA StyleBothwell, S. W., Omileke, D., Patabendige, A., & Spratt, N. J. (2021). CSF Secretion Is Not Altered by NKCC1 Nor TRPV4 Antagonism in Healthy Rats. Brain Sciences, 11(9), 1117. https://doi.org/10.3390/brainsci11091117