
The Current Role of Dexmedetomidine as Neuroprotective Agent: An Updated Review


Abstract
:1. Introduction
- It reviews recent studies in various animal models depicting DEXM’s anti-inflammatory role.
- Moreover, it elaborates the longitudinal studies of pre-and post-clinical trials on DEXM, which prove its ability to modulate apoptotic and necrotic events and also illustrate DEXM’s role in astroglia.
- Finally, the proposed study describes all the viable neuroprotective mechanisms of DEXM against nerve injury, which can support scientists and doctors in future studies.
2. Pharmacology of Dexmedetomidine
2.1. Pharmacokinetics of Dexmedetomidine
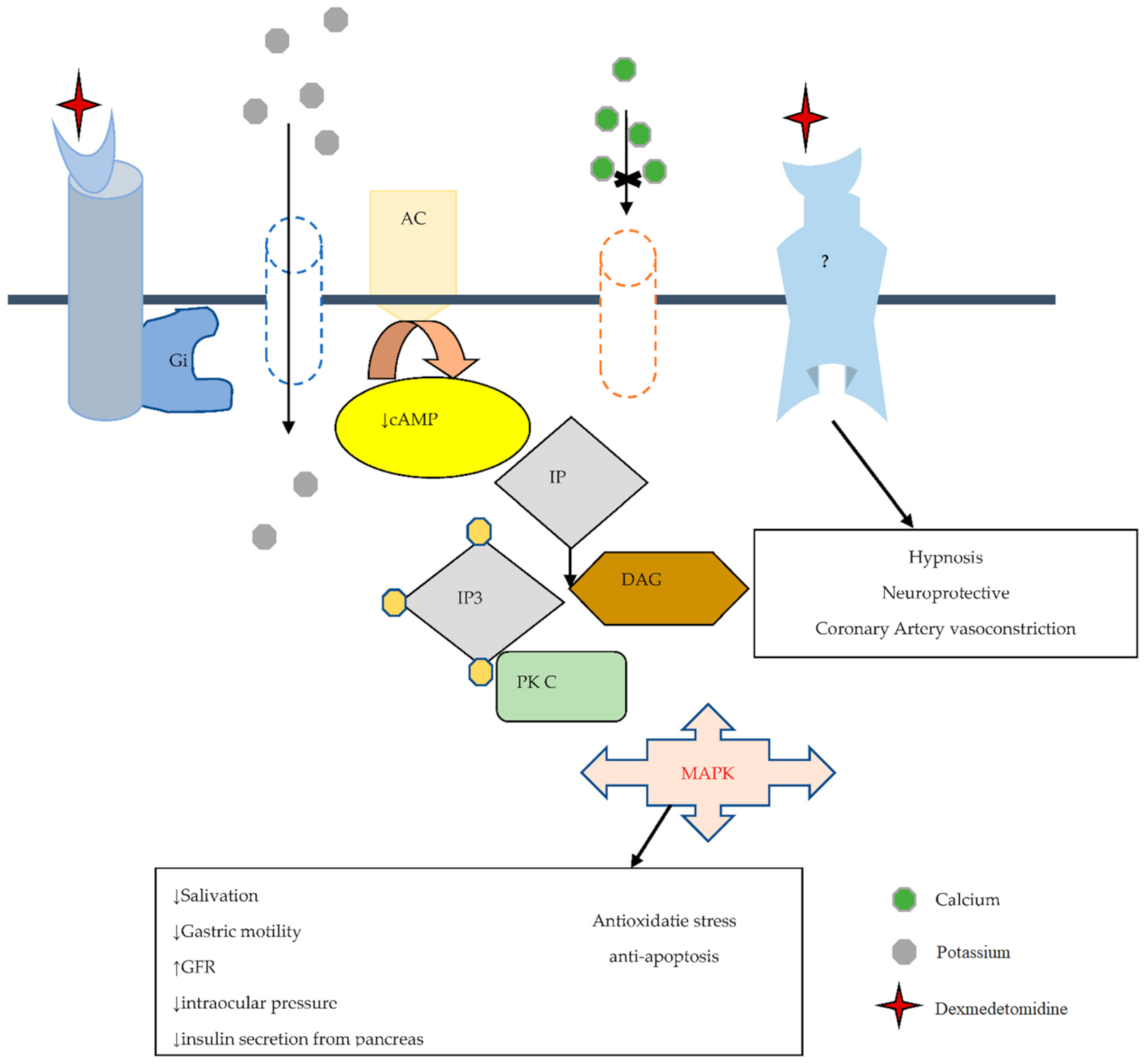
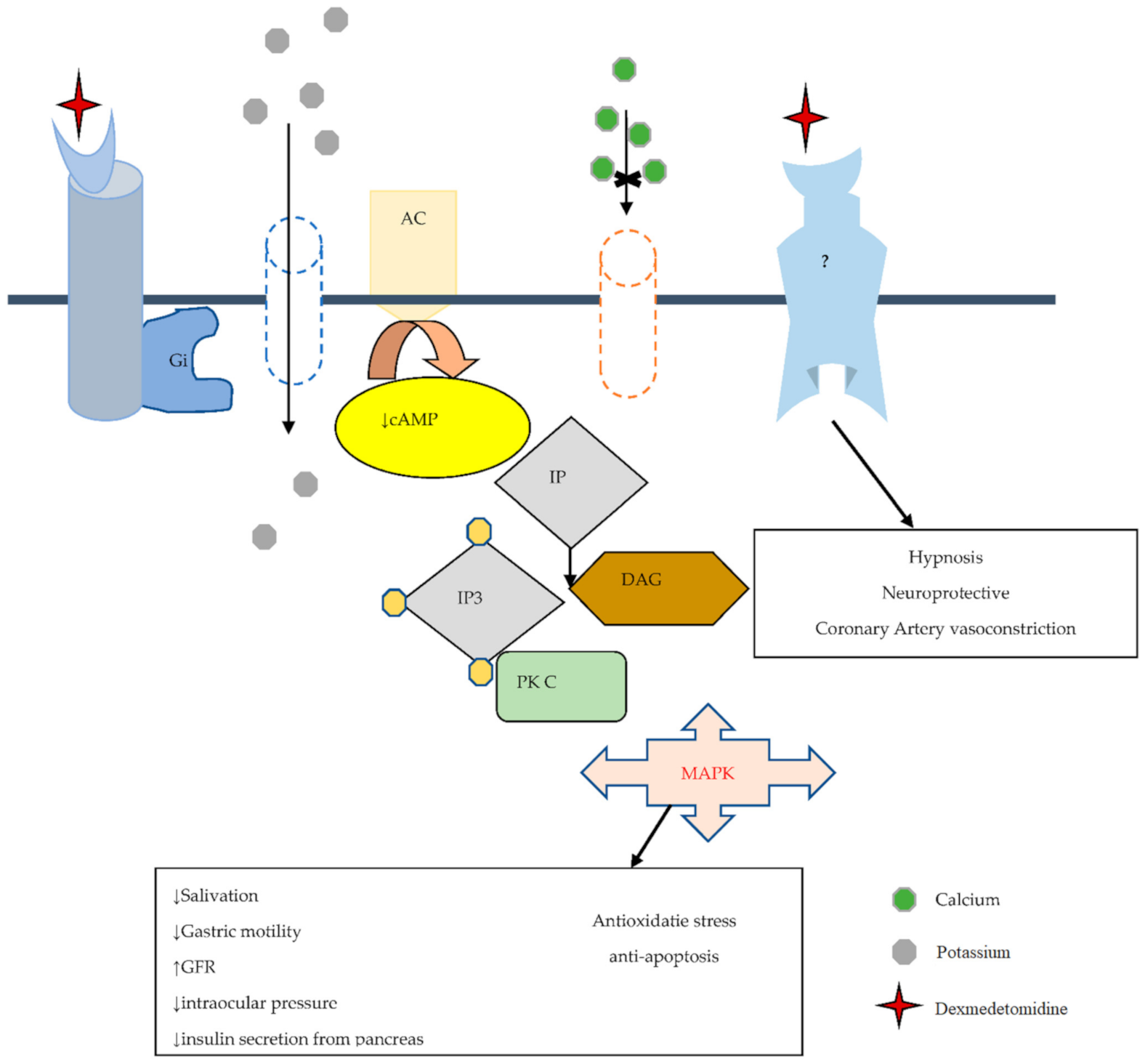
2.2. Pharmacodynamics of Dexmedetomidine
2.3. Adverse Effects of DEXM
2.4. Dexmedetomidine a Selective Alpha 2-Adrenoceptor Agonist
2.5. Dexmedetomidine and Imidazoline Receptor
3. Evidence of Dexmedetomidine as a Neuroprotective Agent
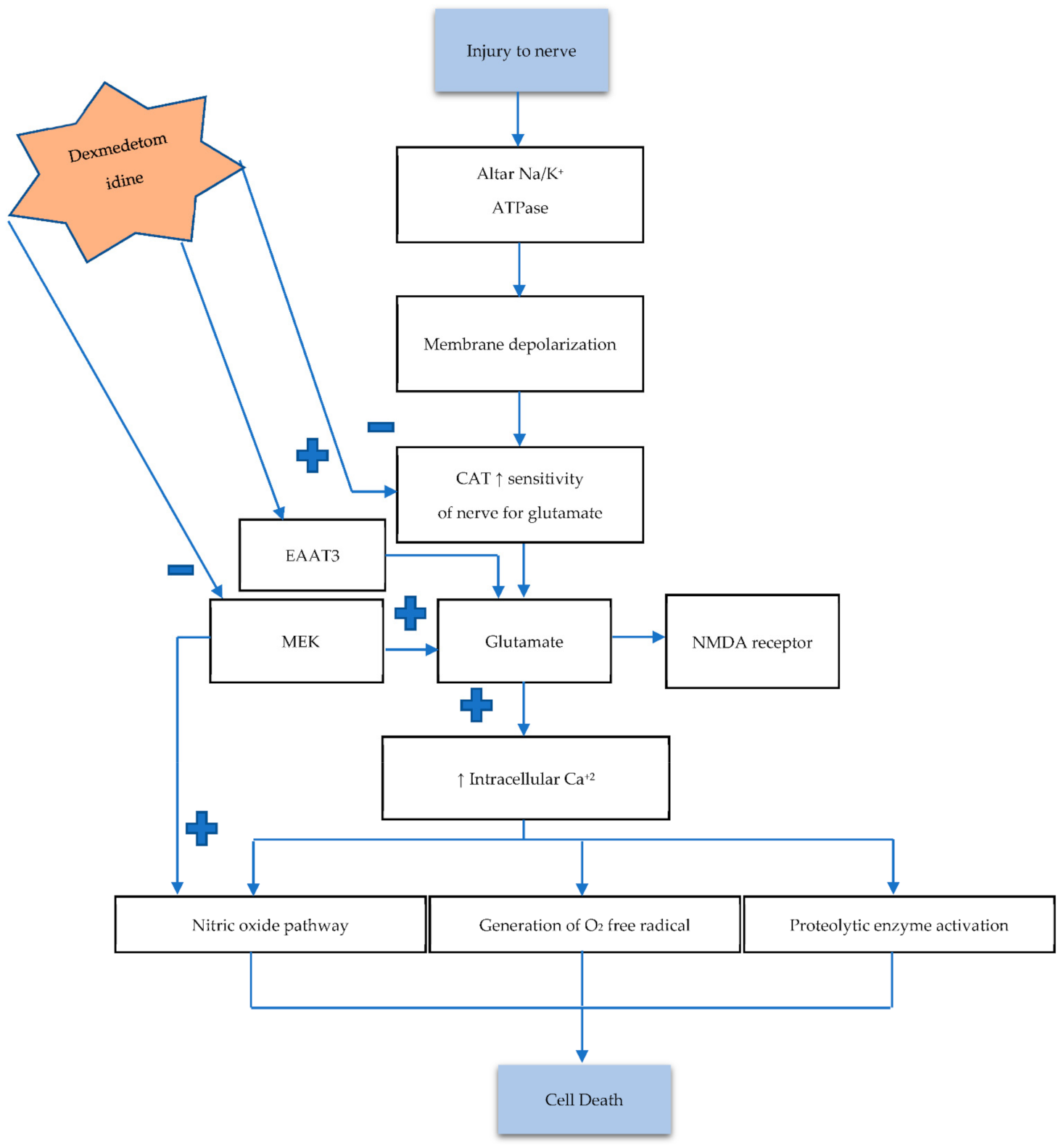
3.1. Role of Dexmedetomidine in Overview Pathogenesis of Neuronal Damage and Death
3.2. The Influence of Dexmedetomidine against Necrosis
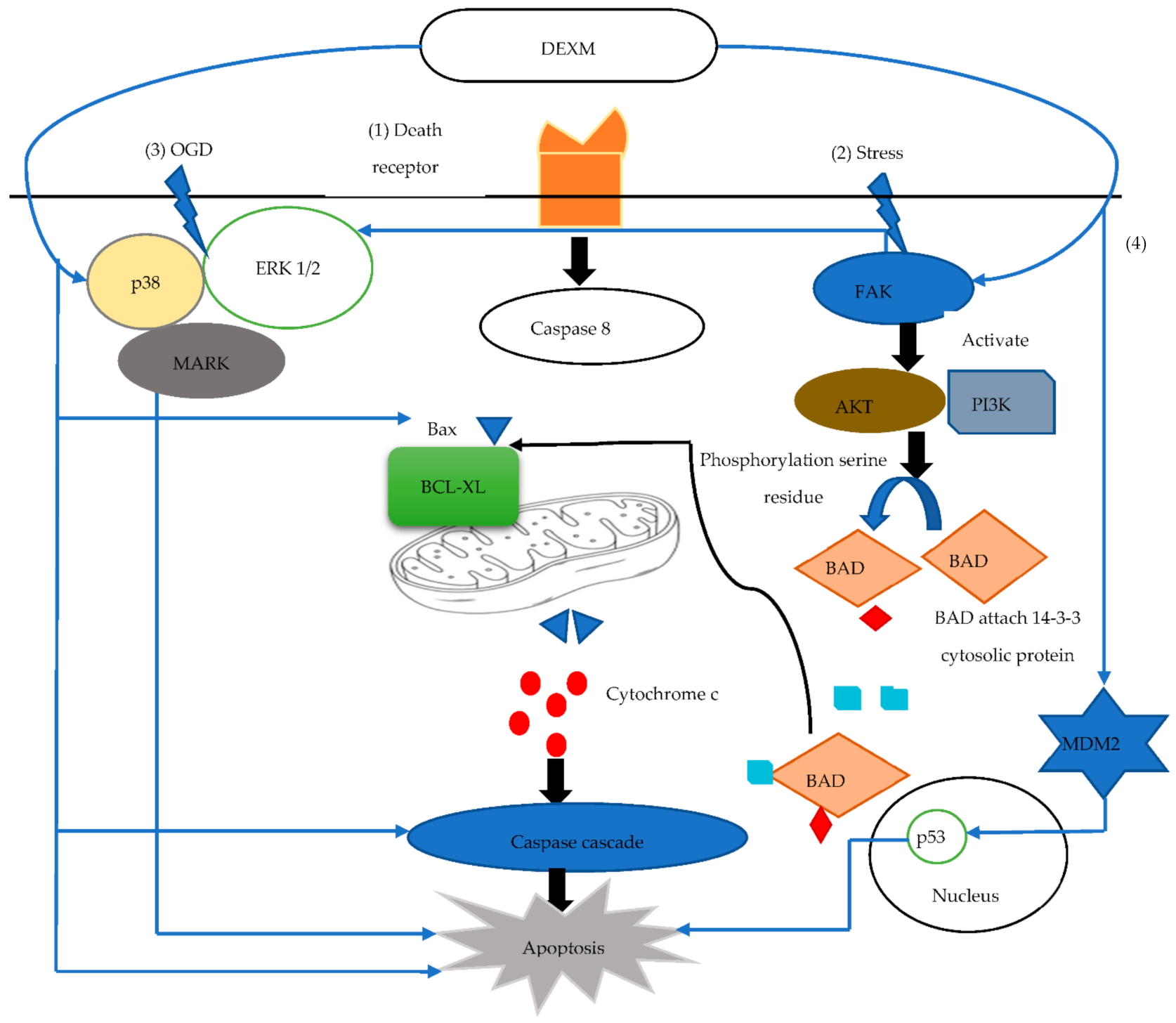
3.3. The Response of Dexmedetomidine against Apoptosis
3.4. The Response of Dexmedetomidine against Immunologic Response
3.5. Clinical Evidence of Dexmedetomidine as Neuroprotective Agent
4. Conclusions
- (i)
- Clinical investigations of DEXM should be designed to verify the efficacy of different steps of the cascade of neuronal damage.
- (ii)
- DEXM improves neuroinflammatory behavior by suppressing inflammatory mediators. It not only controls apoptotic signaling pathways but also decreases oxygen-free radical generation.
- (iii)
- The randomized clinical trials on DEXM suggest its ability to enhance BDNF to protect the brain from ischemic insult.
- (iv)
- DEXM not only improves cognition but also attenuates the duration of postoperative delirium.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Park, H.R.; Lee, G.S.; Kim, I.S.; Chang, J.-C. Brachial plexus injury in adults. Nerve 2017, 3, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Yannascoli, S.M.; Stwalley, D.; Saeed, M.J.; Olsen, M.A.; Dy, C.J. A population-based assessment of depression and anxiety in patients with brachial plexus injuries. J. Hand Surg. 2018, 43, 1136.e1–1136.e9. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; Brimble, M. A review of neuroprotective agents. Curr. Med. Chem. 2004, 11, 2383–2397. [Google Scholar] [CrossRef] [PubMed]
- Chrysostomou, C.; Schmitt, C.G. Dexmedetomidine: Sedation, analgesia and beyond. Expert Opin. Drug Metab. Toxicol. 2008, 4, 619–627. [Google Scholar] [CrossRef]
- Grewal, A. Dexmedetomidine: New avenues. J. Anaesthesiol. Clin. Pharmacol. 2011, 27, 297–302. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, Y.-Q. Advances in the Clinical Use of Dexmedetomidine. Med Recapitul. 2011, 3, 434–437. (In Chinese) [Google Scholar]
- Marwaha, A. Anesthesia Considerations for Tracheal Reconstruction. In Clinical Thoracic Anesthesia; Springer: Singapore, 2020; pp. 181–201. [Google Scholar]
- Keating, G.M. Dexmedetomidine: A review of its use for sedation in the intensive care setting. Drugs 2015, 75, 1119–1130. [Google Scholar] [CrossRef]
- Weerink, M.A.; Struys, M.M.; Hannivoort, L.N.; Barends, C.R.; Absalom, A.R.; Colin, P. Clinical pharmacokinetics and pharmacodynamics of dexmedetomidine. Clin. Pharmacokinet. 2017, 56, 893–913. [Google Scholar] [CrossRef] [Green Version]
- Elgebaly, A.S.; Sabry, M. Sedation effects by dexmedetomidine versus propofol in decreasing duration of mechanical ventilation after open heart surgery. Ann. Card. Anaesth. 2018, 21, 235–242. [Google Scholar] [CrossRef]
- Deiner, S.; Luo, X.; Lin, H.-M.; Sessler, D.I.; Saager, L.; Sieber, F.E.; Lee, H.B.; Sano, M.; Jankowski, C.; Bergese, S.D. Intraoperative infusion of dexmedetomidine for prevention of postoperative delirium and cognitive dysfunction in elderly patients undergoing major elective noncardiac surgery: A randomized clinical trial. JAMA Surg. 2017, 152, e171505. [Google Scholar] [CrossRef]
- Zhao, W.; Hu, Y.; Chen, H.; Wang, X.; Wang, L.; Wang, Y.; Wu, X.; Han, F. The Effect and Optimal Dosage of Dexmedetomidine Plus Sufentanil for Postoperative Analgesia in Elderly Patients with Postoperative Delirium and Early Postoperative Cognitive Dysfunction: A Single-Center, Prospective, Randomized, Double-Blind, Controlled Trial. Front. Neurosci. 2020, 14, 549516. [Google Scholar]
- Lee, S. Dexmedetomidine: Present and future directions. Korean J. Anesthesiol. 2019, 72, 323–330. [Google Scholar] [CrossRef]
- Yoshikawa, Y.; Hirata, N.; Kawaguchi, R.; Tokinaga, Y.; Yamakage, M. Dexmedetomidine maintains its direct cardioprotective effect against ischemia/reperfusion injury in hypertensive hypertrophied myocardium. Anesth. Analg. 2018, 126, 443–452. [Google Scholar] [CrossRef]
- Ma, J.; Chen, Q.; Li, J.; Zhao, H.; Mi, E.; Chen, Y.; Yi, B.; Ning, J.; Ma, D.; Lu, K. Dexmedetomidine-mediated prevention of renal ischemia-reperfusion injury depends in part on cholinergic anti-inflammatory mechanisms. Anesth. Analg. 2020, 130, 1054–1062. [Google Scholar] [CrossRef]
- Zhu, L.; Zhang, Y.; Zhang, Z.; Ding, X.; Gong, C.; Qian, Y. Activation of PI3K/Akt/HIF-1α signaling is involved in lung protection of dexmedetomidine in patients undergoing video-assisted thoracoscopic surgery: A pilot study. Drug Des. Dev. Ther. 2020, 14, 5155–5166. [Google Scholar] [CrossRef]
- Lv, H.; Li, Y.; Cheng, Q.; Chen, J.; Chen, W. Neuroprotective Effects Against Cerebral Ischemic Injury Exerted by Dexmedetomidine via the HDAC5/NPAS4/MDM2/PSD-95 Axis. Mol. Neurobiol. 2021, 58, 1990–2004. [Google Scholar] [CrossRef]
- Anttila, M.; Penttilä, J.; Helminen, A.; Vuorilehto, L.; Scheinin, H. Bioavailability of dexmedetomidine after extravascular doses in healthy subjects. Br. J. Clin. Pharmacol. 2003, 56, 691–693. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; Zheng, X.; Huang, H. Protective effects of dexmedetomidine on brain function of glioma patients undergoing craniotomy resection and its underlying mechanism. Clin. Neurol. Neurosurg. 2016, 146, 105–108. [Google Scholar] [CrossRef]
- Gertler, R.; Brown, H.C.; Mitchell, D.H.; Silvius, E.N. Dexmedetomidine: A novel sedative-analgesic agent. Bayl. Univ. Med Cent. Proc. 2001, 14, 13–21. [Google Scholar] [CrossRef]
- Colin, P.; Hannivoort, L.N.; Eleveld, D.J.; Reyntjens, K.M.; Absalom, A.R.; Vereecke, H.E.; Struys, M.M. Dexmedetomidine pharmacodynamics in healthy volunteers: 2. Haemodynamic profile. BJA Br. J. Anaesth. 2017, 119, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Mantz, J. Dexmedetomidine. Drugs Today 1999, 35, 151–157. [Google Scholar] [CrossRef]
- Yu, S.-B. Dexmedetomidine sedation in ICU. Korean J. Anesthesiol. 2012, 62, 405–411. [Google Scholar] [CrossRef] [Green Version]
- Naik, B.I.; Nemergut, E.C.; Kazemi, A.; Fernández, L.; Cederholm, S.K.; McMurry, T.L.; Durieux, M.E. The effect of dexmedetomidine on postoperative opioid consumption and pain after major spine surgery. Anesth. Analg. 2016, 122, 1646–1653. [Google Scholar] [CrossRef]
- Ling, X.; Zhou, H.; Ni, Y.; Wu, C.; Zhang, C.; Zhu, Z. Does dexmedetomidine have an antiarrhythmic effect on cardiac patients? A meta-analysis of randomized controlled trials. PLoS ONE 2018, 13, e0193303. [Google Scholar] [CrossRef] [PubMed]
- Botero Posada, L.F.; Arias Jiménez, J.M.; Vasseur Arboleda, J.P. The use of dexmedetomidine (DXM) for implanting a cardiac resynchronization device: Is it really safe? Colomb. J. Anestesiol. 2011, 39, 425–431. [Google Scholar] [CrossRef] [Green Version]
- Seyrek, M.; Halici, Z.; Yildiz, O.; Ulusoy, H.B. Interaction between dexmedetomidine and α-adrenergic receptors: Emphasis on vascular actions. J. Cardiothorac. Vasc. Anesth. 2011, 25, 856–862. [Google Scholar] [CrossRef]
- Afonso, J.; Reis, F. Dexmedetomidine: Current role in anesthesia and intensive care. Braz. J. Anesthesiol. 2012, 62, 118–133. [Google Scholar] [CrossRef] [Green Version]
- Murrell, J.C.; Hellebrekers, L.J. Medetomidine and dexmedetomidine: A review of cardiovascular effects and antinociceptive properties in the dog. Vet. Anaesth. Analg. 2005, 32, 117–127. [Google Scholar] [CrossRef]
- Arcangeli, A.; D’alo, C.; Gaspari, R. Dexmedetomidine use in general anaesthesia. Curr. Drug Targets 2009, 10, 687–695. [Google Scholar] [CrossRef]
- Bylund, D.B. Subtypes of α1-and α2-adrenergic receptors. FASEB J. 1992, 6, 832–839. [Google Scholar] [CrossRef]
- Ciccarelli, M.; Sorriento, D.; Coscioni, E.; Iaccarino, G.; Santulli, G. Adrenergic receptors. Endocrinol. Heart Health Dis. 2017, 285–315. [Google Scholar]
- Kim, H.; Choi, S.-H.; Ha, S.-H.; Chang, W.-S.; Heo, G.-A.; Jeong, J.; Min, K.T. Haemodynamic changes and incisional bleeding after scalp infiltration of dexmedetomidine with lidocaine in neurosurgical patients. Anaesth. Crit. Care Pain Med. 2019, 38, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Liu, Z.; Kaindl, J.; Maeda, S.; Zhao, J.; Sun, X.; Xu, J.; Gmeiner, P.; Wang, H.-W.; Kobilka, B.K. Activation of the α 2B adrenoceptor by the sedative sympatholytic dexmedetomidine. Nat. Chem. Biol. 2020, 16, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Hossain, M.; Rajakumaraswamy, N.; Arshad, M.; Sanders, R.D.; Franks, N.P.; Maze, M. Dexmedetomidine produces its neuroprotective effect via the α2A-adrenoceptor subtype. Eur. J. Pharmacol. 2004, 502, 87–97. [Google Scholar] [CrossRef]
- Zhang, Y.; Kimelberg, H.K. Neuroprotection by alpha 2-adrenergic agonists in cerebral ischemia. Curr. Neuropharmacol. 2005, 3, 317–323. [Google Scholar] [CrossRef] [Green Version]
- De Vos, H.; Vauquelin, G.; De Keyser, J.; De Backer, J.P.; Van Liefde, I. Regional Distribution of α2A-and α2B-Adrenoceptor Subtypes in Postmortem Human Brain. J. Neurochem. 1992, 58, 1555–1560. [Google Scholar] [CrossRef]
- Scheinin, M.; Lomasney, J.W.; Hayden-Hixson, D.M.; Schambra, U.B.; Caron, M.G.; Lefkowitz, R.J.; Fremeau Jr, R.T. Distribution of α2-adrenergic receptor subtype gene expression in rat brain. Mol. Brain Res. 1994, 21, 133–149. [Google Scholar] [CrossRef]
- Sinclair, M.D. A review of the physiological effects of α2-agonists related to the clinical use of medetomidine in small animal practice. Can. Vet. J. 2003, 44, 885–897. [Google Scholar]
- Ruffolo Jr, R.R.; Stadel, J.M.; Hieble, J.P. α-adrenoceptors: Recent developments. Med. Res. Rev. 1994, 14, 229–270. [Google Scholar] [CrossRef]
- Khan, Z.; Ferguson, C.; Jones, R. Alpha-2 and imidazoline receptor agonistsTheir pharmacology and therapeutic role. Anaesthesia 1999, 54, 146–165. [Google Scholar] [CrossRef]
- Bousquet, P.; Hudson, A.; García-Sevilla, J.A.; Li, J.-X. Imidazoline receptor system: The past, the present, and the future. Pharmacol. Rev. 2020, 72, 50–79. [Google Scholar] [CrossRef]
- Janke, E.L.; Samra, S. Dexmedetomidine and neuroprotection. Semin. Anesth. Perioper. Med. Pain 2006, 25, 71–76. [Google Scholar] [CrossRef]
- Inagaki, M.; Somei, M.; Oguchi, T.; Ono, R.; Fukutaka, S.; Matsuoka, I.; Tsuji, M.; Oguchi, K. Neuroprotective Effects of Dexmedetomidine against-induced ER-stress via Activity of α2-adrenoceptors and Imidazoline Receptors. AIMS Neurosci. 2016, 3, 237–252. [Google Scholar] [CrossRef]
- Takamatsu, I.; Iwase, A.; Ozaki, M.; Kazama, T.; Wada, K.; Sekiguchi, M. Dexmedetomidine reduces long-term potentiation in mouse hippocampus. J. Am. Soc. Anesthesiol. 2008, 108, 94–102. [Google Scholar] [CrossRef] [Green Version]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Süudhof, T.C. Neurotransmitter release. In Pharmacology of Neurotransmitter Release; Springer: Berlin, Germany, 2008; pp. 1–21. [Google Scholar]
- Südhof, T.C. Calcium control of neurotransmitter release. Cold Spring Harb. Perspect. Biol. 2012, 4, a011353. [Google Scholar] [CrossRef]
- Degos, V.; Charpentier, T.L.; Chhor, V.; Brissaud, O.; Lebon, S.; Schwendimann, L.; Bednareck, N.; Passemard, S.; Mantz, J.; Gressens, P. Neuroprotective effects of dexmedetomidine against glutamate agonist-induced neuronal cell death are related to increased astrocyte brain-derived neurotrophic factor expression. Anesthesiology 2013, 118, 1123–1132. [Google Scholar] [CrossRef]
- Zhao, Y.; He, J.; Yu, N.; Jia, C.; Wang, S. Mechanisms of dexmedetomidine in neuropathic pain. Front. Neurosci. 2020, 14, 330. [Google Scholar] [CrossRef]
- Noch, E.; Khalili, K. Molecular mechanisms of necrosis in glioblastoma: The role of glutamate excitotoxicity. Cancer Biol. Ther. 2009, 8, 1791–1797. [Google Scholar] [CrossRef]
- Ji, X.; Guo, Y.; Zhou, G.; Wang, Y.; Zhang, J.; Wang, Z.; Wang, Q. Dexmedetomidine protects against high mobility group box 1-induced cellular injury by inhibiting pyroptosis. Cell Biol. Int. 2019, 43, 651–657. [Google Scholar] [CrossRef]
- Chiu, K.-M.; Lin, T.-Y.; Lu, C.-W.; Wang, S.-J. Inhibitory effect of glutamate release from rat cerebrocortical nerve terminals by α2 adrenoceptor agonist dexmedetomidine. Eur. J. Pharmacol. 2011, 670, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Do, S.-H.; Park, S.-J.; Shin, H.-J.; Paik, H.-S.; Zuo, Z.; Yoon, H.-J.; Ryu, J.-H. Dexmedetomidine increases the activity of excitatory amino acid transporter type 3 expressed in Xenopus oocytes: The involvement of protein kinase C and phosphatidylinositol 3-kinase. Eur. J. Pharmacol. 2014, 738, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shan, Y.; Tang, Z.; Gao, L.; Liu, H. Neuroprotective effects of dexmedetomidine against isoflurane-induced neuronal injury via glutamate regulation in neonatal rats. Drug Des. Dev. Ther. 2019, 13, 153–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, M.; Ling, X.; Song, R.; Gao, X.; Liang, Z.; Fang, F.; Cang, J. Upregulation of GLT-1 via PI3K/Akt pathway contributes to neuroprotection induced by dexmedetomidine. Front. Neurol. 2019, 10, 1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Zhao, H.; Wang, D.; Ma, D. Dexmedetomidine alleviates LPS-induced pyroptosis in astrocytes in vitro. Br. J. Anaesth. 2018, 120, e8–e9. [Google Scholar] [CrossRef]
- Sun, Y.-B.; Zhao, H.; Mu, D.-L.; Zhang, W.; Cui, J.; Wu, L.; Alam, A.; Wang, D.-X.; Ma, D. Dexmedetomidine inhibits astrocyte pyroptosis and subsequently protects the brain in in vitro and in vivo models of sepsis. Cell Death Dis. 2019, 10, 167. [Google Scholar] [CrossRef] [Green Version]
- Han, J.-H.; Kim, D.-O.; Yi, J.-W.; Park, S.-W.; Kang, W.-J.; Choi, Y.-K.; Kim, S.-H.; Ko, I.-G.; Jin, J.-J.; Kim, S.-E. Dexmedetomidine, α2-adrenoceptor agonist, does not induce apoptosis in the brachial plexus of rats. Anim. Cells Syst. 2014, 18, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Thomasova, D.; Bruns, H.A.; Kretschmer, V.; Ebrahim, M.; Romoli, S.; Liapis, H.; Kotb, A.M.; Endlich, N.; Anders, H.-J. Murine double minute-2 prevents p53-overactivation-related cell death (podoptosis) of podocytes. J. Am. Soc. Nephrol. 2015, 26, 1513–1523. [Google Scholar] [CrossRef] [Green Version]
- Dahmani, S.; Rouelle, D.; Gressens, P.; Mantz, J. Effects of dexmedetomidine on hippocampal focal adhesion kinase tyrosine phosphorylation in physiologic and ischemic conditions. J. Am. Soc. Anesthesiol. 2005, 103, 969–977. [Google Scholar] [CrossRef]
- Zhang, F.; Ding, T.; Yu, L.; Zhong, Y.; Dai, H.; Yan, M. Dexmedetomidine protects against oxygen–glucose deprivation-induced injury through the I2 imidazoline receptor-PI3K/AKT pathway in rat C6 glioma cells. J. Pharm. Pharmacol. 2012, 64, 120–127. [Google Scholar] [CrossRef]
- Ding, X.-D.; Zheng, N.-N.; Cao, Y.-Y.; Zhao, G.-Y.; Zhao, P. Dexmedetomidine preconditioning attenuates global cerebral ischemic injury following asphyxial cardiac arrest. Int. J. Neurosci. 2016, 126, 249–256. [Google Scholar] [CrossRef]
- Pugliese, A.M.; Latini, S.; Corradetti, R.; Pedata, F. Brief, repeated, oxygen-glucose deprivation episodes protect neurotransmission from a longer ischemic episode in the in vitro hippocampus: Role of adenosine receptors. Br. J. Pharmacol. 2003, 140, 305–314. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.; Wang, S.; Wen, X.; Han, X.-R.; Wang, Y.-J.; Zhou, X.-M.; Zhang, M.-H.; Wu, D.-M.; Lu, J.; Zheng, Y.-L. Dexmedetomidine exerts neuroprotective effect via the activation of the PI3K/Akt/mTOR signaling pathway in rats with traumatic brain injury. Biomed. Pharmacother. 2017, 95, 885–893. [Google Scholar] [CrossRef]
- Zhu, Y.-M.; Wang, C.-C.; Chen, L.; Qian, L.-B.; Ma, L.-L.; Yu, J.; Zhu, M.-H.; Wen, C.-Y.; Yu, L.-N.; Yan, M. Both PI3K/Akt and ERK1/2 pathways participate in the protection by dexmedetomidine against transient focal cerebral ischemia/reperfusion injury in rats. Brain Res. 2013, 1494, 1–8. [Google Scholar] [CrossRef]
- Wang, K.; Zhu, Y. Dexmedetomidine protects against oxygen-glucose deprivation/reoxygenation injury-induced apoptosis via the p38 MAPK/ERK signalling pathway. J. Int. Med Res. 2018, 46, 675–686. [Google Scholar] [CrossRef]
- Tanabe, K.; Kozawa, O.; Iida, H. Midazolam suppresses interleukin-1β-induced interleukin-6 release from rat glial cells. J. Neuroinflamm. 2011, 8, 68. [Google Scholar] [CrossRef] [Green Version]
- Tanabe, K.; Kozawa, O.; Iida, H. cAMP/PKA enhances interleukin-1β-induced interleukin-6 synthesis through STAT3 in glial cells. Cell. Signal. 2016, 28, 19–24. [Google Scholar] [CrossRef]
- Zhu, Y.-J.; Peng, K.; Meng, X.-W.; Ji, F.-H. Attenuation of neuroinflammation by dexmedetomidine is associated with activation of a cholinergic anti-inflammatory pathway in a rat tibial fracture model. Brain Res. 2016, 1644, 1–8. [Google Scholar] [CrossRef]
- Ge, Y.; Li, Q.; Nie, Y.; Gao, J.; Luo, K.; Fang, X.; Wang, C. Dexmedetomidine improves cognition after carotid endarterectomy by inhibiting cerebral inflammation and enhancing brain-derived neurotrophic factor expression. J. Int. Med Res. 2019, 47, 2471–2482. [Google Scholar] [CrossRef]
- Tanabe, K.; Matsushima-Nishiwaki, R.; Kozawa, O.; Iida, H. Dexmedetomidine suppresses interleukin-1β-induced interleukin-6 synthesis in rat glial cells. Int. J. Mol. Med. 2014, 34, 1032–1038. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wang, J.; Qian, W.; Zhao, J.; Sun, L.; Qian, Y.; Xiao, H. Dexmedetomidine inhibits tumor necrosis factor-alpha and interleukin 6 in lipopolysaccharide-stimulated astrocytes by suppression of c-Jun N-terminal kinases. Inflammation 2014, 37, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Kim, H.-C.; Lee, S.; Ryu, H.-G.; Park, Y.-H.; Kim, J.H.; Lim, Y.-J.; Park, H.-P. Dexmedetomidine confers neuroprotection against transient global cerebral ischemia/reperfusion injury in rats by inhibiting inflammation through inactivation of the TLR-4/NF-κB pathway. Neurosci. Lett. 2017, 649, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, H.; Zhang, L.; Wang, G.; Zhang, M.; Yu, Y. Neuroprotection of dexmedetomidine against cerebral ischemia-reperfusion injury in rats: Involved in inhibition of NF-κB and inflammation response. Biomol. Ther. 2017, 25, 383–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, H.; Hu, B.; Li, Z.; Li, J. Dexmedetomidine controls systemic cytokine levels through the cholinergic anti-inflammatory pathway. Inflammation 2014, 37, 1763–1770. [Google Scholar] [CrossRef]
- Giacobbo, B.L.; Doorduin, J.; Klein, H.C.; Dierckx, R.A.; Bromberg, E.; de Vries, E.F. Brain-derived neurotrophic factor in brain disorders: Focus on neuroinflammation. Mol. Neurobiol. 2019, 56, 3295–3312. [Google Scholar] [CrossRef] [Green Version]
- Bindra, A.; Kaushal, A.; Prabhakar, H.; Chaturvedi, A.; Chandra, P.S.; Tripathi, M.; Subbiah, V.; Sathianathan, S.; Banerjee, J.; Prakash, C. Neuroprotective role of dexmedetomidine in epilepsy surgery: A preliminary study. Neurol. India 2019, 67, 163–168. [Google Scholar] [CrossRef]
- Liu, Y.; Ma, L.; Gao, M.; Guo, W.; Ma, Y. Dexmedetomidine reduces postoperative delirium after joint replacement in elderly patients with mild cognitive impairment. Aging Clin. Exp. Res. 2016, 28, 729–736. [Google Scholar] [CrossRef]
- Yang, L.; Xu, J.-M.; Jiang, X.; Ruan, W.; Cui, Y.; He, L. Effect of dexmedetomidine on plasma brain-derived neurotrophic factor: A double-blind, randomized and placebo-controlled study. Upsala J. Med Sci. 2013, 118, 235–239. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Alpha 2 Receptor Subtype | Anatomical Location | Functions | References |
|---|---|---|---|
| α2a | CNS including the brain, spinal cord, and locus coeruleus | Regulates the phase of awareness produces anesthetic, sympatholytic responses, helps in arousal, and vigilance | [36,37,38] |
| α2b | Liver, spleen, heart, Thalamus, smooth muscle cells of the blood vessel. | Vasoconstrictive. | [39,40] |
| α2c | Hippocampus, locus coeruleus, especially in basal ganglia, platelets. | Control anxiolytic and analgesic effects, hypnotic-sedative actions | [38,39,40] |
| References | Mechanisms | Animal Model | Marker | DEXM Effect |
|---|---|---|---|---|
| [55] | Excitotoxicity | Neonatal rats randomly allocated to 4 groups (n = 36 each) | Glutamate | ↑ EAAT3 expression, ↓ nerve sensitivity for glutamate. |
| [59] | Apoptosis | Adult male Sprague Dawley rats randomly allocated to 4 groups (n = 10 each) | Bcl2, FAK | Upregulates anti-apoptotic marker |
| [17] | Adult male C57BL/6 J mice n = 80 | MDM2, p53 pathway | ↓ p53 activity via Mdm2 | |
| [62] | Rat glioma C6 cells Cell culture in Dulbecco’s modified Eagle’s Medium (DMEM) | PI3k/AKT | Phosphorylates AKT, upregulates PI3k expression | |
| [61] | Male Sprague-Dawley rats n = unknown | Caspase 3 | Upregulates FAK that activates IAP that attenuate caspase 3 | |
| [72] | Inflammatory mediator | Rat C6 glioma cells. Cell culture in DMEM | IL-1ꞵ induces IL-6 release | ↓ IL-1ꞵ induces IL-6 release independently adenyl cyclase cAMP pathway |
| [71] | Long–Evans female rats | Systemic cytokines TNF-α, NF-kB, IL-1ꞵ, IL-18 | Significantly decreases systemic cytokines level | |
| [58] | Stepsis | Human astrocyte 1321N1 cells and rat neuron PC12 cells. Cell culture in DMEM | LHD, NLRP3, ASC, Caspase 1 inducing IL-1ꞵ, IL-18 | Inhibits NL RP3 inflammasome assembly |
| [43] | Astroglia | Astrocytes, Cell culture | Astrocytes | Decrease over activated astrocytes |
| Condition | Number of Cases | Outcome | References |
|---|---|---|---|
| Brain protection in patients undergoing craniotomy resection of glioma. | 60 cases | DEXM stabilized hemodynamics, attenuated inflammation, and inhibited the generation of free radicals. | [19] |
| Cerebroprotection during epilepsy surgery. | 19 cases | Treatment with DEXM low S100b level | [78] |
| Improves cognition after carotid endarterectomy. | 49 cases | DEXM was neuroprotective in the stroke model by reducing TNF-α and IL-6 and enhancing BNDF. | [71] |
| Reduce postoperative delirium (POD) in elderly patients with mild cognitive impairment (MCI) after joint replacement surgery. | 80 cases | DEXM treatment significantly improved POD MCI in elderly patients. | [79] |
| A double-blind, randomized, and placebo-controlled study. | 40 cases | DEXM enhances plasma concentrations of BDNF caused by the anesthetic agent. | [80] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liaquat, Z.; Xu, X.; Zilundu, P.L.M.; Fu, R.; Zhou, L. The Current Role of Dexmedetomidine as Neuroprotective Agent: An Updated Review. Brain Sci. 2021, 11, 846. https://doi.org/10.3390/brainsci11070846
Liaquat Z, Xu X, Zilundu PLM, Fu R, Zhou L. The Current Role of Dexmedetomidine as Neuroprotective Agent: An Updated Review. Brain Sciences. 2021; 11(7):846. https://doi.org/10.3390/brainsci11070846
Chicago/Turabian StyleLiaquat, Zaara, Xiaoying Xu, Prince Last Mudenda Zilundu, Rao Fu, and Lihua Zhou. 2021. "The Current Role of Dexmedetomidine as Neuroprotective Agent: An Updated Review" Brain Sciences 11, no. 7: 846. https://doi.org/10.3390/brainsci11070846
APA StyleLiaquat, Z., Xu, X., Zilundu, P. L. M., Fu, R., & Zhou, L. (2021). The Current Role of Dexmedetomidine as Neuroprotective Agent: An Updated Review. Brain Sciences, 11(7), 846. https://doi.org/10.3390/brainsci11070846