
Systems Biology Reveals S-Nitrosylation-Dependent Regulation of Mitochondrial Functions in Mice with Shank3 Mutation Associated with Autism Spectrum Disorder



Abstract
1. Introduction
2. Methods and Materials
2.1. Cortical Tissue Preparation for MS Analysis
2.2. MS Analysis
2.3. Bioinformatics and Systems Biology Analysis
3. Results
3.1. Systems Biology Analysis of the S-Nitroso-Proteome in the ASD Mouse Model
3.2. Protein–Protein Interaction Network of the Mitochondrial SNO Proteins
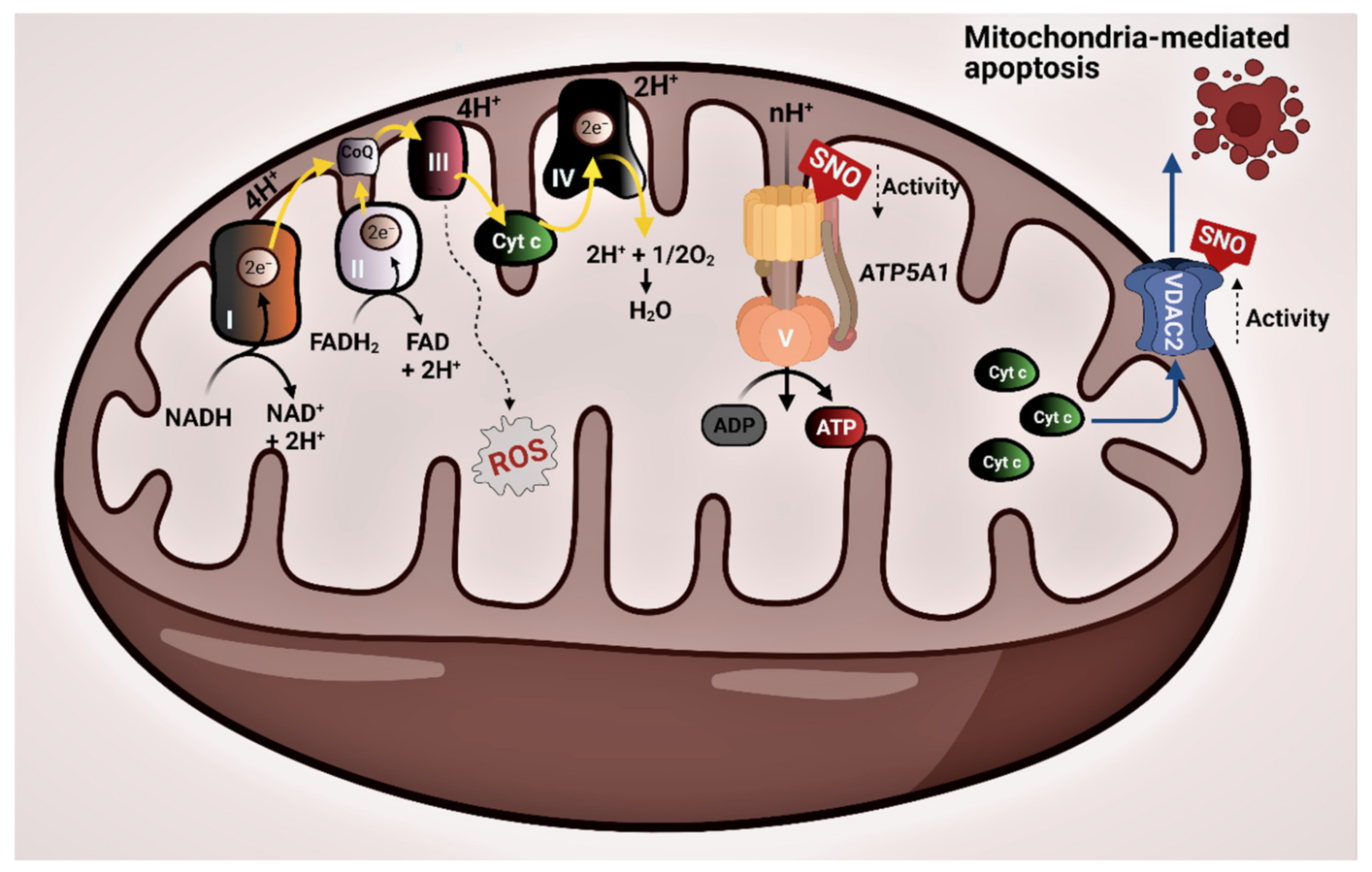
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shen, L.; Zhang, K.; Feng, C.; Chen, Y.; Li, S.; Iqbal, J.; Liao, L.; Zhao, Y.; Zhai, J. iTRAQ-Based Proteomic Analysis Reveals Protein Profile in Plasma from Children with Autism. Proteom. Clin. Appl. 2018, 12, e1700085. [Google Scholar] [CrossRef]
- Lord, C.; Elsabbagh, M.; Baird, G.; Veenstra-Vanderweele, J. Autism spectrum disorder. Lancet 2018, 392, 508–520. [Google Scholar] [CrossRef]
- Amal, H.; Barak, B.; Bhat, V.; Gong, G.; Joughin, B.A.; Wang, X.; Wishnok, J.S.; Feng, G.; Tannenbaum, S.R. Shank3 mutation in a mouse model of autism leads to changes in the S-nitroso-proteome and affects key proteins involved in vesicle release and synaptic function. Mol. Psychiatry 2018, 25, 1835–1848. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, P.; Feng, G. SHANK proteins: Roles at the synapse and in autism spectrum disorder. Nat. Rev. Neurosci. 2017, 18, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Kaiser, T.; Monteiro, P.; Zhang, X.; Van der Goes, M.S.; Wang, D.; Barak, B.; Zeng, M.; Li, C.; Lu, C.; et al. Mice with Shank3 Mutations Associated with ASD and Schizophrenia Display Both Shared and Distinct Defects. Neuron 2016, 89, 147–162. [Google Scholar] [CrossRef]
- Griffiths, K.K.; Levy, R.J. Evidence of Mitochondrial Dysfunction in Autism: Biochemical Links, Genetic-Based Associations, and Non-Energy-Related Mechanisms. Oxid. Med. Cell. Longev. 2017, 2017, 4314025. [Google Scholar] [CrossRef]
- Hsiao, E.Y. Gastrointestinal Issues in Autism Spectrum Disorder. Harv. Rev. Psychiatry 2014, 22, 104–111. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Frye, R.E. Evidence linking oxidative stress, mitochondrial dysfunction, and inflammation in the brain of individuals with autism. Front. Physiol. 2014, 5, 150. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.E. Mitochondrial Dysfunction in Autism Spectrum Disorder: Unique Abnormalities and Targeted Treatments. Semin. Pediatr. Neurol. 2020, 35, 100829. [Google Scholar] [CrossRef]
- Frye, R.E.; Rossignol, D.A. Mitochondrial dysfunction can connect the diverse medical symptoms associated with autism spectrum disorders. Pediatr. Res. 2011, 69, 41r–47r. [Google Scholar] [CrossRef]
- Citrigno, L.; Muglia, M.; Qualtieri, A.; Spadafora, P.; Cavalcanti, F.; Pioggia, G.; Cerasa, A. The Mitochondrial Dysfunction Hypothesis in Autism Spectrum Disorders: Current Status and Future Perspectives. Int. J. Mol. Sci. 2020, 21, 5785. [Google Scholar] [CrossRef]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Valenti, D.; de Bari, L.; De Filippis, B.; Henrion-Caude, A.; Vacca, R.A. Mitochondrial dysfunction as a central actor in intellectual disability-related diseases: An overview of Down syndrome, autism, Fragile X and Rett syndrome. Neurosci. Biobehav. Rev. 2014, 46, 202–217. [Google Scholar] [CrossRef]
- Brorson, J.R.; Schumacker, P.T.; Zhang, H. Nitric oxide acutely inhibits neuronal energy production. The Committees on Neurobiology and Cell Physiology. J. Neurosci. 1999, 19, 147–158. [Google Scholar] [CrossRef]
- Tripathi, M.K.; Kartawy, M.; Amal, H. The role of nitric oxide in brain disorders: Autism spectrum disorder and other psychiatric, neurological, and neurodegenerative disorders. Redox Biol. 2020, 34, 101567. [Google Scholar] [CrossRef] [PubMed]
- Bredt, D.S.; Snyder, S.H. Nitric oxide: A physiologic messenger molecule. Annu. Rev. Biochem. 1994, 63, 175–195. [Google Scholar] [CrossRef]
- Khaliulin, I.; Kartawy, M.; Amal, H. Sex Differences in Biological Processes and Nitrergic Signaling in Mouse Brain. Biomedicines 2020, 8, 124. [Google Scholar] [CrossRef] [PubMed]
- Kartawy, M.; Khaliulin, I.; Amal, H. Systems biology reveals reprogramming of the S-nitroso-proteome in the cortical and striatal regions of mice during aging process. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Hamoudi, W.; von Lendenfeld, F.; Kartawy, M.; Mencer, S.; Suloh, H.; Khaliulin, I.; Amal, H. Regional Differences in S-Nitrosylation in the Cortex, Striatum, and Hippocampus of Juvenile Male Mice. J. Mol. Neurosci. 2021, 1–10. [Google Scholar] [CrossRef]
- Amal, H.; Gong, G.; Gjoneska, E.; Lewis, S.M.; Wishnok, J.S.; Tsai, L.-H.; Tannenbaum, S.R. S-nitrosylation of E3 ubiquitin-protein ligase RNF213 alters non-canonical Wnt/Ca+2 signaling in the P301S mouse model of tauopathy. Transl. Psychiatry 2019, 9, 44. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Lipton, S.A. Protein S-Nitrosylation as a Therapeutic Target for Neurodegenerative Diseases. Trends Pharm. Sci. 2016, 37, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Prikhodko, O.A.; Pirie, E.; Nagar, S.; Akhtar, M.W.; Oh, C.-K.; McKercher, S.R.; Ambasudhan, R.; Okamoto, S.-i.; Lipton, S.A. Aberrant protein S-nitrosylation contributes to the pathophysiology of neurodegenerative diseases. Neurobiol. Dis. 2015, 84, 99–108. [Google Scholar] [CrossRef]
- Nakamura, T.; Tu, S.; Akhtar, M.W.; Sunico, C.R.; Okamoto, S.-i.; Lipton, S.A. Aberrant protein S-nitrosylation in neurodegenerative diseases. Neuron 2013, 78, 596–614. [Google Scholar] [CrossRef]
- Nasyrova, R.F.; Ivashchenko, D.V.; Ivanov, M.V.; Neznanov, N.G. Role of nitric oxide and related molecules in schizophrenia pathogenesis: Biochemical, genetic and clinical aspects. Front. Physiol. 2015, 6, 139. [Google Scholar] [CrossRef]
- Chauhan, A.; Gu, F.; Essa, M.M.; Wegiel, J.; Kaur, K.; Brown, W.T.; Chauhan, V. Brain region-specific deficit in mitochondrial electron transport chain complexes in children with autism. J. Neurochem. 2011, 117, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Foley, T.D.; Koval, K.S.; Olsen, S.H.; Gallagher, A.G.; Dennis, E.R. Protein S-Nitrosylation: Possible Links between Psychophysiological Stress and Neurodegeneration. Free Radic. Biol. Med. 2017, 112, 73–74. [Google Scholar] [CrossRef]
- Rizza, S.; Cardaci, S.; Montagna, C.; Di Giacomo, G.; De Zio, D.; Bordi, M.; Maiani, E.; Campello, S.; Borreca, A.; Puca, A.A. S-nitrosylation drives cell senescence and aging in mammals by controlling mitochondrial dynamics and mitophagy. Proc. Natl. Acad. Sci. USA 2018, 115, E3388–E3397. [Google Scholar] [CrossRef]
- Yao, D.; Gu, Z.; Nakamura, T.; Shi, Z.-Q.; Ma, Y.; Gaston, B.; Palmer, L.A.; Rockenstein, E.M.; Zhang, Z.; Masliah, E.; et al. Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proc. Natl. Acad. Sci. USA 2004, 101, 10810–10814. [Google Scholar] [CrossRef]
- Anitha, A.; Nakamura, K.; Thanseem, I.; Matsuzaki, H.; Miyachi, T.; Tsujii, M.; Iwata, Y.; Suzuki, K.; Sugiyama, T.; Mori, N. Downregulation of the expression of mitochondrial electron transport complex genes in autism brains. Brain Pathol. 2013, 23, 294–302. [Google Scholar] [CrossRef]
- Tang, G.; Rios, P.G.; Kuo, S.-H.; Akman, H.O.; Rosoklija, G.; Tanji, K.; Dwork, A.; Schon, E.A.; Dimauro, S.; Goldman, J.; et al. Mitochondrial abnormalities in temporal lobe of autistic brain. Neurobiol. Dis. 2013, 54, 349–361. [Google Scholar] [CrossRef]
- Chang, A.H.; Sancheti, H.; Garcia, J.; Kaplowitz, N.; Cadenas, E.; Han, D. Respiratory substrates regulate S-nitrosylation of mitochondrial proteins through a thiol-dependent pathway. Chem. Res. Toxicol. 2014, 27, 794–804. [Google Scholar] [CrossRef]
- Siddiqui, M.F.; Elwell, C.; Johnson, M.H. Mitochondrial Dysfunction in Autism Spectrum Disorders. Autism Open Access 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Hirrlinger, J. Glutathione pathways in the brain. Biol. Chem. 2003, 384, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Cha, M.; Lee, B.H. Neuroprotective Effect of Antioxidants in the Brain. Int. J. Mol. Sci. 2020, 21, 7152. [Google Scholar] [CrossRef]
- Bjørklund, G.; Meguid, N.A.; El-Bana, M.A.; Tinkov, A.A.; Saad, K.; Dadar, M.; Hemimi, M.; Skalny, A.V.; Hosnedlová, B.; Kizek, R.; et al. Oxidative Stress in Autism Spectrum Disorder. Mol. Neurobiol. 2020, 57, 2314–2332. [Google Scholar] [CrossRef] [PubMed]
- Nadeem, A.; Ahmad, S.F.; Attia, S.M.; Al-Ayadhi, L.Y.; Al-Harbi, N.O.; Bakheet, S.A. Dysregulated enzymatic antioxidant network in peripheral neutrophils and monocytes in children with autism. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 88, 352–359. [Google Scholar] [CrossRef]
- Eshraghi, R.S.; Deth, R.C.; Mittal, R.; Aranke, M.; Kay, S.-I.S.; Moshiree, B.; Eshraghi, A.A. Early disruption of the microbiome leading to decreased antioxidant capacity and epigenetic changes: Implications for the rise in autism. Front. Cell. Neurosci. 2018, 12, 256. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, S.; Liu, X.; Zheng, Y.; Li, L.; Meng, S. Oxytocin improves animal behaviors and ameliorates oxidative stress and inflammation in autistic mice. Biomed. Pharmacother. 2018, 107, 262–269. [Google Scholar] [CrossRef]
- Mayer, B.; Oberbauer, R. Mitochondrial regulation of apoptosis. News Physiol. Sci. 2003, 18, 89–94. [Google Scholar] [CrossRef]
- Wei, H.; Alberts, I.; Li, X. The apoptotic perspective of autism. Int. J. Dev. Neurosci. 2014, 36, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Amaral, D.G.; Schumann, C.M.; Nordahl, C.W. Neuroanatomy of autism. Trends Neurosci. 2008, 31, 137–145. [Google Scholar] [CrossRef]
- Geschwind, D.H.; Levitt, P. Autism spectrum disorders: Developmental disconnection syndromes. Curr. Opin. Neurobiol. 2007, 17, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Asp. Med. 2010, 31, 227–285. [Google Scholar] [CrossRef] [PubMed]
- Zahid, S.; Khan, R.; Oellerich, M.; Ahmed, N.; Asif, A.R. Differential S-nitrosylation of proteins in Alzheimer’s disease. Neuroscience 2014, 256, 126–136. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Accession ID | Protein’s Name |
|---|---|
| Q03265 | ATP synthase subunit alpha, mitochondrial |
| Q99KI0 | Aconitate hydratase, mitochondrial |
| P05202 | Aspartate aminotransferase, mitochondrial |
| P30275 | Creatine kinase U-type, mitochondrial |
| P39053 | Dynamin-1 |
| P51881 | ADP/ATP translocase 2 |
| P63328 | Serine/threonine-protein phosphatase 2B catalytic subunit alpha isoform |
| P18760 | Cofilin-1 |
| Q80VP2 | Spermatogenesis-associated protein 7 homolog |
| Q8BMK0 | Centrosomal protein of 85 kDa |
| P48754 | Breast cancer type 1 susceptibility protein homolog |
| Q499E0 | Protein FAM5C |
| D3Z7P3 | Glutaminase kidney isoform, mitochondrial |
| O70337 | Bcl-2-interacting killer |
| Q3U7U3 | F-box only protein 7 |
| Q924D0 | Reticulon-4-interacting protein 1, mitochondrial |
| P62897 | Cytochrome c, somatic |
| P51612 | DNA repair protein complementing XP-C cells homolog |
| Q8CBB9 | Radical S-adenosyl methionine domain-containing protein 2 |
| P56564 | Excitatory amino acid transporter 1 |
| O70372 | Telomerase reverse transcriptase |
| Q9CPQ1 | Cytochrome c oxidase subunit 6C |
| P50171 | Estradiol 17-beta-dehydrogenase 8 |
| Q9R1M5 | NACHT, LRR and PYD domains-containing protein 5 |
| Q8BH59 | Calcium-binding mitochondrial carrier protein Aralar1 |
| P62908 | 40S ribosomal protein S3 |
| Q02566 | Myosin-6 |
| Q03137 | Ephrin type-A receptor 4 |
| P0CG49 | Polyubiquitin-B |
| O55042 | Alpha-synuclein |
| P97450 | ATP synthase-coupling factor 6, mitochondrial |
| P48962 | ADP/ATP translocase 1 |
| P08249 | Malate dehydrogenase, mitochondrial |
| P28184 | Metallothionein-3 |
| P11531 | Dystrophin |
| P70296 | Phosphatidylethanolamine-binding protein 1 |
| P00520 | Tyrosine-protein kinase ABL1 |
| P56391 | Cytochrome c oxidase subunit 6B1 |
| D3YXS5 | Kinesin-like protein KLP6 |
| P12787 | Cytochrome c oxidase subunit 5A, mitochondrial |
| Q05A80 | Caprin-2 |
| Q149F5 | Transmembrane protein 71 |
| Q9D8P4 | 39S ribosomal protein L17, mitochondrial |
| P00015 | Cytochrome c, testis-specific |
| Q9DAQ9 | Spermatogenesis-associated protein 19, mitochondrial |
| Q8BX70 | Vacuolar protein sorting-associated protein 13C |
| Q99MD6 | Thioredoxin reductase 3 (Fragment) |
| Q60575 | Kinesin-like protein KIF1B |
| Q6K1E7 | Gametogenetin-binding protein 1 |
| Q8R4U6 | DNA topoisomerase I, mitochondrial |
| Q99K67 | Alpha-aminoadipic semialdehyde synthase, mitochondrial |
| Q924H5 | DNA repair protein RAD51 homolog 3 |
| Q60930 | Voltage-dependent anion-selective channel protein 2 |
| P11798 | Calcium/calmodulin-dependent protein kinase type II subunit alpha |
| Q8CAS9 | Poly [ADP-ribose] polymerase 9 |
| Q9QZM2 | DNA polymerase subunit gamma-2, mitochondrial |
| Q8CGY8 | UDP-N-acetylglucosamine--peptide N-acetylglucosaminyltransferase 110 kDa subunit |
| Q9CZW5 | Mitochondrial import receptor subunit TOM70 |
| Q60855 | Receptor-interacting serine/threonine-protein kinase 1 |
| Q62425 | NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 4 |
| Q80Y44 | Probable ATP-dependent RNA helicase DDX10 |
| Q925I1 | ATPase family AAA domain-containing protein 3 |
| Q6ZWQ0 | Nesprin-2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kartawy, M.; Khaliulin, I.; Amal, H. Systems Biology Reveals S-Nitrosylation-Dependent Regulation of Mitochondrial Functions in Mice with Shank3 Mutation Associated with Autism Spectrum Disorder. Brain Sci. 2021, 11, 677. https://doi.org/10.3390/brainsci11060677
Kartawy M, Khaliulin I, Amal H. Systems Biology Reveals S-Nitrosylation-Dependent Regulation of Mitochondrial Functions in Mice with Shank3 Mutation Associated with Autism Spectrum Disorder. Brain Sciences. 2021; 11(6):677. https://doi.org/10.3390/brainsci11060677
Chicago/Turabian StyleKartawy, Maryam, Igor Khaliulin, and Haitham Amal. 2021. "Systems Biology Reveals S-Nitrosylation-Dependent Regulation of Mitochondrial Functions in Mice with Shank3 Mutation Associated with Autism Spectrum Disorder" Brain Sciences 11, no. 6: 677. https://doi.org/10.3390/brainsci11060677
APA StyleKartawy, M., Khaliulin, I., & Amal, H. (2021). Systems Biology Reveals S-Nitrosylation-Dependent Regulation of Mitochondrial Functions in Mice with Shank3 Mutation Associated with Autism Spectrum Disorder. Brain Sciences, 11(6), 677. https://doi.org/10.3390/brainsci11060677