
A Computational Model for Pain Processing in the Dorsal Horn Following Axonal Damage to Receptor Fibers


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
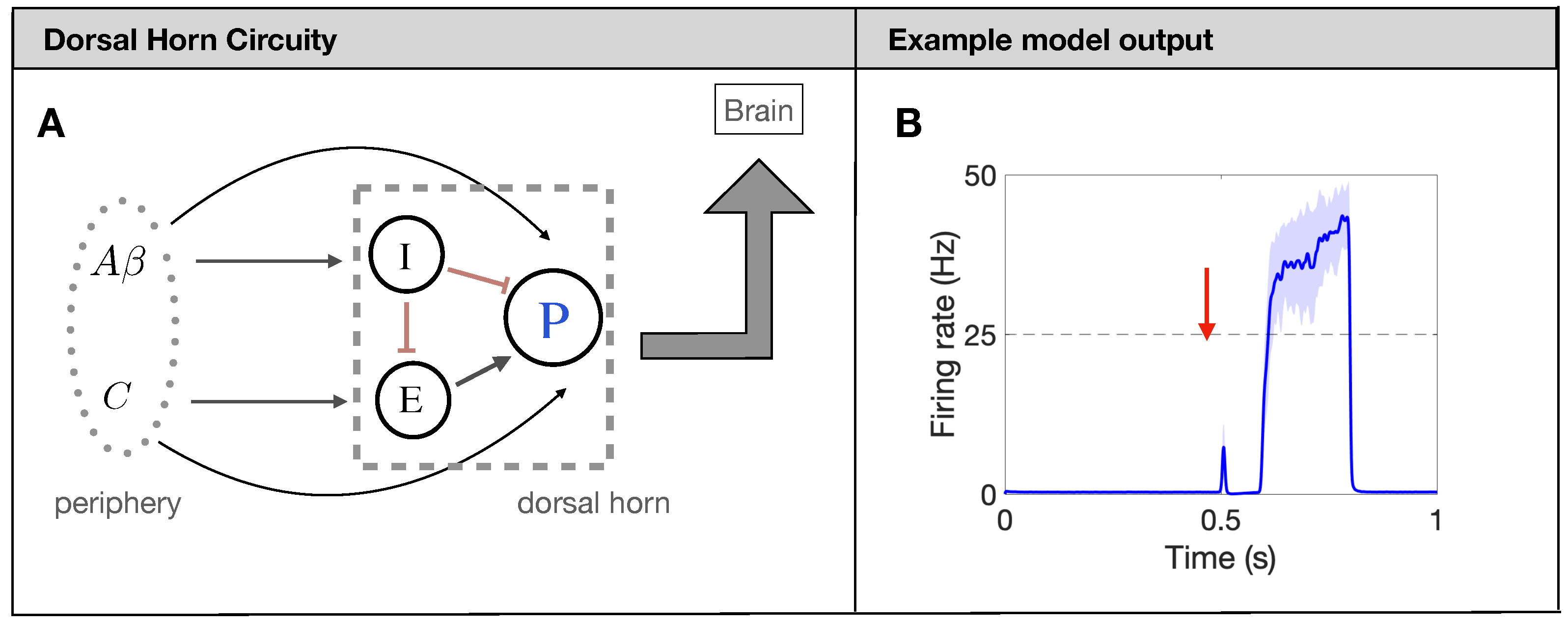
2.1. Overview of Spinal Cord Model for Pain Processing
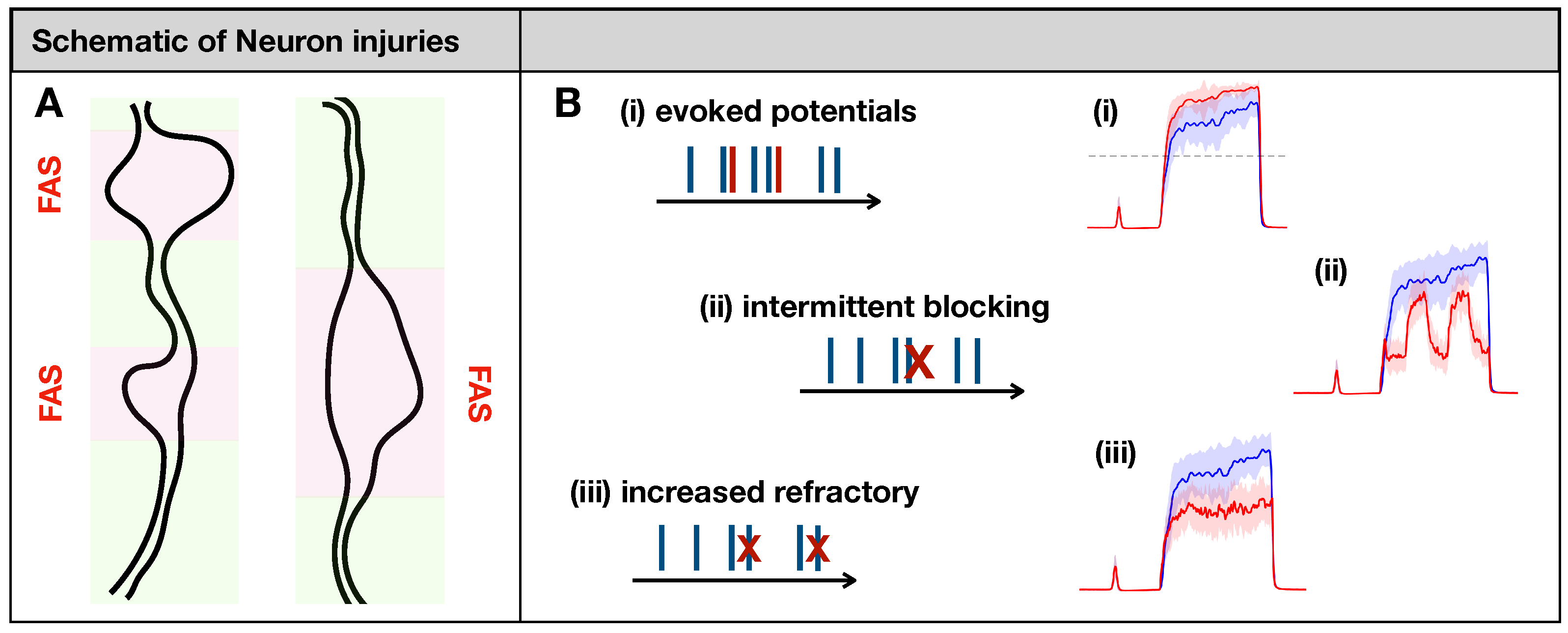
2.2. Modeling Effects of Neuronal Injury to Spike-Train Activity
- (i)
- Evoking potentials: In this rule, a single input spike triggers the formation of k additional spikes.
- (ii)
- Intermittent blocking: In this rule, the spike train switches between (total) blocking and normal conduction periodically (with period ).
- (iii)
- Increasing refractoriness: In this rule, consecutive spikes may be deleted if the inter-spike interval between them is below . This effectively increases the refractory period of the spike train.
2.3. Injury Protocols for Receptor Fibers
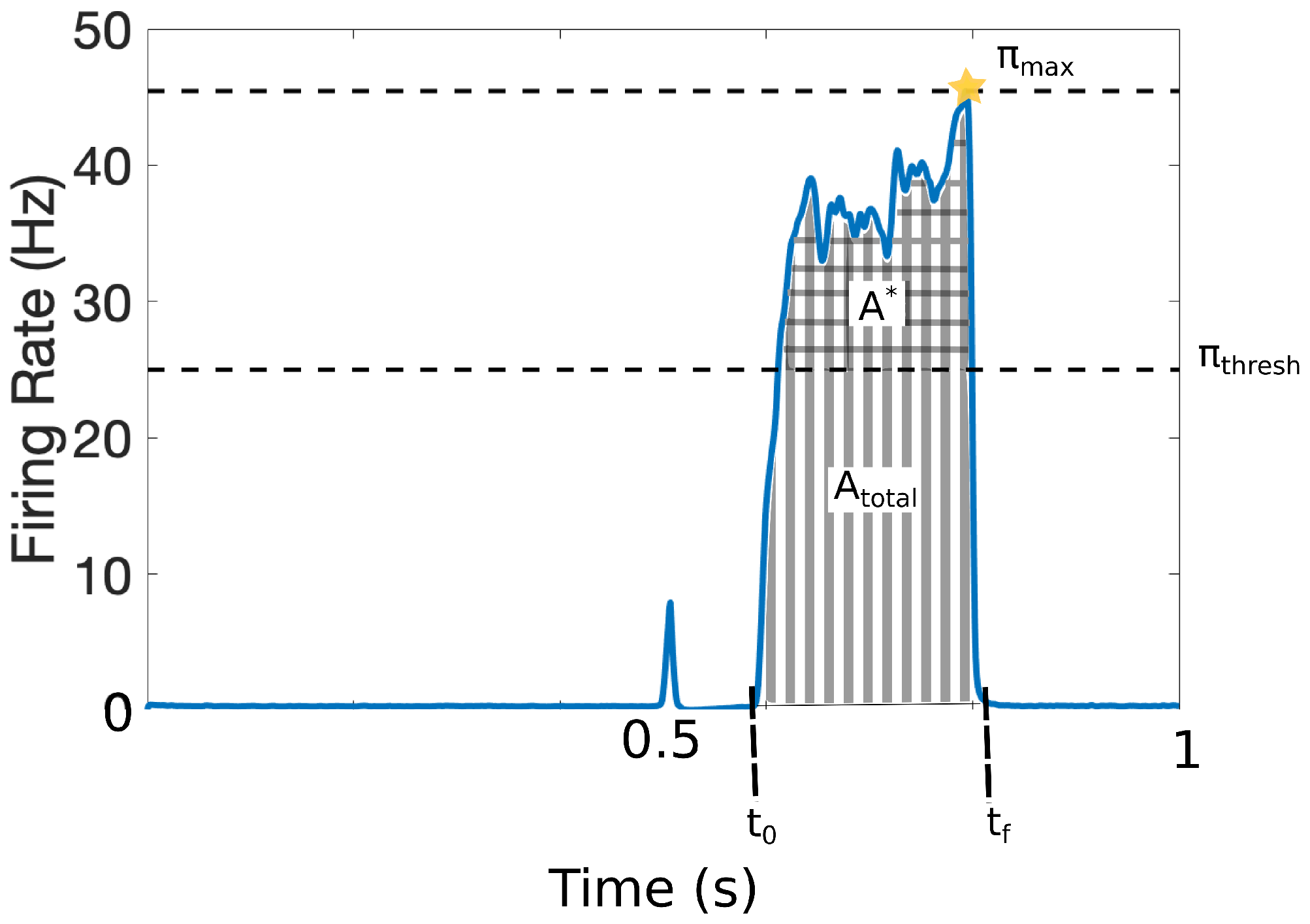
2.4. Quantitative Markers for Pain Response
3. Results
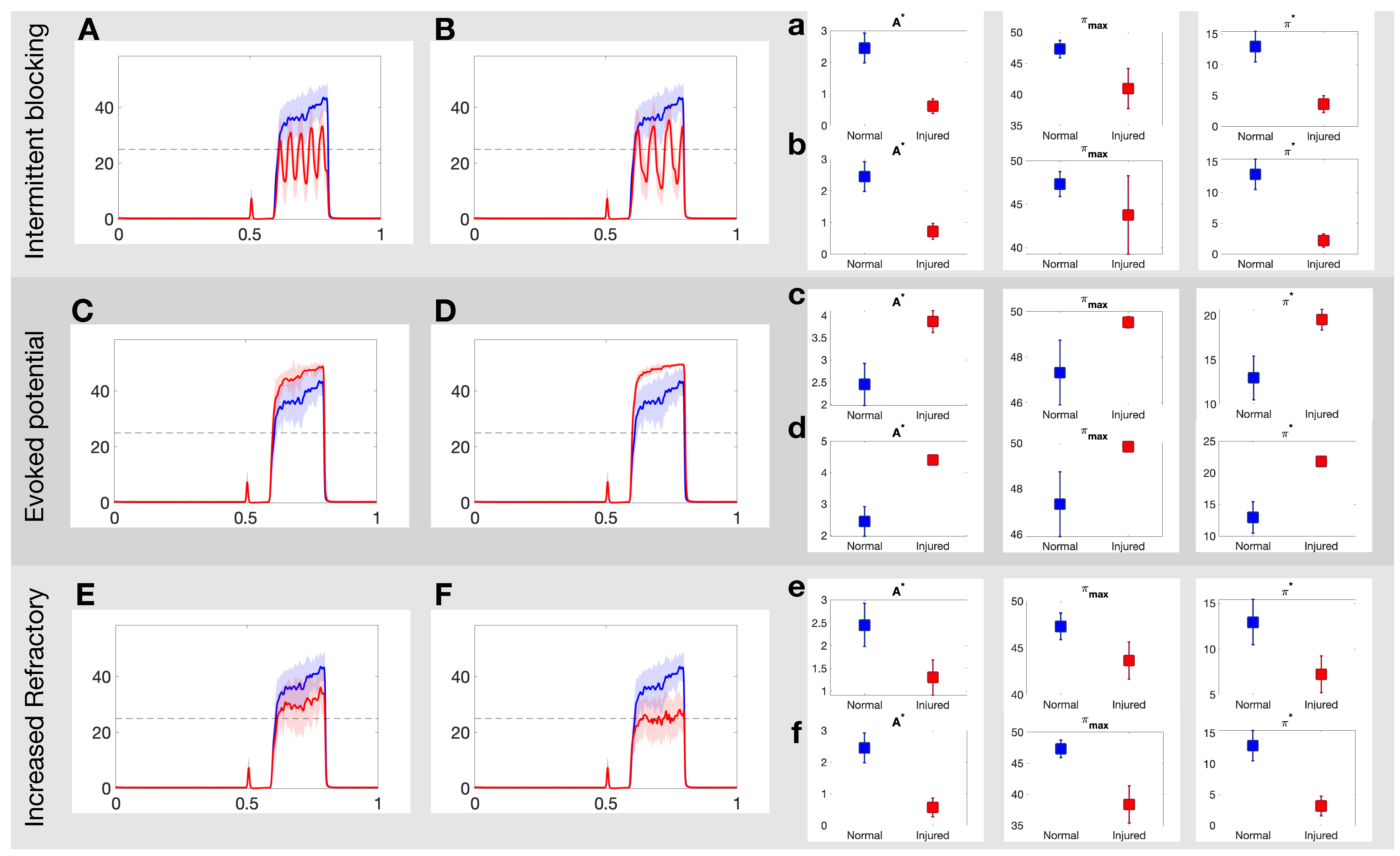
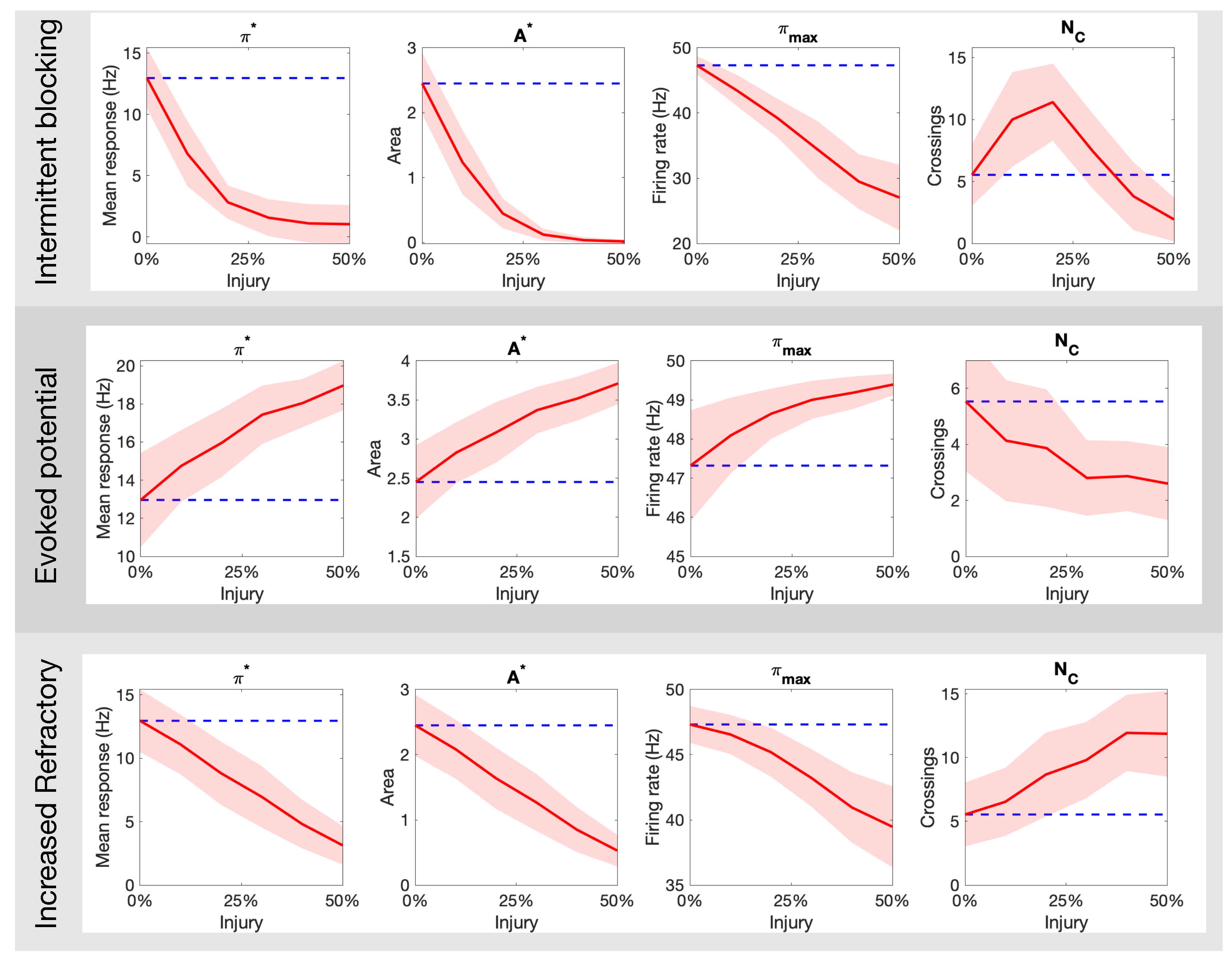
3.1. Effects of Different Injuries on C Fibers
3.2. Optimal Spike Delays Parameters for A Fibers
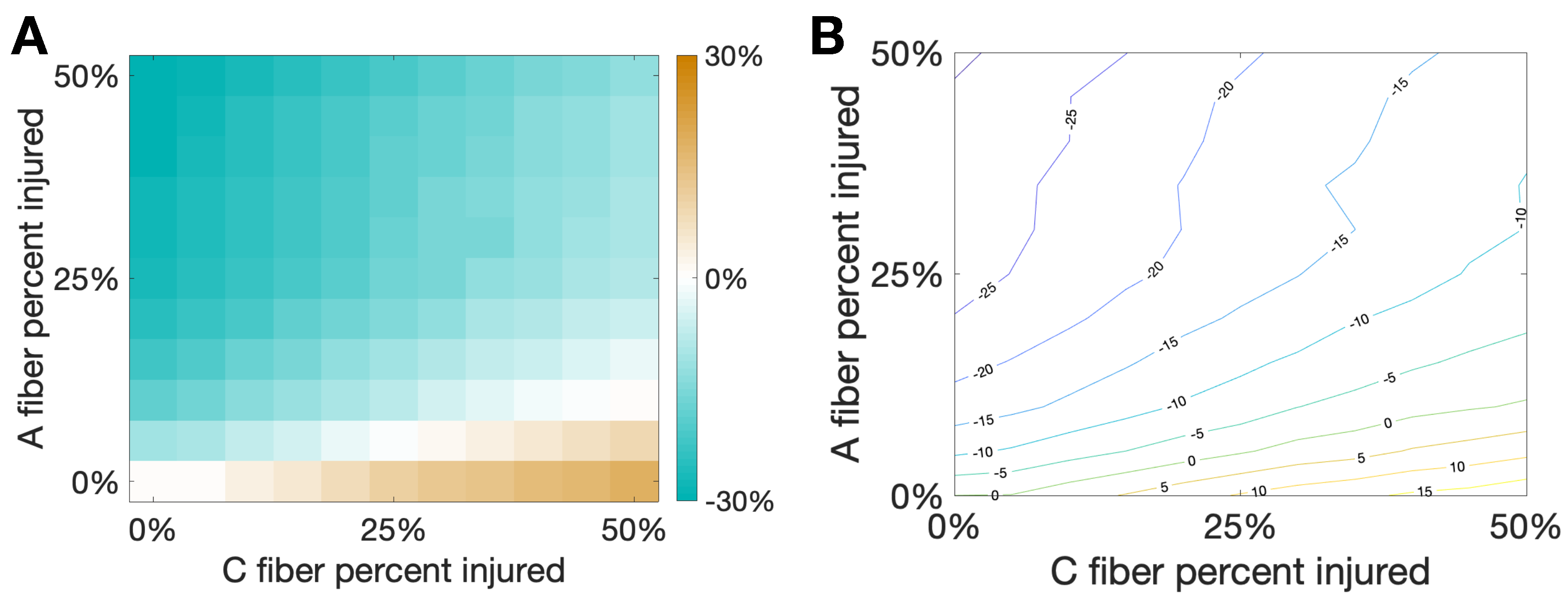
3.3. Damaging Both A and C Fibers
4. Discussion and Conclusions
4.1. Overview of Results
4.2. Connection to Neuropathic Pain
4.3. Limitations and Future Work
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DH | Dorsal Horn |
| FAS | Focal Axonal Swelling |
Appendix A. More Details on the Spinal-Cord Model
References
- Loeser, J.D.; Melzack, R. Pain: An overview. Lancet 1999, 353, 1607–1609. [Google Scholar] [CrossRef]
- Garland, E.L. Pain processing in the human nervous system: A selective review of nociceptive and biobehavioral pathways. Prim. Care. 2012, 39, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Millan, M.J. Descending control of pain. Prog. Neurobiol. 2002, 66, 355–474. [Google Scholar] [CrossRef]
- Todd, A.J. Neuronal circuitry for pain processing in the dorsal horn. Nat. Rev. Neurosci. 2010, 11, 823–836. [Google Scholar] [CrossRef]
- Zhang, T.C.; Janik, J.J.; Grill, W.M. Modeling effects of spinal cord stimulation on wide-dynamic range dorsal horn neurons: Influence of stimulation frequency and GABAergic inhibition. J. Neurophysiol. 2014, 112, 552–567. [Google Scholar] [CrossRef] [PubMed]
- Britton, N.F.; Skevington, S.M. A mathematical model of the gate control theory of pain. J. Theor. Biol. 1989, 137, 91–105. [Google Scholar] [CrossRef]
- Aguiar, P.; Sousa, M.; Lima, D. NMDA Channels Together with L-Type Calcium Currents and Calcium-Activated Nonspecific Cationic Currents Are Sufficient to Generate Windup in WDR Neurons. J. Neurophys. 2010, 104, 1155–1166. [Google Scholar] [CrossRef]
- Le Franc, Y.; Le Masson, G. Multiple firing patterns in deep dorsal horn neurons of the spinal cord: Computational analysis of mechanisms and functional implications. J. Neurophysiol. 2010, 104, 1978–1996. [Google Scholar] [CrossRef] [PubMed]
- Melzack, R.; Wall, P.D. Pain mechanisms: A new theory. Science 1965, 150, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Mendell, L.M. Constructing and deconstructing the gate theory of pain. Pain 2014, 155, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Moayedi, M.; Davis, K.D. Theories of pain: From specificity to gate control. J. Neurophysiol. 2013, 109, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Crodelle, J.A.; Piltz, S.H.; Hagenauer, M.H.; Booth, V. Investigating circadian rhythmicity in pain sensitivity using a neural circuit model for spinal cord processing of pain. In Women in Mathematical Biology; Springer: Cham, Switzerland, 2016. [Google Scholar]
- Crodelle, J.; Piltz, S.H.; Hagenauer, M.H.; Booth, V. Modeling the daily rhythm of human pain processing in the dorsal horn. PLoS Comput. Biol. 2019, 15, e1007106. [Google Scholar] [CrossRef]
- Maia, P.D.; Raj, A.; Kutz, J.N. Slow-gamma frequencies are optimally guarded against neurodegenerative diseases and traumatic brain injury: Consequences for neural encoding and working memory. J. Comp. Neurosci. 2019, 47, 1–16. [Google Scholar] [CrossRef]
- Maia, P.D.; Hemphill, M.A.; Zehnder, B.; Zhang, C.; Parker, K.K.; Kutz, J.N. Diagnostic tools for evaluating the impact of focal axonal swellings arising in neurodegenerative diseases and/or traumatic brain injury. J. Neurosci. Methods 2015, 253, 233–243. [Google Scholar] [CrossRef]
- Maia, P.D.; Kutz, J.N. Compromised axonal functionality after neurodegeneration, concussion and/or traumatic brain injury. J. Comp. Neurosci. 2014, 27, 317–332. [Google Scholar] [CrossRef]
- Maia, P.D.; Kutz, J.N. Identifying critical regions for spike propagation in axon segments. J. Comp. Neurosci. 2014, 36, 141–155. [Google Scholar] [CrossRef]
- Lusch, B.; Weholt, J.; Maia, P.D.; Kutz, J.N. Modeling cognitive deficits following neurodegenerative diseases and traumatic brain injuries with deep convolutional neural networks. Brain Cogn. 2018, 123, 154–164. [Google Scholar] [CrossRef]
- Weber, M.; Maia, P.D.; Kutz, J.N. Estimating memory deterioration rates following neurodegeneration and traumatic brain injuries in a Hopfield Network Model. Front. Neurosci. 2017, 11, 623. [Google Scholar]
- Morris, M.; Maia, P.D.; Kutz, J.N. Preventing neurodegenerative memory loss in Hopfield neuronal networks using cerebral organoids or external microelectronics. Comput. Math. Methods Med. 2017, 2017, 6102494. [Google Scholar]
- Maia, P.D.; Kutz, J.N. Reaction time impairments in decision-making networks as a diagnostic marker for traumatic brain injury and neurodegenerative diseases. J. Comp. Neurosci. 2017, 42, 323–347. [Google Scholar] [CrossRef]
- Kunert, J.; Maia, P.D.; Kutz, J.N. Functionality and robustness of injured connectomic dynamics in C. elegans: Linking behavioral deficits to neural circuit damage. PLoS Comp. Biol. 2017, 13, e1005261. [Google Scholar] [CrossRef] [PubMed]
- Rudy, S.; Maia, P.D.; Kutz, J.N. Cognitive and behavioral deficits arising from neurodegeneration and traumatic brain injury: A model for the underlying role of focal axonal swellings in neuronal networks with plasticity. J. Syst. Int. Neurosci. 2016. [Google Scholar] [CrossRef]
- Caro, X.J.; Galbraith, R.G.; Winter, E.F. Evidence of peripheral large nerve involvement in fibromyalgia: A retrospective review of EMG and nerve conduction findings in 55 FM subjects. Eur. J. Rheumatol. 2018, 5, 104–110. [Google Scholar] [CrossRef]
- Zimmermann, M. Pathobiology of neuropathic pain. Eur. J. Pharmacol. 2001, 429, 23–37. [Google Scholar] [CrossRef]
- Wilson, H.R.; Cowan, J.D. Excitatory and inhibitory interactions in localized populations of model neurons. Biophys. J. 1972, 12, 1–24. [Google Scholar] [CrossRef]
- Ermentrout, G.B.; Terman, D.H. Mathematical Foundations of Neuroscience; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Reeve, A.J.; Walker, K.; Urban, L.; Fox, A. Excitatory effects of galanin in the spinal cord of intact, anaesthetized rats. Neurosci. Lett. 2000, 295, 25–28. [Google Scholar] [CrossRef]
- Hulse, R.; Wynick, D.; Donaldson, L.F. Intact cutaneous C fibre afferent properties in mechanical and cold neuropathic allodynia. Eur. J. Pain 2010, 14, 565.e1–565.e10. [Google Scholar] [CrossRef]
- Peyronnard, J.M.; Charron, L.F.; Lavoie, J.; Messier, J.P. Motor, sympathetic and sensory innervation of rat skeletal muscles. Brain Res. 1986, 373, 288–302. [Google Scholar] [CrossRef]
- Le Bars, D.; Gozariu, M.; Cadden, S.W. Animal models of nociception. Pharmacol. Rev. 2001, 53, 597–652. [Google Scholar]
- Smith, K.J. Conduction properties of central demyelinated and remyelinated axons, and their relation to symptom production in demyelinating disorders. Eye 1994, 8, 224–237. [Google Scholar] [CrossRef]
- Gu, C. Rapid and reversible development of axonal varicosities: A new form of neural plasticity. Front. Mol. Neurosci. 2021, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Axonal pathology in traumatic brain injury. Exp. Neurol. 2013, 246, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic pain. Nat. Rev. Dis. Prim. 2017, 3, 17002. [Google Scholar] [CrossRef] [PubMed]
- Finnerup, N.B.; Haroutounian, S.; Kamerman, P.; Baron, R.; Bennett, D.L.; Bouhassira, D.; Cruccu, G.; Freeman, R.; Hansson, P.; Nurmikko, T.; et al. Neuropathic pain: An updated grading system for research and clinical practice. Pain 2016, 157, 1599–1606. [Google Scholar] [CrossRef]
- Serra, J.; Bostock, H.; Solà, R.; Aleu, J.; García, E.; Cokic, B.; Navarro, X.; Quiles, C. Microneurographic identification of spontaneous activity in C-nociceptors in neuropathic pain states in humans and rats. Pain 2012, 153, 42–55. [Google Scholar] [CrossRef]
- Kleggetveit, I.P.; Namer, B.; Schmidt, R.; Helås, T.; Rückel, M.; Ørstavik, K.; Schmelz, M.; Jørum, E. High spontaneous activity of C-nociceptors in painful polyneuropathy. Pain 2012, 153, 2040–2047. [Google Scholar] [CrossRef]
- Tesfaye, S.; Boulton, A.J.; Dickenson, A.H. Mechanisms and management of diabetic painful distal symmetrical polyneuropathy. Diabetes Care 2013, 36, 2456–2465. [Google Scholar] [CrossRef]
- Fields, H.L.; Rowbotham, M.; Baron, R. Postherpetic neuralgia: Irritable nociceptors and deafferentation. Neurobiol. Dis. 1998, 5, 209–227. [Google Scholar] [CrossRef] [PubMed]
- Woolf, C.J. Central sensitization: Implications for the diagnosis and treatment of pain. Pain 2011, 152, S2–S15. [Google Scholar] [CrossRef]
- Baron, R.; Hans, G.; Dickenson, A.H. Peripheral input and its importance for central sensitization. Ann. Neurol. 2013, 74, 630–636. [Google Scholar] [CrossRef]
- Stavros, K.; Simpson, D.M. Understanding the etiology and management of HIV-associated peripheral neuropathy. Curr. HIV/AIDS Rep. 2014, 11, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.; Dworkin, R.H.; Haroun, O.M.; Lockwood, D.N.; Rice, A.S. Acute and chronic pain associated with leprosy. Pain 2015, 156, 998–1002. [Google Scholar] [CrossRef] [PubMed]
- Duan, B.; Cheng, L.; Ma, Q. Spinal circuits transmitting mechanical pain and itch. Neurosci. Bull. 2018, 34, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Peirs, C.; Dallel, R.; Todd, A.J. Recent advances in our understanding of the organization of dorsal horn neuron populations and their contribution to cutaneous mechanical allodynia. J. Neural Transm. 2020, 127, 505–525. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crodelle, J.; Maia, P.D. A Computational Model for Pain Processing in the Dorsal Horn Following Axonal Damage to Receptor Fibers. Brain Sci. 2021, 11, 505. https://doi.org/10.3390/brainsci11040505
Crodelle J, Maia PD. A Computational Model for Pain Processing in the Dorsal Horn Following Axonal Damage to Receptor Fibers. Brain Sciences. 2021; 11(4):505. https://doi.org/10.3390/brainsci11040505
Chicago/Turabian StyleCrodelle, Jennifer, and Pedro D. Maia. 2021. "A Computational Model for Pain Processing in the Dorsal Horn Following Axonal Damage to Receptor Fibers" Brain Sciences 11, no. 4: 505. https://doi.org/10.3390/brainsci11040505
APA StyleCrodelle, J., & Maia, P. D. (2021). A Computational Model for Pain Processing in the Dorsal Horn Following Axonal Damage to Receptor Fibers. Brain Sciences, 11(4), 505. https://doi.org/10.3390/brainsci11040505