
Final Exon Frameshift Biallelic PTPN23 Variants Are Associated with Microcephalic Complex Hereditary Spastic Paraplegia

 ,
,
Abstract
1. Introduction
2. Materials and Methods
Genetic Studies
3. Results
3.1. Clinical Findings
3.2. Genetic Findings
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ruano, L.; Melo, C.; Silva, M.C.; Coutinho, P. The global epidemiology of hereditary ataxia and spastic paraplegia: A systematic review of prevalence studies. Neuroepidemiology 2014, 42, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Harding, A.E. Classification of the hereditary ataxias and paraplegias. Lancet 1983, 1, 1151–1155. [Google Scholar] [CrossRef] [PubMed]
- Genomics England. Hereditary Spastic Paraplegia (Version 1.219) 2021. Available online: https://panelapp.genomicsengland.co.uk/panels/165/ (accessed on 11 April 2021).
- Kara, E.; Tucci, A.; Manzoni, C.; Lynch, D.S.; Elpidorou, M.; Bettencourt, C.; Chelban, V.; Manole, A.; Hamed, S.A.; Haridy, N.A.; et al. Genetic and phenotypic characterization of complex hereditary spastic paraplegia. Brain J. Neurol. 2016, 139, 1904–1918. [Google Scholar] [CrossRef] [PubMed]
- Laurá, M.; Pipis, M.; Rossor, A.M.; Reilly, M.M. Charcot-Marie-Tooth disease and related disorders: An evolving landscape. Curr. Opin. Neurol. 2019, 32, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Husedzinovic, A.; Neumann, B.; Reymann, J.; Draeger-Meurer, S.; Chari, A.; Erfle, H.; Fischer, U.; Gruss, O.J. The catalytically inactive tyrosine phosphatase HD-PTP/PTPN23 is a novel regulator of SMN complex localization. Mol. Biol. Cell. 2015, 26, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Tonks, N.K. Protein tyrosine phosphatases--from housekeeping enzymes to master regulators of signal transduction. FEBS J. 2013, 280, 346–378. [Google Scholar] [CrossRef]
- Tonks, N.K. Protein tyrosine phosphatases: From genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 2006, 7, 833–846. [Google Scholar] [CrossRef]
- Cao, L.; Zhang, L.; Ruiz-Lozano, P.; Yang, Q.; Chien, K.R.; Graham, R.M.; Zhou, M. A novel putative protein-tyrosine phosphatase contains a BRO1-like domain and suppresses Ha-ras-mediated transformation. J. Biol. Chem. 1998, 273, 21077–21083. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, J.E.; Heynen-Genel, S.; Suyama, E.; Ono, K.; Lee, K.; Ideker, T.; Aza-Blanc, P.; Gleeson, J.G. Functional genomic screen for modulators of ciliogenesis and cilium length. Nature 2010, 464, 1048–1051. [Google Scholar] [CrossRef]
- Gingras, M.C.; Zhang, Y.L.; Kharitidi, D.; Barr, A.J.; Knapp, S.; Tremblay, M.L.; Pause, A. HD-PTP is a catalytically inactive tyrosine phosphatase due to a conserved divergence in its phosphatase domain. PLoS ONE 2009, 4, e5105. [Google Scholar] [CrossRef]
- Doyotte, A.; Mironov, A.; McKenzie, E.; Woodman, P. The Bro1-related protein HD-PTP/PTPN23 is required for endosomal cargo sorting and multivesicular body morphogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 6308–6313. [Google Scholar] [CrossRef]
- Laver, T.W.; Franco, E.D.; Johnson, M.B.; Patel, K.; Ellard, S.; Weedon, M.N.; Flanagan, S.E.; Wakeling, M.N. SavvyCNV: Genome-wide CNV calling from off-target reads. BioRxiv 2019, 617605. [Google Scholar] [CrossRef]
- Bend, R.; Cohen, L.; Carter, M.T.; Lyons, M.J.; Niyazov, D.; Mikati, M.A.; Rojas, S.K.; Person, R.E.; Si, Y.; Wentzensen, I.M.; et al. Phenotype and mutation expansion of the PTPN23 associated disorder characterized by neurodevelopmental delay and structural brain abnormalities. Eur. J. Hum. Genet. 2020, 28, 76–87. [Google Scholar] [CrossRef]
- Alazami, A.M.; Patel, N.; Shamseldin, H.E.; Anazi, S.; Al-Dosari, M.S.; Alzahrani, F.; Hijazi, H.; Alshammari, M.; Aldahmesh, M.A.; Salih, M.A.; et al. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep. 2015, 10, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Trujillano, D.; Bertoli-Avella, A.M.; Kumar Kandaswamy, K.; Weiss, M.E.; Köster, J.; Marais, A.; Paknia, O.; Schröder, R.; Garcia-Aznar, J.M.; Werber, M.; et al. Clinical exome sequencing: Results from 2819 samples reflecting 1000 families. Eur. J. Hum. Genet. 2017, 25, 176–182. [Google Scholar] [CrossRef]
- Sowada, N.; Hashem, M.O.; Yilmaz, R.; Hamad, M.; Kakar, N.; Thiele, H.; Arold, S.T.; Bode, H.; Alkuraya, F.S.; Borck, G. Mutations of PTPN23 in developmental and epileptic encephalopathy. Hum. Genet. 2017, 136, 1455–1461. [Google Scholar] [CrossRef] [PubMed]
- Smigiel, R.; Landsberg, G.; Schilling, M.; Rydzanicz, M.; Pollak, A.; Walczak, A.; Stodolak, A.; Stawinski, P.; Mierzewska, H.; Sasiadek, M.M.; et al. Developmental epileptic encephalopathy with hypomyelination and brain atrophy associated with PTPN23 variants affecting the assembly of UsnRNPs. Eur. J. Hum. Genet. 2018, 26, 1502–1511. [Google Scholar] [CrossRef]
- Windpassinger, C.; Auer-Grumbach, M.; Irobi, J.; Patel, H.; Petek, E.; Hörl, G.; Malli, R.; Reed, J.A.; Dierick, I.; Verpoorten, N.; et al. Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome. Nat. Genet. 2004, 36, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Tanase, C.A. Histidine domain-protein tyrosine phosphatase interacts with Grb2 and GrpL. PLoS ONE 2010, 5, e14339. [Google Scholar] [CrossRef]
- Zhai, Q.; Fisher, R.D.; Chung, H.Y.; Myszka, D.G.; Sundquist, W.I.; Hill, C.P. Structural and functional studies of ALIX interactions with YPX(n)L late domains of HIV-1 and EIAV. Nat. Struct. Mol. Biol. 2008, 15, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Sasin, J.; Bottini, N.; Friedberg, I.; Friedberg, I.; Osterman, A.; Godzik, A.; Hunter, T.; Dixon, J.; Mustelin, T. Protein Tyrosine Phosphatases in the Human Genome. Cell 2004, 117, 699–711. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, M.; Mehler, E.L.; Goldberg, R.; Zampino, G.; Brunner, H.G.; Kremer, H.; Van der Burgt, I.; Crosby, A.H.; Ion, A.; Jeffery, S.; et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet. 2001, 29, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.C.; Kalidas, K.; Crosby, A.H.; Jeffery, S.; Patton, M.A. The natural history of Noonan syndrome: A long-term follow-up study. Arch. Dis. Child. 2007, 92, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Farazi Fard, M.A.; Rebelo, A.P.; Buglo, E.; Nemati, H.; Dastsooz, H.; Gehweiler, I.; Reich, S.; Reichbauer, J.; Quintáns, B.; Ordóñez-Ugalde, A.; et al. Truncating Mutations in UBAP1 Cause Hereditary Spastic Paraplegia. Am. J. Hum. Genet. 2019, 104, 767–773. [Google Scholar] [CrossRef]
- Reid, E.; Connell, J.; Edwards, T.L.; Duley, S.; Brown, S.E.; Sanderson, C.M. The hereditary spastic paraplegia protein spastin interacts with the ESCRT-III complex-associated endosomal protein CHMP1B. Hum. Mol. Genet. 2004, 14, 19–38. [Google Scholar] [CrossRef]
- Connell, J.W.; Allison, R.J.; Rodger, C.E.; Pearson, G.; Zlamalova, E.; Reid, E. ESCRT-III-associated proteins and spastin inhibit protrudin-dependent polarised membrane traffic. Cell. Mol. Life Sci. 2019. [Google Scholar] [CrossRef]
- Ciccarelli, F.D.; Proukakis, C.; Patel, H.; Cross, H.; Azam, S.; Patton, M.A.; Bork, P.; Crosby, A.H. The identification of a conserved domain in both spartin and spastin, mutated in hereditary spastic paraplegia. Genomics 2003, 81, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Cross, H.; Proukakis, C.; Hershberger, R.; Bork, P.; Ciccarelli, F.D.; Patton, M.A.; McKusick, V.A.; Crosby, A.H. SPG20 is mutated in Troyer syndrome, an hereditary spastic paraplegia. Nat. Genet. 2002, 31, 347–348. [Google Scholar] [CrossRef] [PubMed]
- Renvoisé, B.; Parker, R.L.; Yang, D.; Bakowska, J.C.; Hurley, J.H.; Blackstone, C. SPG20 protein spartin is recruited to midbodies by ESCRT-III protein Ist1 and participates in cytokinesis. Mol. Biol. Cell. 2010, 21, 3293–3303. [Google Scholar] [CrossRef]
- Proukakis, C.; Cross, H.; Patel, H.; Patton, M.A.; Valentine, A.; Crosby, A.H. Troyer syndrome revisited. A clinical and radiological study of a complicated hereditary spastic paraplegia. J. Neurol. 2004, 251, 1105–1110. [Google Scholar] [CrossRef] [PubMed]


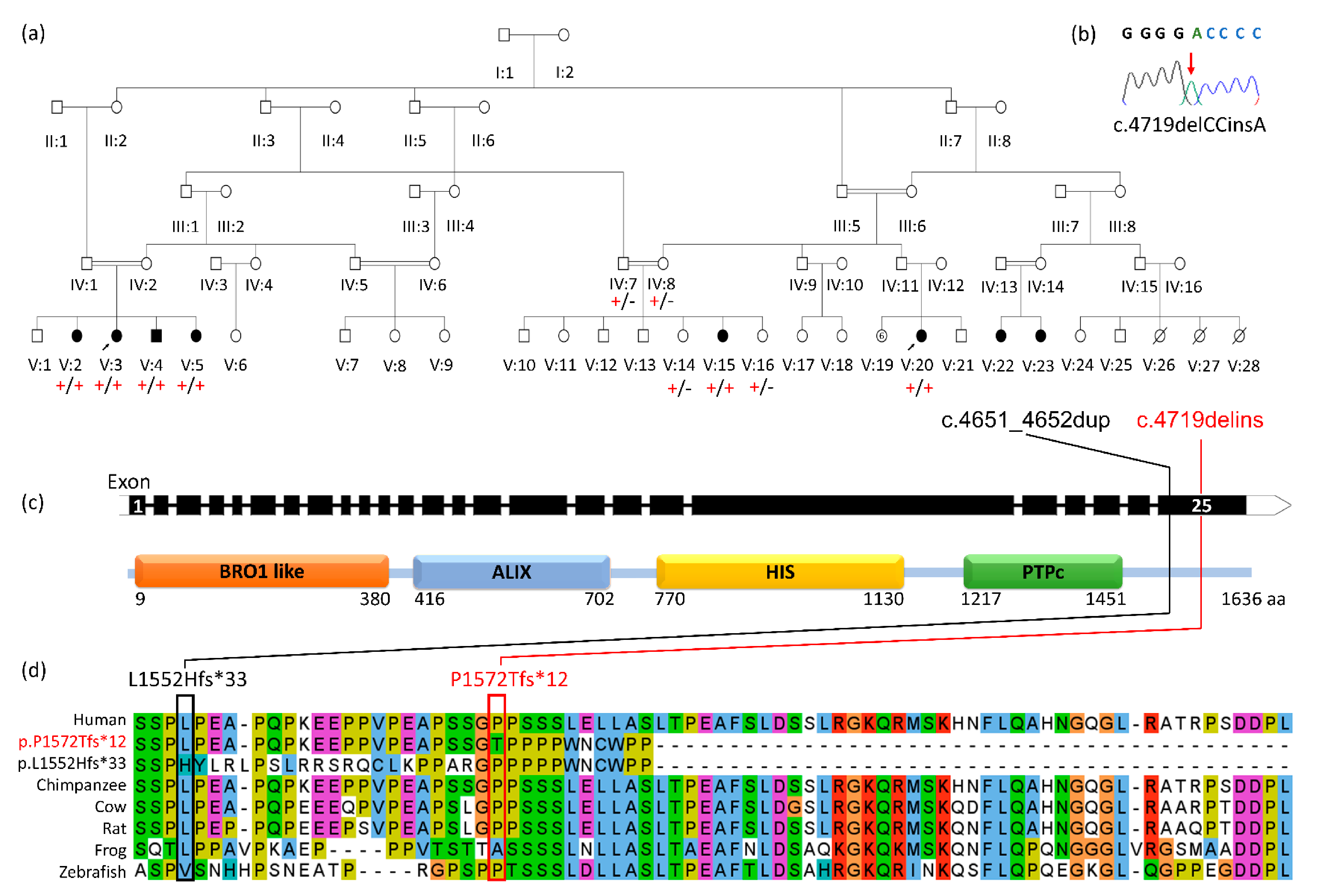{kind=link}
| Reference | V:2 | V:3 | V:4 | V:5 | V:15 | V:20 | Bend et al. Patient 6. [14] |
|---|---|---|---|---|---|---|---|
| Genotype | +/+ | +/+ | +/+ | +/+ | +/+ | +/+ | p.(Leu1552Hisfs*33) /p.(Leu1552Hisfs*33) |
| Sex, Age last seen | F, 17y10m | F, 14y2m | M, 22y4m | F, 10y1m | F, 16y11m | F, 25y1m | F, 11y |
| Age of onset | 6y | 6y | 4-5y | 6y | 7y | 7y | NK |
| OFC (cm) [SD1] | 50.5 [−3.8] | 50.8 [−3.1] | NK | 49.2 [−3.6] | 52 [−2.5] | 50.8 [−3.4] | microcephaly |
| Height (cm) [SD1] | 152 [−1.9] | 143 [−2.7] | 169 [−1.3] | 134.5 [−0.7] | 152 [−1.9] | 152 [−2.0] | NK |
| Weight (kg) [SD1] | 66 [+0.9] | 59 [+0.9] | 66 [−0.6] | NK | NK | 55 [−0.4] | NK |
| Dev. impairment | ✓mild | ✓mild | ✘ university | ✓mild | ✓mild | ✘ university | ✓no speech |
| Toe walking | ✘ | ✓ | ✓ | ✓ | ✓ | ✓ | NK |
| Speech delay | ✘ | ✘ | ✘ | ✘ | ✘ | ✘ | NK |
| Upper limb neurology | normal | normal | normal | normal | normal | normal | NK |
| Lower limb spasticity | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Lower limb DTRs | +++ | +++ | +++ | +++ | +++ | +++ | NK |
| Babinski reflex | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | NK |
| Hypo/paraesthesia | ✘ | ✓episodic | ✓episodic | ✓episodic | ✓ | ✓ | NK |
| Light touch sensation | normal | normal | normal | normal | normal | normal | NK |
| Pain sensation | normal | normal | normal | normal | normal | normal | NK |
| Seizures | ✘ | ✘ | ✘ | ✘ | ✘ | ✘ | ✘ normal EEG |
| Bulbar features | ✘ | ✘ | ✘ | ✘ | ✘ | ✘ | NK |
| Sphincter dysfunction | ✘ | ✘ | ✘ | ✘ | ✘ | ✘ | NK |
| Optic atrophy | NK | NK | NK | NK | NK | NK | ✓& strabismus |
| Horizontal nystagmus | ✓ | ✓ | ✓ | ✓ | ✘ | ✓ | NK |
| MRI brain | NP | normal | normal | NP | NP | NP | enlarged lateral ventricle, delayed myelination |
| Other | DDH, dysphagia | constipation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalaf-Nazzal, R.; Fasham, J.; Ubeyratna, N.; Evans, D.J.; Leslie, J.S.; Warner, T.T.; Al-Hijawi, F.; Alshaer, S.; Baker, W.; Turnpenny, P.D.; et al. Final Exon Frameshift Biallelic PTPN23 Variants Are Associated with Microcephalic Complex Hereditary Spastic Paraplegia. Brain Sci. 2021, 11, 614. https://doi.org/10.3390/brainsci11050614
Khalaf-Nazzal R, Fasham J, Ubeyratna N, Evans DJ, Leslie JS, Warner TT, Al-Hijawi F, Alshaer S, Baker W, Turnpenny PD, et al. Final Exon Frameshift Biallelic PTPN23 Variants Are Associated with Microcephalic Complex Hereditary Spastic Paraplegia. Brain Sciences. 2021; 11(5):614. https://doi.org/10.3390/brainsci11050614
Chicago/Turabian StyleKhalaf-Nazzal, Reham, James Fasham, Nishanka Ubeyratna, David J. Evans, Joseph S. Leslie, Thomas T. Warner, Fida’ Al-Hijawi, Shurouq Alshaer, Wisam Baker, Peter D. Turnpenny, and et al. 2021. "Final Exon Frameshift Biallelic PTPN23 Variants Are Associated with Microcephalic Complex Hereditary Spastic Paraplegia" Brain Sciences 11, no. 5: 614. https://doi.org/10.3390/brainsci11050614
APA StyleKhalaf-Nazzal, R., Fasham, J., Ubeyratna, N., Evans, D. J., Leslie, J. S., Warner, T. T., Al-Hijawi, F., Alshaer, S., Baker, W., Turnpenny, P. D., Baple, E. L., & Crosby, A. H. (2021). Final Exon Frameshift Biallelic PTPN23 Variants Are Associated with Microcephalic Complex Hereditary Spastic Paraplegia. Brain Sciences, 11(5), 614. https://doi.org/10.3390/brainsci11050614