
Crosstalk between 17β-Estradiol and TGF-β Signaling Modulates Glioblastoma Progression



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatments
2.2. Western Blotting
2.3. Immunofluorescence
2.4. Cell Morphology
2.5. RT-qPCR
2.6. Statistical Analysis
3. Results
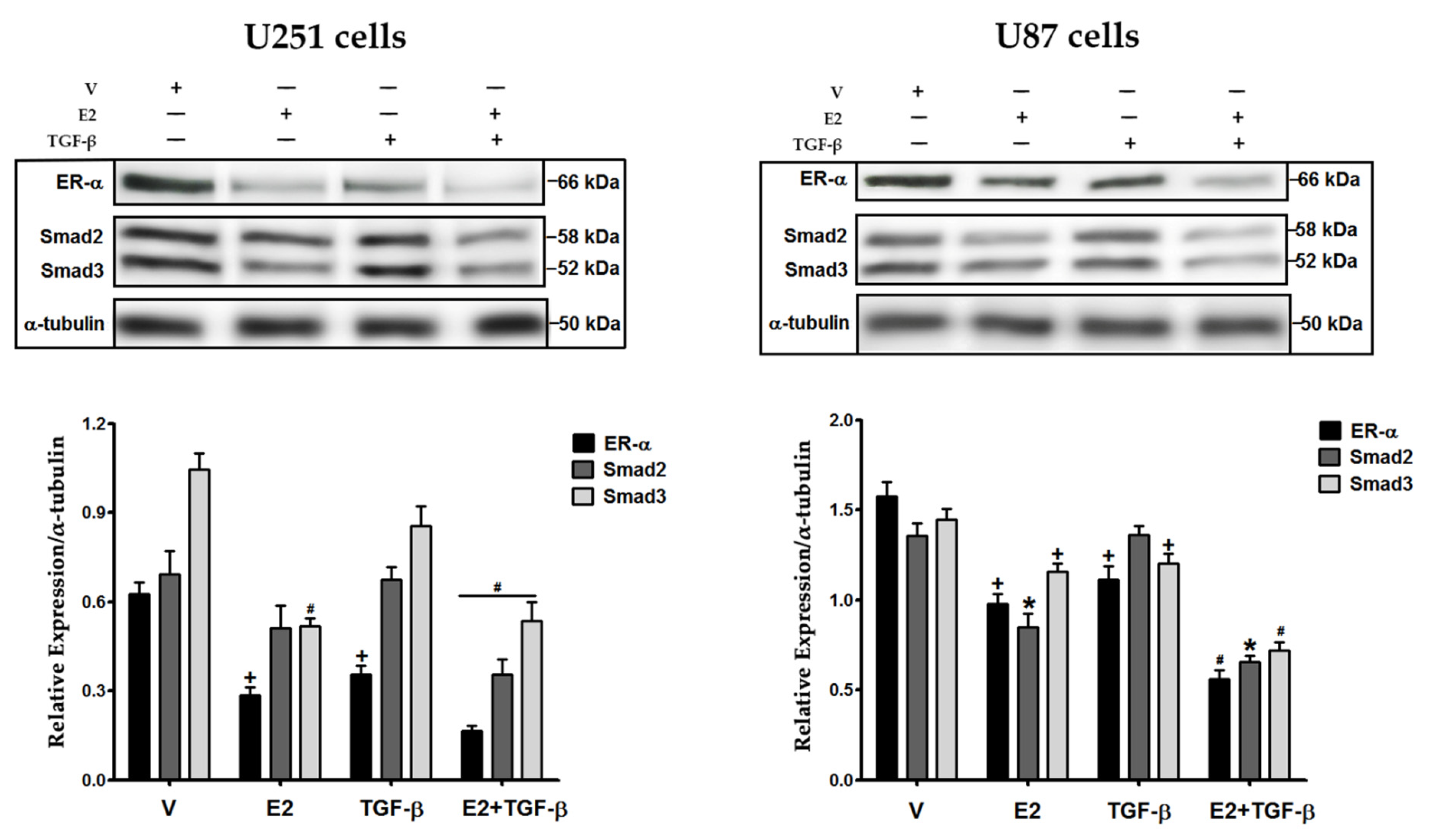
3.1. E2 and TGF-β Decreased the Expression of ER-α, Smad2, and Smad3 Proteins
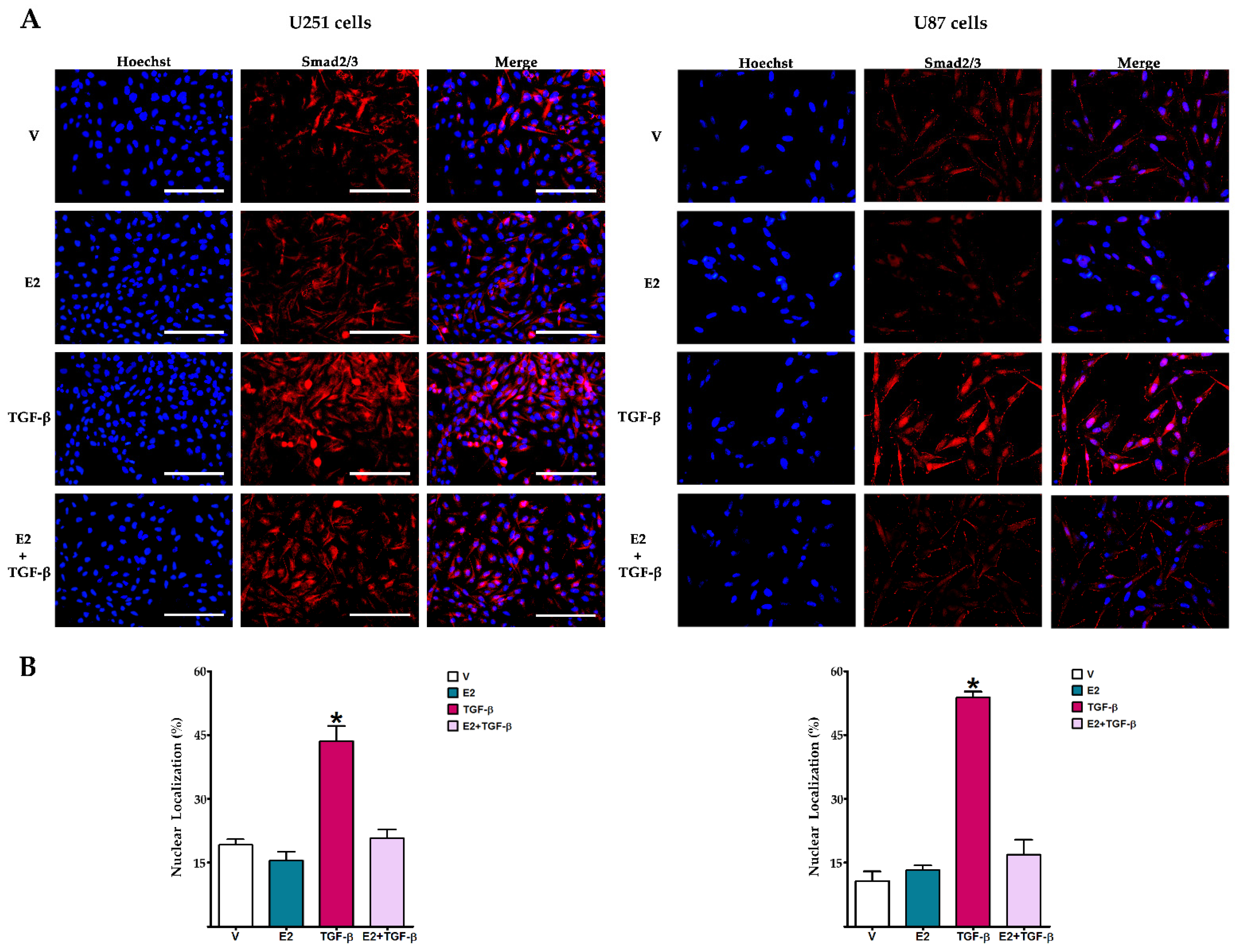
3.2. E2 Decreased Smad2 Phosphorylation and Subsequent Nuclear Translocation of Smad2/3 Complex Induced by TGF-β
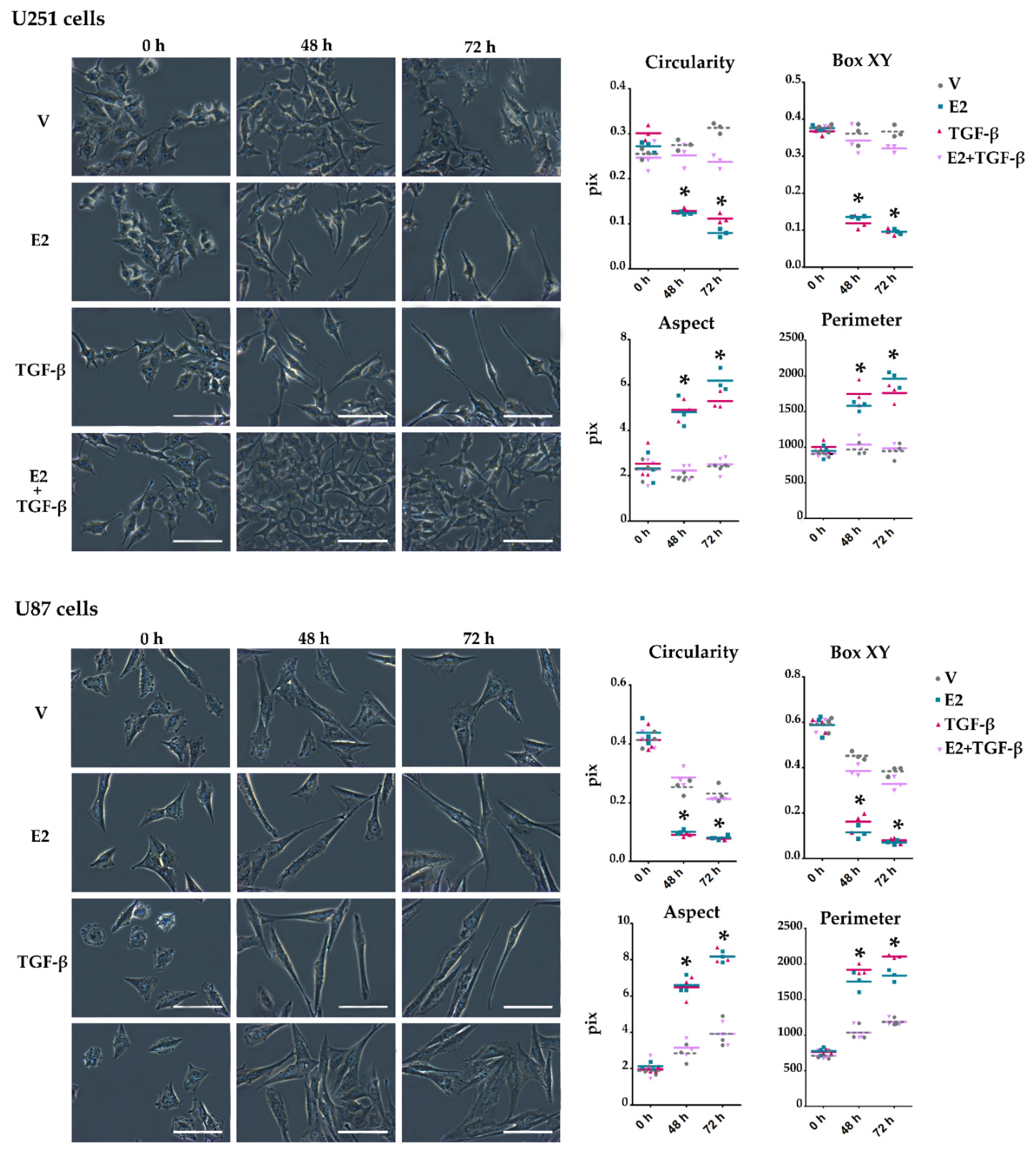
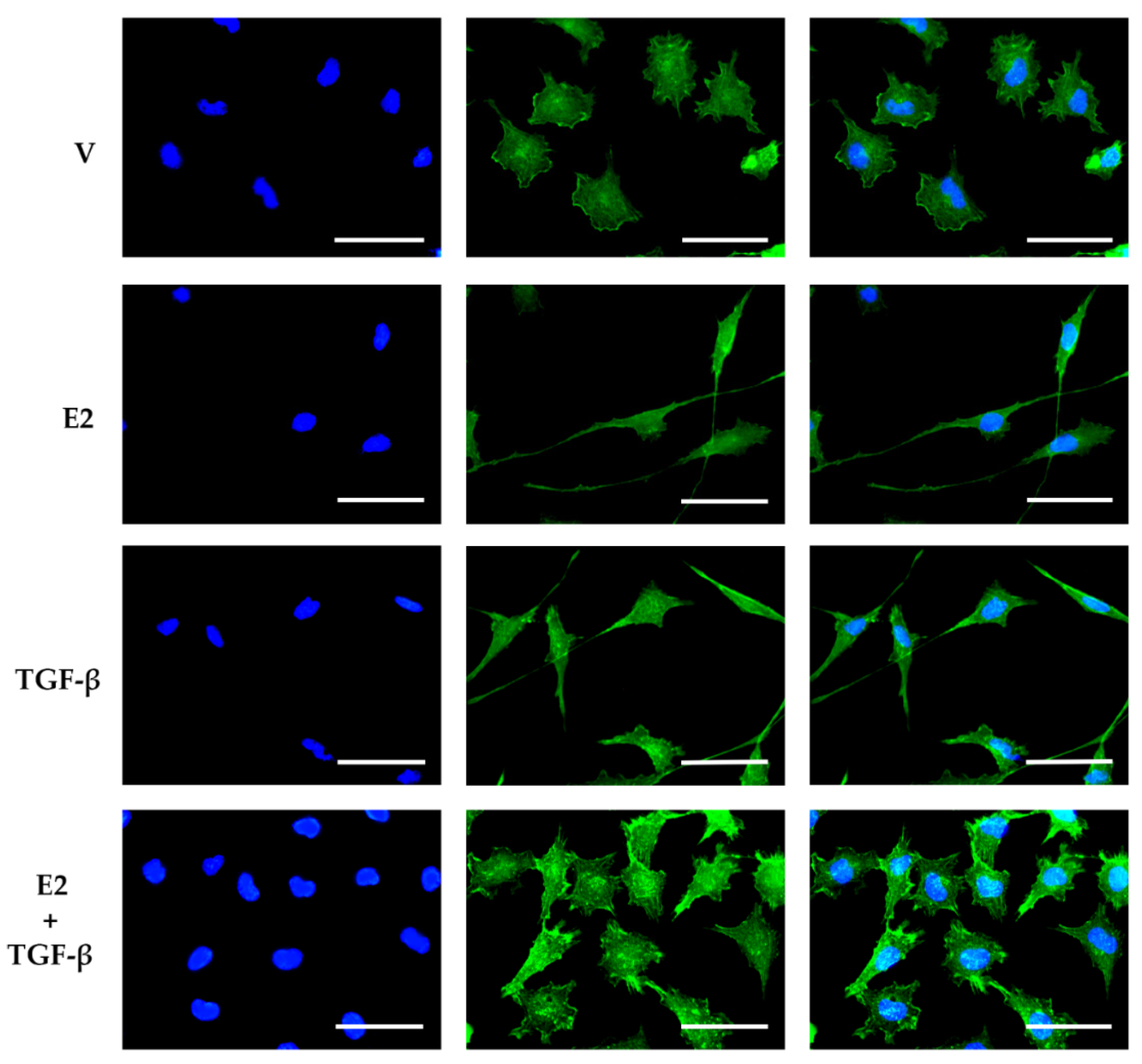
3.3. E2 and TGF-β Mutually Inhibited Their Effects on Morphological Changes Induced by Actin Filament Reorganization in GBM-Derived Cells
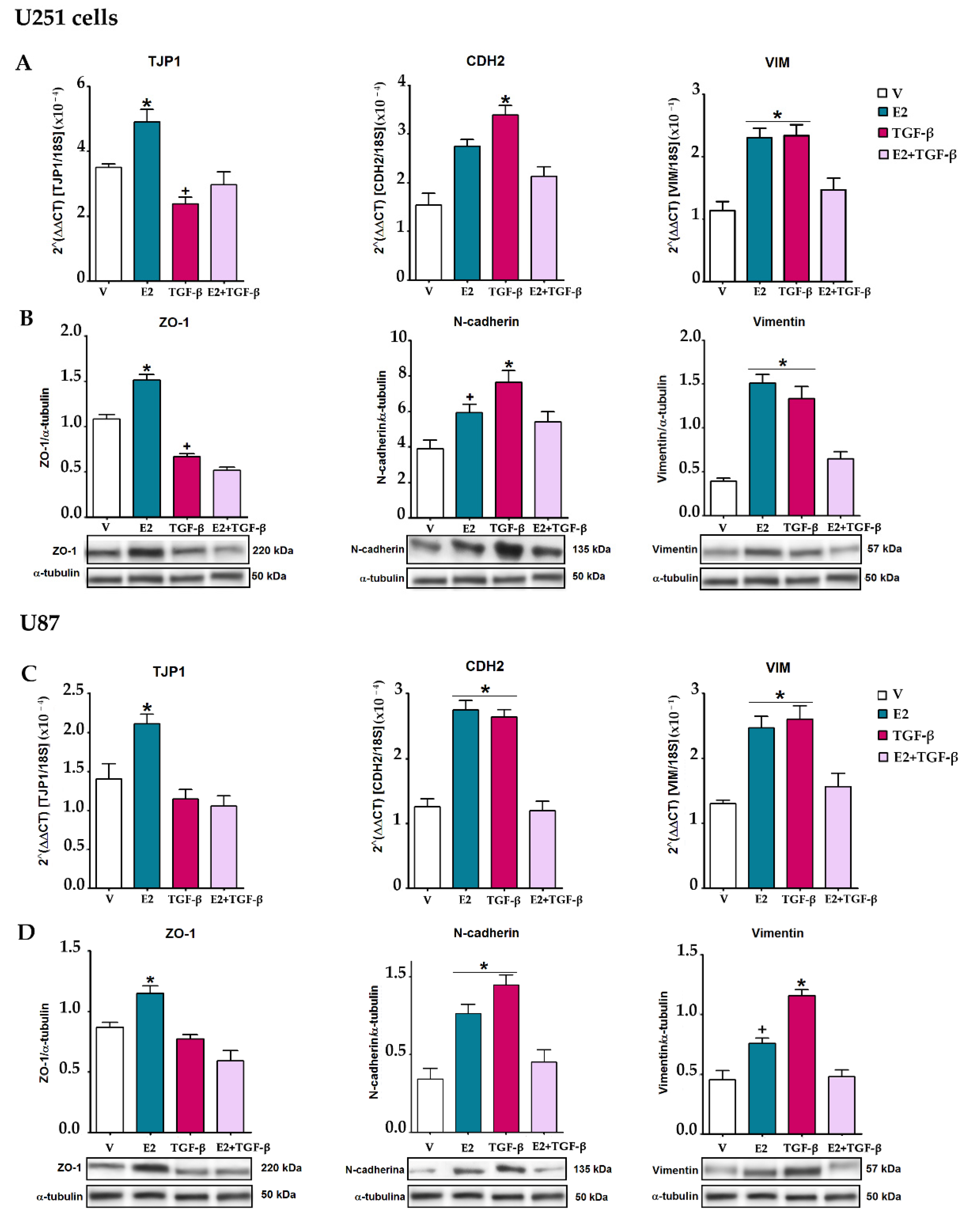
3.4. E2 and TGF-β Mutually Inhibited Their Effects on EMT Markers Expression
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lapointe, S.; Perry, A.; Butowski, N.A. Primary brain tumours in adults. Lancet 2018, 392, 432–446. [Google Scholar] [CrossRef]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [PubMed]
- Stoyanov, G.S.; Dzhenkov, D.; Ghenev, P.; Iliev, B.; Enchev, Y.; Tonchev, A.B. Cell biology of glioblastoma multiforme: From basic science to diagnosis and treatment. Med. Oncol. 2018, 35, 27. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro Oncol. 2019, 21, v1–v100. [Google Scholar] [CrossRef] [PubMed]
- Reifenberger, G.; Wirsching, H.G.; Knobbe-Thomsen, C.B.; Weller, M. Advances in the molecular genetics of gliomas—Implications for classification and therapy. Nat. Rev. Clin. Oncol. 2017, 14, 434–452. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Farhood, B.; Khodamoradi, E.; Hoseini-Ghahfarokhi, M.; Motevaseli, E.; Mirtavoos-Mahyari, H.; Eleojo Musa, A.; Najafi, M. TGF-β in radiotherapy: Mechanisms of tumor resistance and normal tissues injury. Pharmacol. Res. 2020, 155, 104745. [Google Scholar] [CrossRef]
- Montemurro, N.; Anania, Y.; Cagnazzo, F.; Perrini, P. Survival outcomes in patients with recurrent glioblastoma treated with Laser Interstitial Thermal Therapy (LITT): A systematic review. Clin. Neurol. Neurosurg. 2020, 195, 105942. [Google Scholar] [CrossRef]
- Vollmann-Zwerenz, A.; Leidgens, V.; Feliciello, G.; Klein, C.A.; Hau, P. Tumor Cell Invasion in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 1932. [Google Scholar] [CrossRef]
- Vehlow, A.; Cordes, N. Invasion as target for therapy of glioblastoma multiforme. Biochim. Biophys. Acta 2013, 1836, 236–244. [Google Scholar] [CrossRef]
- Tzanakakis, G.; Kavasi, R.M.; Voudouri, K.; Berdiaki, A.; Spyridaki, I.; Tsatsakis, A.; Nikitovic, D. Role of the extracellular matrix in cancer-associated epithelial to mesenchymal transition phenomenon. Dev. Dyn. 2018, 247, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Cuddapah, V.A.; Robel, S.; Watkins, S.; Sontheimer, H. A neurocentric perspective on glioma invasion. Nat. Rev. Neurosci. 2014, 15, 455–465. [Google Scholar] [CrossRef]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef]
- Carro, M.S.; Lim, W.K.; Alvarez, M.J.; Bollo, R.J.; Zhao, X.; Snyder, E.Y.; Sulman, E.P.; Anne, S.L.; Doetsch, F.; Colman, H.; et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature 2010, 463, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Freije, W.A.; Castro-Vargas, F.E.; Fang, Z.; Horvath, S.; Cloughesy, T.; Liau, L.M.; Mischel, P.S.; Nelson, S.F. Gene expression profiling of gliomas strongly predicts survival. Cancer Res. 2004, 64, 6503–6510. [Google Scholar] [CrossRef]
- Kahlert, U.D.; Nikkhah, G.; Maciaczyk, J. Epithelial-to-mesenchymal(-like) transition as a relevant molecular event in malignant gliomas. Cancer Lett. 2013, 331, 131–138. [Google Scholar] [CrossRef]
- Tso, C.L.; Shintaku, P.; Chen, J.; Liu, Q.; Liu, J.; Chen, Z.; Yoshimoto, K.; Mischel, P.S.; Cloughesy, T.F.; Liau, L.M.; et al. Primary glioblastomas express mesenchymal stem-like properties. Mol. Cancer Res. 2006, 4, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.V.; Conroy, S.; Tomar, T.; Eggens-Meijer, E.; Bhat, K.; Copray, S.; Walenkamp, A.M.; Boddeke, E.; Balasubramanyian, V.; Wagemakers, M.; et al. TGF-β is an inducer of ZEB1-dependent mesenchymal transdifferentiation in glioblastoma that is associated with tumor invasion. Cell Death Dis. 2014, 5, e1443. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, W.; Li, Y.; Alvarez, A.; Li, Z.; Wang, Y.; Song, L.; Lv, D.; Nakano, I.; Hu, B.; et al. SHP-2-upregulated ZEB1 is important for PDGFRα-driven glioma epithelial-mesenchymal transition and invasion in mice and humans. Oncogene 2016, 35, 5641–5652. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.V.; Chang, J.P.; Parachoniak, C.A.; Pandika, M.M.; Aghi, M.K.; Meyronet, D.; Isachenko, N.; Fouse, S.D.; Phillips, J.J.; Cheresh, D.A.; et al. VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell 2012, 22, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, S.; Li, X.; Jin, S.; Xiong, F.; Wang, X. The critical role of EGF-β-catenin signaling in the epithelial-mesenchymal transition in human glioblastoma. OncoTargets Ther. 2017, 10, 2781–2789. [Google Scholar] [CrossRef]
- Kahlert, U.D.; Maciaczyk, D.; Doostkam, S.; Orr, B.A.; Simons, B.; Bogiel, T.; Reithmeier, T.; Prinz, M.; Schubert, J.; Niedermann, G.; et al. Activation of canonical WNT/β-catenin signaling enhances in vitro motility of glioblastoma cells by activation of ZEB1 and other activators of epithelial-to-mesenchymal transition. Cancer Lett. 2012, 325, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, H.; Mu, X.; Cui, J.; Peng, Z. Dysregulation of Fra1 expression by Wnt/β-catenin signalling promotes glioma aggressiveness through epithelial-mesenchymal transition. Biosci Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.V.; Conroy, S.; Pavlov, K.; Sontakke, P.; Tomar, T.; Eggens-Meijer, E.; Balasubramaniyan, V.; Wagemakers, M.; den Dunnen, W.F.; Kruyt, F.A. Hypoxia enhances migration and invasion in glioblastoma by promoting a mesenchymal shift mediated by the HIF1α-ZEB1 axis. Cancer Lett. 2015, 359, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Rahimpour, S.; Nesvick, C.L.; Zhang, X.; Ma, J.; Zhang, M.; Zhang, G.; Wang, L.; Yang, C.; Hong, C.S.; et al. Activation of hypoxia signaling induces phenotypic transformation of glioma cells: Implications for bevacizumab antiangiogenic therapy. Oncotarget 2015, 6, 11882–11893. [Google Scholar] [CrossRef]
- Shan, Y.; He, X.; Song, W.; Han, D.; Niu, J.; Wang, J. Role of IL-6 in the invasiveness and prognosis of glioma. Int. J. Clin. Exp. Med. 2015, 8, 9114–9120. [Google Scholar] [PubMed]
- Hernández-Vega, A.M.; Del Moral-Morales, A.; Zamora-Sánchez, C.J.; Piña-Medina, A.G.; González-Arenas, A.; Camacho-Arroyo, I. Estradiol Induces Epithelial to Mesenchymal Transition of Human Glioblastoma Cells. Cells 2020, 9, 1930. [Google Scholar] [CrossRef]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Itoh, S.; Itoh, F.; Goumans, M.J.; Ten Dijke, P. Signaling of transforming growth factor-beta family members through Smad proteins. Eur. J. Biochem. 2000, 267, 6954–6967. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Souchelnytskyi, S.; Heldin, C.H. Smad regulation in TGF-beta signal transduction. J. Cell Sci. 2001, 114, 4359–4369. [Google Scholar] [CrossRef]
- Massagué, J. How cells read TGF-beta signals. Nat. Rev. Mol. Cell Biol. 2000, 1, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Luo, K. Signaling Cross Talk between TGF-β/Smad and Other Signaling Pathways. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef]
- Itoh, Y.; Saitoh, M.; Miyazawa, K. Smad3-STAT3 crosstalk in pathophysiological contexts. Acta Biochim. Biophys. Sin. (Shanghai) 2018, 50, 82–90. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, Y.; Mao, H.; Chen, W.; Luo, N.; Zhou, Q.; Chen, W.; Yu, X. A crosstalk between the Smad and JNK signaling in the TGF-β-induced epithelial-mesenchymal transition in rat peritoneal mesothelial cells. PLoS ONE 2012, 7, e32009. [Google Scholar] [CrossRef] [PubMed]
- Band, A.M.; Laiho, M. Crosstalk of TGF-β and estrogen receptor signaling in breast cancer. J. Mammary Gland Biol. Neoplasia 2011, 16, 109–115. [Google Scholar] [CrossRef]
- Matsuda, T.; Yamamoto, T.; Muraguchi, A.; Saatcioglu, F. Cross-talk between transforming growth factor-beta and estrogen receptor signaling through Smad3. J. Biol. Chem. 2001, 276, 42908–42914. [Google Scholar] [CrossRef] [PubMed]
- Malek, D.; Gust, R.; Kleuser, B. 17-Beta-estradiol inhibits transforming-growth-factor-beta-induced MCF-7 cell migration by Smad3-repression. Eur. J. Pharmacol. 2006, 534, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Ito, I.; Hanyu, A.; Wayama, M.; Goto, N.; Katsuno, Y.; Kawasaki, S.; Nakajima, Y.; Kajiro, M.; Komatsu, Y.; Fujimura, A.; et al. Estrogen inhibits transforming growth factor beta signaling by promoting Smad2/3 degradation. J. Biol. Chem. 2010, 285, 14747–14755. [Google Scholar] [CrossRef]
- Dueñas Jiménez, J.M.; Candanedo Arellano, A.; Santerre, A.; Orozco Suárez, S.; Sandoval Sánchez, H.; Feria Romero, I.; López-Elizalde, R.; Alonso Venegas, M.; Netel, B.; de la Torre Valdovinos, B.; et al. Aromatase and estrogen receptor alpha mRNA expression as prognostic biomarkers in patients with astrocytomas. J. Neurooncol. 2014, 119, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Khalid, H.; Yasunaga, A.; Kishikawa, M.; Shibata, S. Immunohistochemical expression of the estrogen receptor-related antigen (ER-D5) in human intracranial tumors. Cancer 1995, 75, 2571–2578. [Google Scholar] [CrossRef]
- Batistatou, A.; Stefanou, D.; Goussia, A.; Arkoumani, E.; Papavassiliou, A.G.; Agnantis, N.J. Estrogen receptor beta (ERbeta) is expressed in brain astrocytic tumors and declines with dedifferentiation of the neoplasm. J. Cancer Res. Clin. Oncol. 2004, 130, 405–410. [Google Scholar] [CrossRef]
- Sareddy, G.R.; Nair, B.C.; Gonugunta, V.K.; Zhang, Q.G.; Brenner, A.; Brann, D.W.; Tekmal, R.R.; Vadlamudi, R.K. Therapeutic significance of estrogen receptor β agonists in gliomas. Mol. Cancer Ther. 2012, 11, 1174–1182. [Google Scholar] [CrossRef]
- González-Arenas, A.; Hansberg-Pastor, V.; Hernández-Hernández, O.T.; González-García, T.K.; Henderson-Villalpando, J.; Lemus-Hernández, D.; Cruz-Barrios, A.; Rivas-Suárez, M.; Camacho-Arroyo, I. Estradiol increases cell growth in human astrocytoma cell lines through ERα activation and its interaction with SRC-1 and SRC-3 coactivators. Biochim. Biophys. Acta 2012, 1823, 379–386. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Bradshaw, A.; Wickremsekera, A.; Tan, S.T.; Peng, L.; Davis, P.F.; Itinteang, T. Cancer Stem Cell Hierarchy in Glioblastoma Multiforme. Front. Surg. 2016, 3, 21. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef]
- Fane, M.; Weeraratna, A.T. How the ageing microenvironment influences tumour progression. Nat. Rev. Cancer 2020, 20, 89–106. [Google Scholar] [CrossRef]
- Joyce, J.A. Therapeutic targeting of the tumor microenvironment. Cancer Cell 2005, 7, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Kjellman, C.; Olofsson, S.P.; Hansson, O.; Von Schantz, T.; Lindvall, M.; Nilsson, I.; Salford, L.G.; Sjögren, H.O.; Widegren, B. Expression of TGF-beta isoforms, TGF-beta receptors, and SMAD molecules at different stages of human glioma. Int. J. Cancer 2000, 89, 251–258. [Google Scholar] [CrossRef]
- Jordan, J.D.; Landau, E.M.; Iyengar, R. Signaling networks: The origins of cellular multitasking. Cell 2000, 103, 193–200. [Google Scholar] [CrossRef]
- Kolch, W.; Halasz, M.; Granovskaya, M.; Kholodenko, B.N. The dynamic control of signal transduction networks in cancer cells. Nat. Rev. Cancer 2015, 15, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Rowland, M.A.; Fontana, W.; Deeds, E.J. Crosstalk and competition in signaling networks. Biophys. J. 2012, 103, 2389–2398. [Google Scholar] [CrossRef] [PubMed]
- Rowland, M.A.; Greenbaum, J.M.; Deeds, E.J. Crosstalk and the evolvability of intracellular communication. Nat. Commun. 2017, 8, 16009. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Han, J.; Alvarez-Breckenridge, C.A.; Wang, Q.E.; Yu, J. TGF-β signaling and its targeting for glioma treatment. Am. J. Cancer Res. 2015, 5, 945–955. [Google Scholar] [PubMed]
- Jennings, M.T.; Pietenpol, J.A. The role of transforming growth factor beta in glioma progression. J. Neurooncol. 1998, 36, 123–140. [Google Scholar] [CrossRef]
- Bruna, A.; Darken, R.S.; Rojo, F.; Ocaña, A.; Peñuelas, S.; Arias, A.; Paris, R.; Tortosa, A.; Mora, J.; Baselga, J.; et al. High TGFbeta-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell 2007, 11, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Merzak, A.; McCrea, S.; Koocheckpour, S.; Pilkington, G.J. Control of human glioma cell growth, migration and invasion in vitro by transforming growth factor beta 1. Br. J. Cancer 1994, 70, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Hawse, J.R.; Subramaniam, M.; Ingle, J.N.; Oursler, M.J.; Rajamannan, N.M.; Spelsberg, T.C. Estrogen-TGFbeta cross-talk in bone and other cell types: Role of TIEG, Runx2, and other transcription factors. J. Cell Biochem. 2008, 103, 383–392. [Google Scholar] [CrossRef]
- Smith, L.C.; Moreno, S.; Robertson, L.; Robinson, S.; Gant, K.; Bryant, A.J.; Sabo-Attwood, T. Transforming growth factor beta1 targets estrogen receptor signaling in bronchial epithelial cells. Respir. Res. 2018, 19, 160. [Google Scholar] [CrossRef]
- Stope, M.B.; Popp, S.L.; Knabbe, C.; Buck, M.B. Estrogen receptor alpha attenuates transforming growth factor-beta signaling in breast cancer cells independent from agonistic and antagonistic ligands. Breast Cancer Res. Treat. 2010, 120, 357–367. [Google Scholar] [CrossRef]
- Wu, L.; Wu, Y.; Gathings, B.; Wan, M.; Li, X.; Grizzle, W.; Liu, Z.; Lu, C.; Mao, Z.; Cao, X. Smad4 as a transcription corepressor for estrogen receptor alpha. J. Biol. Chem. 2003, 278, 15192–15200. [Google Scholar] [CrossRef] [PubMed]
- Hardee, M.E.; Marciscano, A.E.; Medina-Ramirez, C.M.; Zagzag, D.; Narayana, A.; Lonning, S.M.; Barcellos-Hoff, M.H. Resistance of glioblastoma-initiating cells to radiation mediated by the tumor microenvironment can be abolished by inhibiting transforming growth factor-β. Cancer Res. 2012, 72, 4119–4129. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Kleber, S.; Röhrich, M.; Timke, C.; Han, N.; Tuettenberg, J.; Martin-Villalba, A.; Debus, J.; Peschke, P.; Wirkner, U.; et al. Blockade of TGF-β signaling by the TGFβR-I kinase inhibitor LY2109761 enhances radiation response and prolongs survival in glioblastoma. Cancer Res. 2011, 71, 7155–7167. [Google Scholar] [CrossRef]
- Tran, T.T.; Uhl, M.; Ma, J.Y.; Janssen, L.; Sriram, V.; Aulwurm, S.; Kerr, I.; Lam, A.; Webb, H.K.; Kapoun, A.M.; et al. Inhibiting TGF-beta signaling restores immune surveillance in the SMA-560 glioma model. Neuro-oncology 2007, 9, 259–270. [Google Scholar] [CrossRef]
- Birch, J.L.; Coull, B.J.; Spender, L.C.; Watt, C.; Willison, A.; Syed, N.; Chalmers, A.J.; Hossain-Ibrahim, M.K.; Inman, G.J. Multifaceted transforming growth factor-beta (TGFβ) signalling in glioblastoma. Cell Signal 2020, 72, 109638. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Lei, B.; Xiang, W.; Wang, H.; Feng, W.; Liu, Y.; Qi, S. Differences in Protein Expression between the U251 and U87 Cell Lines. Turk Neurosurg. 2017, 27, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Patil, V.; Pal, J.; Somasundaram, K. Elucidating the cancer-specific genetic alteration spectrum of glioblastoma derived cell lines from whole exome and RNA sequencing. Oncotarget 2015, 6, 43452–43471. [Google Scholar] [CrossRef] [PubMed]
- Borrás, M.; Hardy, L.; Lempereur, F.; el Khissiin, A.H.; Legros, N.; Gol-Winkler, R.; Leclercq, G. Estradiol-induced down-regulation of estrogen receptor. Effect of various modulators of protein synthesis and expression. J. Steroid Biochem. Mol. Biol. 1994, 48, 325–336. [Google Scholar] [CrossRef]
- Hatsumi, T.; Yamamuro, Y. Downregulation of estrogen receptor gene expression by exogenous 17beta-estradiol in the mammary glands of lactating mice. Exp. Biol. Med. 2006, 231, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Shi, H. Regulation of Estrogen Receptor α Expression in the Hypothalamus by Sex Steroids: Implication in the Regulation of Energy Homeostasis. Int. J. Endocrinol. 2015, 2015, 949085. [Google Scholar] [CrossRef]
- Stoica, A.; Saceda, M.; Fakhro, A.; Solomon, H.B.; Fenster, B.D.; Martin, M.B. The role of transforming growth factor-beta in the regulation of estrogen receptor expression in the MCF-7 breast cancer cell line. Endocrinology 1997, 138, 1498–1505. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández-Vega, A.M.; Camacho-Arroyo, I. Crosstalk between 17β-Estradiol and TGF-β Signaling Modulates Glioblastoma Progression. Brain Sci. 2021, 11, 564. https://doi.org/10.3390/brainsci11050564
Hernández-Vega AM, Camacho-Arroyo I. Crosstalk between 17β-Estradiol and TGF-β Signaling Modulates Glioblastoma Progression. Brain Sciences. 2021; 11(5):564. https://doi.org/10.3390/brainsci11050564
Chicago/Turabian StyleHernández-Vega, Ana M., and Ignacio Camacho-Arroyo. 2021. "Crosstalk between 17β-Estradiol and TGF-β Signaling Modulates Glioblastoma Progression" Brain Sciences 11, no. 5: 564. https://doi.org/10.3390/brainsci11050564
APA StyleHernández-Vega, A. M., & Camacho-Arroyo, I. (2021). Crosstalk between 17β-Estradiol and TGF-β Signaling Modulates Glioblastoma Progression. Brain Sciences, 11(5), 564. https://doi.org/10.3390/brainsci11050564