VEGF Upregulates EGFR Expression to Stimulate Chemotactic Behaviors in the rMC-1 Model of Müller Glia


Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Gene Expression via Reverse Transcription Quantitative Polymerase Chain Reaction (qPCR)
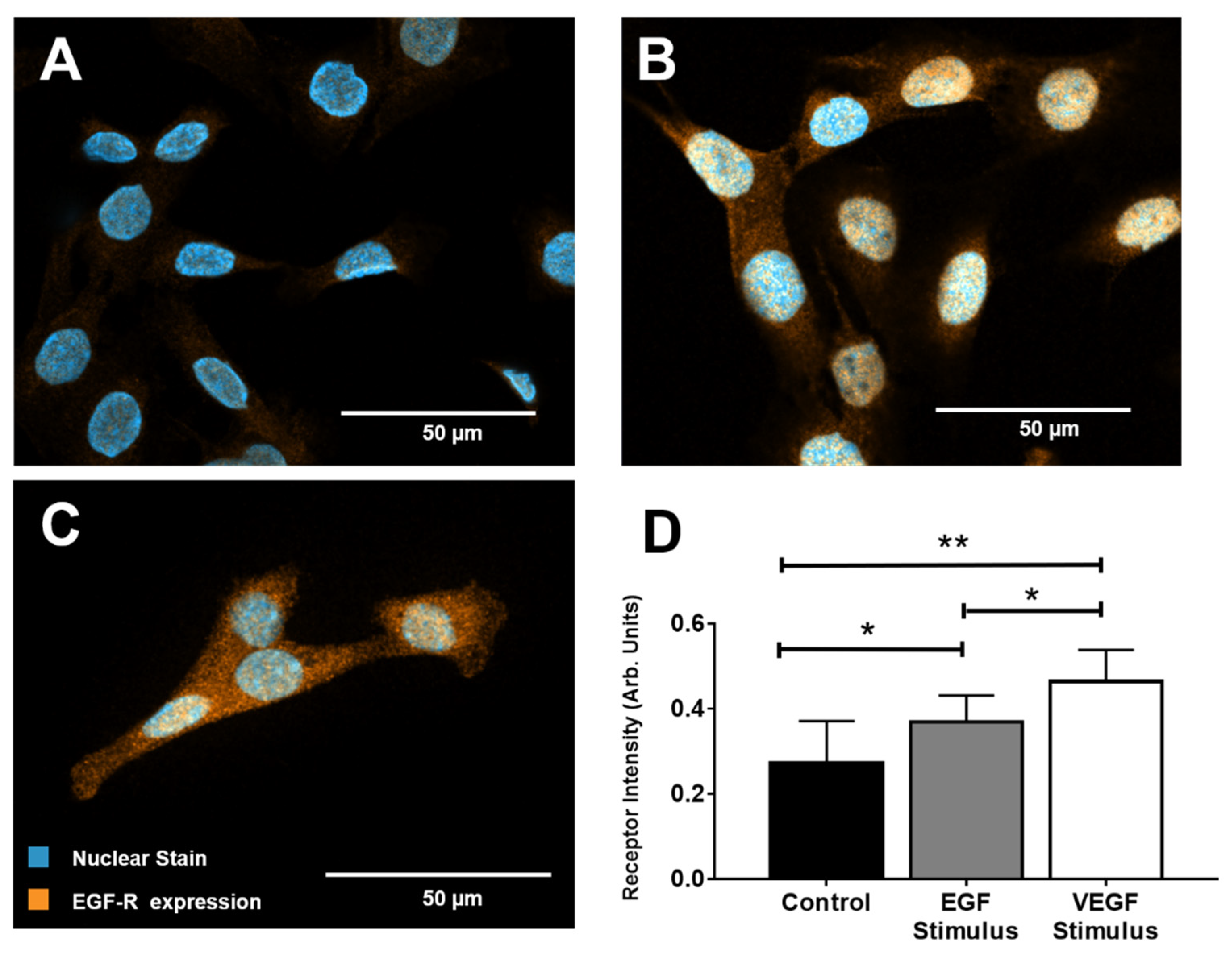
2.3. Immunocytochemical Staining (ICC)
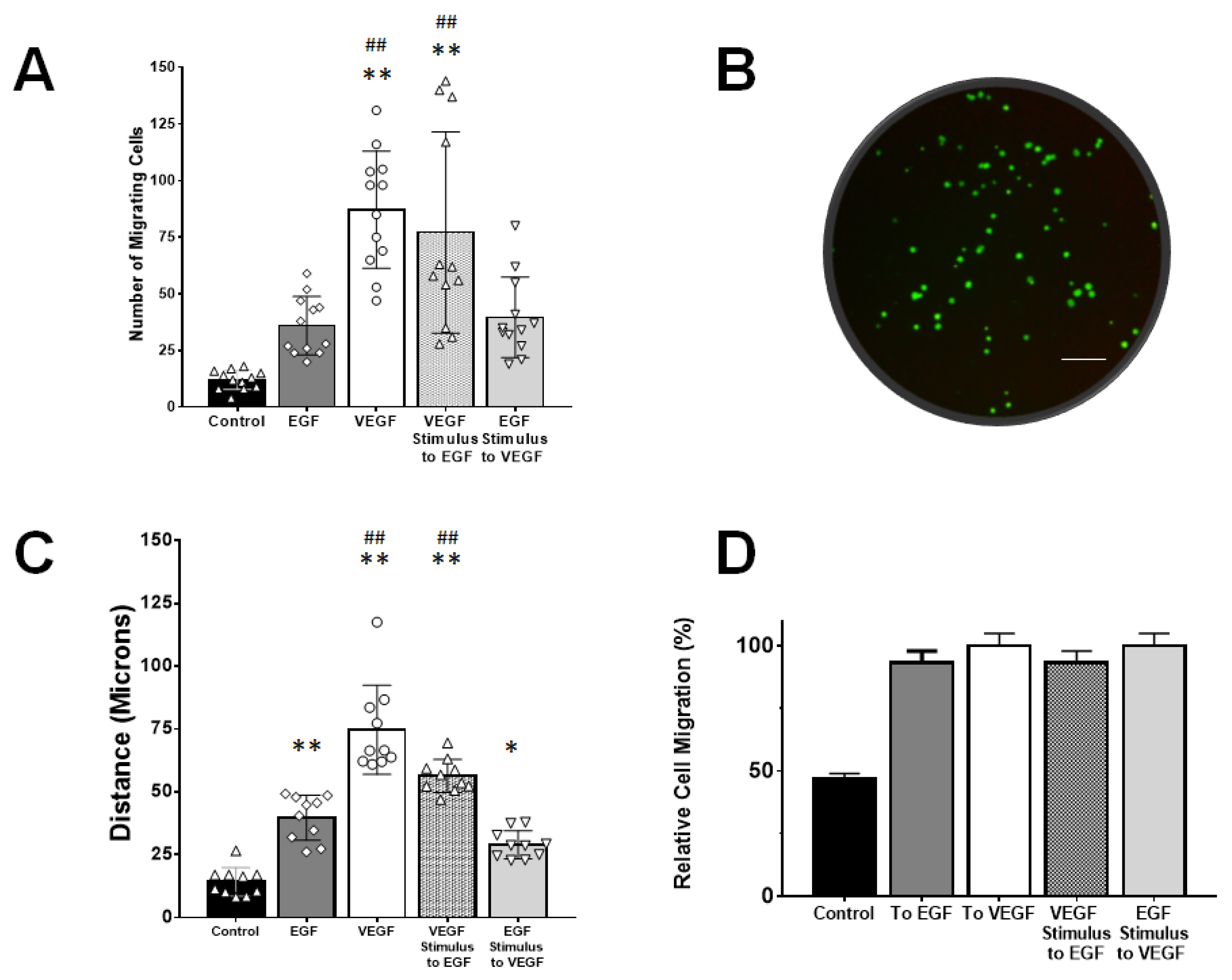
2.4. Transwell Assays
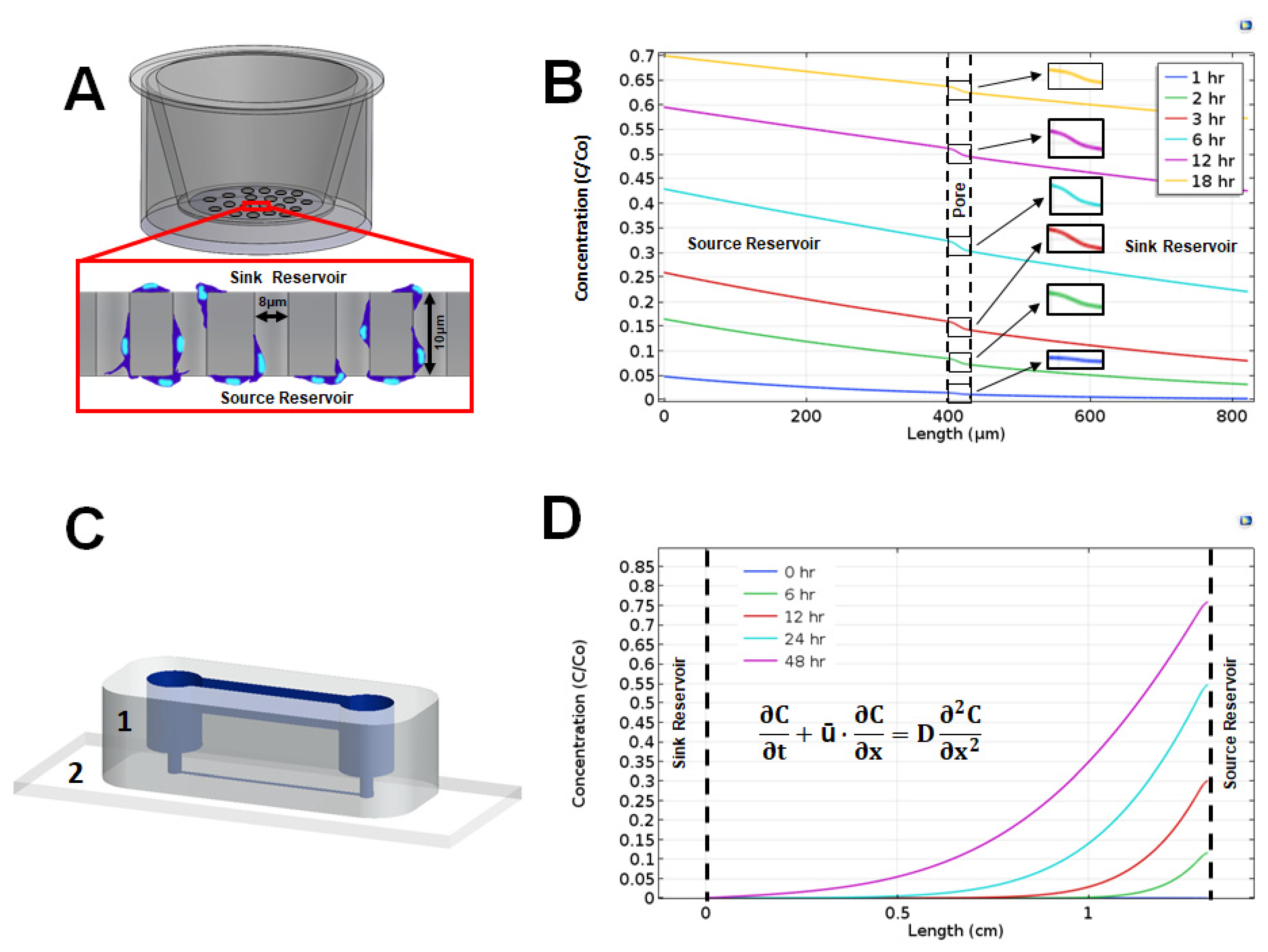
2.5. Overview of the gLL Microfluidic System
2.6. Measurement of Cell Migration in the gLL
2.7. Computational Modeling
2.8. Imaging and Software
2.9. Statistical Analysis
3. Results
3.1. Gene Expression via qPCR
3.2. EGF Receptor Expression
3.3. Extracellular Signaling Fields
3.4. rMC-1 Chemotactic Responses to Signaling from EGF and VEGF
4. Discussion
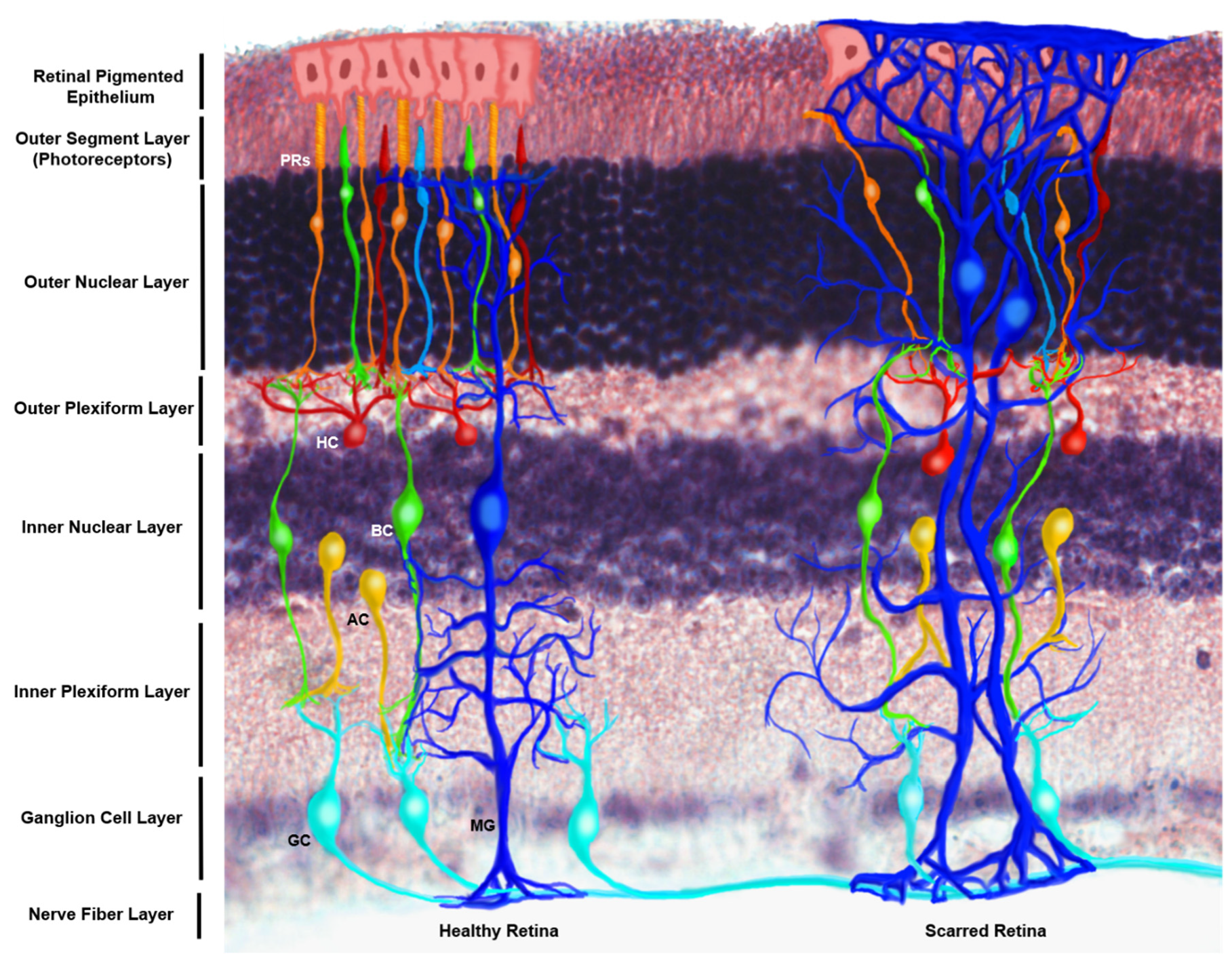
4.1. rMC-1 Cells as an In Vitro Model of Müller Glia Cells
4.2. VEGF-Targeted Therapies in Retina
4.3. Relative Receptor Expression
4.4. Müller Glia Migration Ability within the In Vivo Retina
4.5. Exogenous Signaling Fields of VEGF
4.6. VEGF-Augmented Chemotactic Responses to EGF Signaling
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Swenor, B.K.; Lee, M.J.; Varadaraj, V.; E Whitson, H.; Ramulu, P.Y. Aging With Vision Loss: A Framework for Assessing the Impact of Visual Impairment on Older Adults. Gerontolofgist 2019. [Google Scholar] [CrossRef]
- Peña, J.; Vazquez, M. Reducing health disparities in adult vision loss via interfaces with emerging technology. Eye 2018, 33, 532–533. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Uchino, M.; Sastry, S.M.; Schaumberg, D.A. Age-related macular degeneration and the incidence of cardiovascular disease: A systematic review and meta-analysis. PLoS ONE 2014, 9, e89600. [Google Scholar] [CrossRef]
- Seitz, R.; Ohlmann, A.; Tamm, E.R. The role of Müller glia and microglia in glaucoma. Cell Tissue Res. 2013, 353, 339–345. [Google Scholar] [CrossRef]
- McDowell, R.E.; Barabas, P.; Augustine, J.; Chevallier, O.; McCarron, P.; Chen, M.; McGeown, J.G.; Curtis, T.M. Müller glial dysfunction during diabetic retinopathy in rats is reduced by the acrolein-scavenging drug, 2-hydrazino-4,6-dimethylpyrimidine. Diabetologia 2018, 61, 2654–2667. [Google Scholar] [CrossRef]
- Wohl, S.G.; Jorstad, N.L.; Levine, E.M.; Reh, T.A. Müller glial microRNAs are required for the maintenance of glial homeostasis and retinal architecture. Nat. Commun. 2017, 8, 1603. [Google Scholar] [CrossRef]
- Sorrentino, F.S.; Allkabes, M.; Salsini, G.; Bonifazzi, C.; Perri, P. The importance of glial cells in the homeostasis of the retinal microenvironment and their pivotal role in the course of diabetic retinopathy. Life Sci. 2016, 162, 54–59. [Google Scholar] [CrossRef]
- Li, X.; Liu, J.; Hoh, J.; Liu, J. Müller cells in pathological retinal angiogenesis. Transl. Res. 2019, 207, 96–106. [Google Scholar] [CrossRef]
- Simó, R.; Villarroel, M.; Corraliza, L.; Hernández, C.; Garcia-Ramírez, M. The Retinal Pigment Epithelium: Something More than a Constituent of the Blood-Retinal Barrier—Implications for the Pathogenesis of Diabetic Retinopathy. J. Biomed. Biotechnol. 2010, 2010, 1–15. [Google Scholar] [CrossRef]
- Tzatzalos, E.; Smith, S.M.; Doh, S.T.; Hao, H.; Li, Y.; Wu, A.; Grumet, M.; Cai, L. A cis-element in the Notch1 locus is involved in the regulation of gene expression in interneuron progenitors. Dev. Boil. 2012, 372, 217–228. [Google Scholar] [CrossRef][Green Version]
- Huang, H.; Gandhi, J.K.; Zhong, X.; Wei, Y.; Gong, J.; Duh, E.J.; Vinores, S.A. TNFalpha is required for late BRB breakdown in diabetic retinopathy, and its inhibition prevents leukostasis and protects vessels and neurons from apoptosis. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1336–1344. [Google Scholar] [CrossRef] [PubMed]
- Simó, R.; Sundstrom, J.M.; Antonetti, D.A.; Simó, R. Ocular Anti-VEGF Therapy for Diabetic Retinopathy: The Role of VEGF in the Pathogenesis of Diabetic Retinopathy. Diabetes Care 2014, 37, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Nentwich, M.M.; Ulbig, M.W. Diabetic retinopathy-ocular complications of diabetes mellitus. World J. Diabetes 2015, 6, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; Munn, L. Leaky vessels? Call Ang1! Nat. Med. 2000, 6, 131–132. [Google Scholar] [CrossRef] [PubMed]
- Hermann, D.M.; Zechariah, A. implications of vascular endothelial growth factor for postischemic neurovascular remodeling. Br. J. Pharmacol. 2009, 29, 1620–1643. [Google Scholar] [CrossRef]
- Storkebaum, E.; Carmeliet, P. VEGF: A critical player in neurodegeneration. J. Clin. Investig. 2004, 113, 14–18. [Google Scholar] [CrossRef]
- Hayakawa, K.; Pham, L.-D.D.; Som, A.T.; Lee, B.J.; Guo, S.; Lo, E.H.; Arai, K. Vascular endothelial growth factor regulates the migration of oligodendrocyte precursor cells. J. Neurosci. 2011, 31, 10666–10670. [Google Scholar] [CrossRef]
- Bai, Y.; Ma, J.-X.; Guo, J.; Wang, J.; Zhu, M.; Chen, Y.; Le, Y.-Z. Müller cell-derived VEGF is a significant contributor to retinal neovascularization. J. Pathol. 2009, 219, 446–454. [Google Scholar] [CrossRef]
- Subirada, P.V.; Paz, M.C.; Ridano, M.E.; Lorenc, V.E.; Vaglienti, M.V.; Barcelona, P.F.; Luna, J.D.; Sánchez, M.C. A journey into the retina: Müller glia commanding survival and death. Eur. J. Neurosci. 2018, 47, 1429–1443. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, H.; Culp, D.; Yang, Z.; Fotheringham, L.; Flannery, J.G.; Hammond, S.; Kafri, T.; Hartnett, M.E. Targeting Müller cell–derived VEGF164 to reduce intravitreal neovascularization in the rat model of retinopathy of prematurity. Investig. Ophthalmol. Vis. Sci. 2014, 55, 824–831. [Google Scholar] [CrossRef]
- Le, Y.-Z. VEGF production and signaling in Müller glia are critical to modulating vascular function and neuronal integrity in diabetic retinopathy and hypoxic retinal vascular diseases. Vis. Res. 2017, 139, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Dong, S.; Zhu, M.; Le, Y.-Z. VEGF as a trophic factor for müller glia in hypoxic retinal diseases. Plant. Promot. Transcr. Factors 2018, 1074, 473–478. [Google Scholar] [CrossRef]
- Emerson, M.V.; Lauer, A.K.; Flaxel, C.; Wilson, D.; Francis, P.J.; Stout, J.T.; Emerson, G.G.; Schlesinger, T.K.; Nolte, S.K.; Klein, M.L. Intravitreal bevacizumab (avastin) treatment of neovascular age-related macular degeneration. Retina 2007, 27, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Schauwvlieghe, A.N.-S.M.; Dijkman, G.; Hooymans, J.M.; Verbraak, F.D.; Hoyng, C.B.; Dijkgraaf, M.G.W.; Peto, T.; Vingerling, J.R.; Schlingemann, R.O. Comparing the effectiveness of bevacizumab to ranibizumab in patients with exudative age-related macular degeneration. The BRAMD study. PLoS ONE 2016, 11, e0153052. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-Y.; Guo, X.-F.; Zhang, S.-D.; He, J.-N.; Sun, C.-Y.; Zou, Y.; Bi, H.-S.; Qu, Y. Comparison of bevacizumab and ranibizumab in age-related macular degeneration: A systematic review and meta-analysis. Int. J. Ophthalmol. 2014, 7, 355–364. [Google Scholar] [PubMed]
- Michels, S.; Rosenfeld, P.; Puliafito, C.; Marcus, E.; Venkatraman, A. systemic bevacizumab (avastin) therapy for neovascular age-related macular degenerationtwelve-week results of an uncontrolled open-label clinical study. Ophthalmology 2005, 112, 1035–1047.e9. [Google Scholar] [CrossRef] [PubMed]
- Pennell, N.A.; Lynch, T.J., Jr. Combined inhibition of the VEGFR and EGFR signaling pathways in the treatment of NSCLC. Oncologist 2009, 14, 399–411. [Google Scholar] [CrossRef]
- Liu, X.; Fan, D. The epithelial-mesenchymal transition and cancer stem cells: Functional and mechanistic links. Curr. Pharm. Des. 2015, 21, 1279–1291. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Inoue, Y.; Shimazawa, M.; Nakamura, S.; Takata, S.; Hashimoto, Y.; Izawa, H.; Masuda, T.; Tsuruma, K.; Sakaue, T.; Nakayama, H.; et al. Both autocrine signaling and paracrine signaling of HB-EGF enhance ocular neovascularization. Arter. Thromb. Vasc. Boil. 2018, 38, 174–185. [Google Scholar] [CrossRef]
- Wan, J.; Ramachandran, R.; Goldman, D. HB-EGF is necessary and sufficient for Müller glia dedifferentiation and retina regeneration. Dev. Cell 2012, 22, 334–347. [Google Scholar] [CrossRef] [PubMed]
- Peña, J.; Dülger, N.; Singh, T.; Zhou, J.; Majeska, R.; Redenti, S.; Vazquez, M. Controlled microenvironments to evaluate chemotactic properties of cultured Müller glia. Exp. Eye Res. 2018, 173, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Peña, J.S.; Robles, D.A.; Zhang, S.; Vazquez, M. A milled microdevice to advance glia-mediated therapies in the adult nervous system. Micromachines 2019, 10, 513. [Google Scholar] [CrossRef]
- Unachukwu, U.; Sauane, M.; Vazquez, M.; Redenti, S. Microfluidic generated EGF-gradients induce chemokinesis of transplantable retinal progenitor cells via the JAK/STAT and PI3Kinase signaling pathways. PLoS ONE 2013, 8, e83906. [Google Scholar] [CrossRef] [PubMed]
- De Hoz, R.; Rojas, B.; Ramírez, A.I.; Salazar, J.J.; Gallego, B.I.; Triviño, A.; Ramírez, J.M. Retinal macroglial responses in health and disease. BioMed Res. Int. 2016, 2016, 1–13. [Google Scholar] [CrossRef]
- Hammes, H.-P.; Federoff, H.J.; Brownlee, M. Nerve growth factor prevents both neuroretinal programmed cell death and capillary pathology in experimental diabetes. Mol. Med. 1995, 1, 527–534. [Google Scholar] [CrossRef]
- Pfeffer, B.A.; Xu, L.; Porter, N.A.; Rao, S.R.; Fliesler, S.J. Differential cytotoxic effects of 7-dehydrocholesterol-derived oxysterols on cultured retina-derived cells: Dependence on sterol structure, cell type, and density. Exp. Eye Res. 2016, 145, 297–316. [Google Scholar] [CrossRef]
- Sarthy, V.P.; Brodjian, S.J.; Dutt, K.; Kennedy, B.N.; French, R.P.; Crabb, J.W. Establishment and characterization of a retinal Müller cell line. Investig. Ophthalmol. Vis. Sci. 1998, 39. [Google Scholar]
- Otteson, D.; Phillips, M.J. A conditional immortalized mouse muller glial cell line expressing glial and retinal stem cell genes. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5991–6000. [Google Scholar] [CrossRef]
- Mishra, S.; Peña, J.; Redenti, S.; Vazquez, M. A novel electro-chemotactic approach to impact the directional migration of transplantable retinal progenitor cells. Exp. Eye Res. 2019, 185, 107688. [Google Scholar] [CrossRef]
- Thakur, A.; Mishra, S.; Peña, J.; Zhou, J.; Redenti, S.; Majeska, R.; Vazquez, M. Collective adhesion and displacement of retinal progenitor cells upon extracellular matrix substrates of transplantable biomaterials. J. Tissue Eng. 2018, 9. [Google Scholar] [CrossRef]
- George, A.; Truskey, F.Y.; David, F.K. Transport. Phenomena in Biological Ssystems; Pearson: London, UK, 2004. [Google Scholar]
- McCutcheon, S.; Unachukwu, U.; Thakur, A.; Majeska, R.; Redenti, S.; Vazquez, M. In Vitro formation of neuroclusters in microfluidic devices and cell migration as a function of stromal-derived growth factor 1 gradients. Cell Adhes. Migr. 2016, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pena, C.D.; Zhang, S.; Majeska, R.J.; Venkatesh, T.; Vazquez, M. Invertebrate retinal progenitors as regenerative models in a microfluidic system. Cells 2019, 8, 1301. [Google Scholar] [CrossRef] [PubMed]
- Pena, C.D.; Zhang, S.; Markey, M.; Venkatesh, T.; Vazquez, M. Collective behaviors of Drosophila-derived retinal progenitors in controlled microenvironments. PLoS ONE 2019, 14, e0226250. [Google Scholar] [CrossRef] [PubMed]
- Kong, Q.; Able, R.A.; Dudu, V.; Vazquez, M. A Microfluidic Device to establish concentration gradients using reagent density differences. J. Biomech. Eng. 2010, 132, 121012. [Google Scholar] [CrossRef]
- Venturoli, D.; Rippe, B. Ficoll and dextran vs. globular proteins as probes for testing glomerular permselectivity: Effects of molecular size, shape, charge, and deformability. Am. J. Physiol. Physiol. 2005, 288, F605–F613. [Google Scholar] [CrossRef]
- Kholodenko, A.L.; Douglas, J.F. Generalized Stokes-Einstein equation for spherical particle suspensions. Phys. Rev. E 1995, 51, 1081–1090. [Google Scholar] [CrossRef]
- Azzarelli, R.; Oleari, R.; Lettieri, A.; André, V.; Cariboni, A. In Vitro, Ex Vivo and In Vivo techniques to study neuronal migration in the developing cerebral cortex. Brain Sci. 2017, 7, 48. [Google Scholar] [CrossRef]
- Stone, N.L.; England, T.J.; O’Sullivan, S.E. A novel transwell blood brain barrier model using primary human cells. Front. Cell. Neurosci. 2019, 13, 230. [Google Scholar] [CrossRef]
- Thompson, K.; Chen, J.; Luo, Q.; Xiao, Y.; Cummins, T.R.; Bhatwadekar, A. Advanced glycation end (AGE) product modification of laminin downregulates Kir4.1 in retinal Müller cells. PLoS ONE 2018, 13, e0193280. [Google Scholar] [CrossRef]
- Zhou, P.; Sun, G.; Meng, X.; Zhai, Y.; Dong, X.; Zhang, X.; Sun, G.; Sun, X. Notoginsenoside R1 ameliorates diabetic retinopathy through PINK1-dependent activation of mitophagy. Cells 2019, 8, 213. [Google Scholar] [CrossRef]
- Kittipassorn, T.; Haydinger, C.D.; Wood, J.P.M.; Mammone, T.; Casson, R.J.; Peet, D. Characterization of the novel spontaneously immortalized rat Müller cell line SIRMu-1. Exp. Eye Res. 2019, 181, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Roque, R.S.; Agarwal, N.; Wordinger, R.J.; Brun, A.-M.; Xue, Y.; Huang, L.C.; Nguyen, L.P.; Shay, J.W. Human Papillomavirus-16 E6/E7 transfected retinal cell line expresses the Müller cell phenotype. Exp. Eye Res. 1997, 64, 519–527. [Google Scholar] [CrossRef]
- Tien, T.; Zhang, J.; Muto, T.; Kim, D.; Sarthy, V.P.; Roy, S. High glucose induces mitochondrial dysfunction in retinal müller cells: Implications for diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 2915–2921. [Google Scholar] [CrossRef]
- Weddell, J.C.; Chen, S.; Imoukhuede, P.I. VEGFR1 promotes cell migration and proliferation through PLCγ and PI3K pathways. NPJ Syst. Boil. Appl. 2017, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yamauchi, M.; Muramatsu, M.; Osawa, T.; Tsuchida, R.; Shibuya, M. RACK1 regulates VEGF/Flt1-mediated cell migration via activation of a PI3K/Akt Pathway*. J. Boil. Chem. 2011, 286, 9097–9106. [Google Scholar] [CrossRef]
- Tremolada, G.; Del Turco, C.; Lattanzio, R.; Maestroni, S.; Maestroni, A.; Bandello, F.; Zerbini, G. The role of angiogenesis in the development of proliferative diabetic retinopathy: Impact of intravitreal anti-VEGF treatment. Exp. Diabetes Res. 2012, 2012, 1–8. [Google Scholar] [CrossRef]
- Gerhardinger, C.; Brown, L.F.; Roy, S.; Mizutani, M.; Zucker, C.L.; Lorenzi, M. Expression of vascular endothelial growth factor in the human retina and in nonproliferative diabetic retinopathy. Am. J. Pathol. 1998, 152, 1453–1462. [Google Scholar]
- Vecino, E.; Rodriguez, F.; Ruzafa, N.; Pereiro, X.; Sharma, S.C. Glia–neuron interactions in the mammalian retina. Prog. Retin. Eye Res. 2016, 51, 1–40. [Google Scholar] [CrossRef]
- Lewis, G.P.; Chapin, E.A.; Luna, G.; Linberg, K.A.; Fisher, S.K. The fate of Müller’s glia following experimental retinal detachment: Nuclear migration, cell division, and subretinal glial scar formation. Mol. Vis. 2010, 16, 1361–1372. [Google Scholar]
- Guo, B.; Wang, Y.; Hui, Y.; Yang, X.; Fan, Q. Effects of anti-VEGF agents on rat retinal Müller glial cells. Mol. Vis. 2010, 16, 793–799. [Google Scholar] [PubMed]
- Becker, S.; Wang, H.; Simmons, A.B.; Suwanmanee, T.; Stoddard, G.J.; Kafri, T.; Hartnett, M.E. Targeted Knockdown of Overexpressed VEGFA or VEGF164 in Müller cells maintains retinal function by triggering different signaling mechanisms. Sci. Rep. 2018, 8, 2003. [Google Scholar] [CrossRef] [PubMed]
- Perrotte, P.; Matsumoto, T.; Inoue, K.; Kuniyasu, H.; Eve, B.Y.; Hicklin, D.J.; Radinsky, R.; Dinney, C.P. Anti-epidermal growth factor receptor antibody C225 inhibits angiogenesis in human transitional cell carcinoma growing orthotopically in nude mice. Clin. Cancer Res. 1999, 5, 257–265. [Google Scholar] [PubMed]
- Grugel, S.; Finkenzeller, G.N.; Weindel, K.; Barleon, B.; Marmé, D. Both v-Ha-Ras and v-Raf stimulate expression of the vascular endothelial growth factor in NIH 3T3 Cells. J. Boil. Chem. 1995, 270, 25915–25919. [Google Scholar] [CrossRef]
- Mothe-Satney, I.; Ballotti, R.; Tartare-Deckert, S.; Kowalski-Chauvel, A.; Van Obberghen, E. Cross talk among tyrosine kinase receptors in PC12 cells: Desensitization of mitogenic epidermal growth factor receptors by the neurotrophic factors, nerve growth factor and basic fibroblast growth factor. Mol. Boil. Cell 1993, 4, 737–746. [Google Scholar] [CrossRef]
- Ye, X.; Ren, H.; Zhang, M.; Sun, Z.; Jiang, A.C.; Xu, G. ERK1/2 signaling pathway in the release of VEGF from Müller cells in diabetes. Investig. Ophthalmol. Vis. Sci. 2012, 53, 3481–3489. [Google Scholar] [CrossRef]
- Ye, X.; Xu, G.; Chang, Q.; Fan, J.; Sun, Z.; Qin, Y.; Jiang, A.C. ERK1/2 signaling pathways involved in VEGF release in diabetic rat retina. Invest. Ophthalmol. Vis. Sci. 2010, 51, 5226–5233. [Google Scholar] [CrossRef]
- Goldman, D. Müller glial cell reprogramming and retina regeneration. Nat. Rev. Neurosci. 2014, 15, 431–442. [Google Scholar] [CrossRef]
- Dyer, M.A.; Cepko, C.L. Control of Müller glial cell proliferation and activation following retinal injury. Nat. Neurosci. 2000, 3, 873–880. [Google Scholar] [CrossRef]
- Bejarano-Escobar, R.; Sánchez-Calderón, H.; Otero-Arenas, J.; Martín-Partido, G.; Francisco-Morcillo, J. Müller glia and phagocytosis of cell debris in retinal tissue. J. Anat. 2017, 231, 471–483. [Google Scholar] [CrossRef]
- Takeda, M.; Takamiya, A.; Jiao, J.; Cho, K.-S.; Trevino, S.G.; Matsuda, T.; Chen, N.F. Alpha-Aminoadipate induces progenitor cell properties of Müller glia in adult mice. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1142–1150. [Google Scholar] [CrossRef] [PubMed]
- Tackenberg, M.A.; Tucker, B.A.; Swift, J.S.; Jiang, C.; Redenti, S.; Greenberg, K.P.; Flannery, J.G.; Reichenbach, A.; Young, M.J. Müller cell activation, proliferation and migration following laser injury. Mol. Vis. 2009, 15, 1886–1896. [Google Scholar] [PubMed]
- Ciasca, G.; Pagliei, V.; Minelli, E.; Palermo, F.; Nardini, M.; Pastore, V.; Papi, M.; Caporossi, A.; De Spirito, M.; Minnella, A.M. Nanomechanical mapping helps explain differences in outcomes of eye microsurgery: A comparative study of macular pathologies. PLoS ONE 2019, 14, e0220571. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.J.; Otteson, D. Differential expression of neuronal genes in Müller glia in two- and three-dimensional cultures. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1439–1449. [Google Scholar] [CrossRef][Green Version]
- Zhang, S.; Zhang, S.; Gong, W.; Zhu, G.; Wang, S.; Wang, Y.; Halim, M.; Wang, K.; Zhou, G.; Liu, Q. Müller cell regulated microglial activation and migration in rats with N-Methyl-N-Nitrosourea-induced retinal degeneration. Front. Mol. Neurosci. 2018, 12, 890. [Google Scholar] [CrossRef]
- Ferreira, M.M.; Dewi, R.E.; Heilshorn, S.C. Microfluidic analysis of extracellular matrix-bFGF crosstalk on primary human myoblast chemoproliferation, chemokinesis, and chemotaxis. Integr. Boil. 2015, 7, 569–579. [Google Scholar] [CrossRef]
- Shamloo, A.; Ma, N.; Poo, M.-M.; Sohn, L.L.; Heilshorn, S.C. Endothelial cell polarization and chemotaxis in a microfluidic device. Lab. Chip 2008, 8, 1292. [Google Scholar] [CrossRef]
- Xu, H.; Heilshorn, S.C. Microfluidic investigation of BDNF-enhanced neural stem cell chemotaxis in CXCL12 gradients. Small 2012, 9, 585–595. [Google Scholar] [CrossRef]
- Bhattacharjee, N.; Folch, A. Large-scale microfluidic gradient arrays reveal axon guidance behaviors in hippocampal neurons. Microsyst. Nanoeng. 2017, 3, 17003. [Google Scholar] [CrossRef]
- Dodson, K.H.; Echevarria, F.; Li, D.; Sappington, R.M.; Edd, F.J. Retina-on-a-chip: A microfluidic platform for point access signaling studies. Biomed. Microdevices 2015, 17, 114. [Google Scholar] [CrossRef]
- Penn, J.S.; Madan, A.; Caldwell, R.; Bartoli, M.; Hartnett, M.; Caldwell, R. Vascular endothelial growth factor in eye disease. Prog. Retin. Eye Res. 2008, 27, 331–371. [Google Scholar] [CrossRef] [PubMed]
- DeMaio, L.; Antonetti, D.A.; Scaduto, R.C.; Gardner, T.W.; Tarbell, J.M. VEGF increases paracellular transport without altering the solvent-drag reflection coefficient. Microvasc. Res. 2004, 68, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, X.; Elliott, M.H.; Zhu, M.; Le, Y.-Z. Müller cell-derived VEGF is essential for diabetes-induced retinal inflammation and vascular leakage. Diabetes 2010, 59, 2297–2305. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence (5′–3′) | M.W.(bp) |
|---|---|---|
| FGF2 | F: GAACCGGTACCTGGCTATGA | 182 |
| R: CCGTTTTGGATCCGAGTTTA | ||
| NRP-1 | F: GCTACCCTCATTCTTACCATCC | 119 |
| R: GCAGTCTCTGTCCTCCAAATC | ||
| FGFR3 | F: CTGTATGTGCTGGTGGAGTATG | 98 |
| R: CTGCAGGCATCAAAGGAGTAA | ||
| EGFR | F: GCTGTGCGATTTAGCAACAA | 146 |
| R: GGACAGCTCGGATCACATTT | ||
| Nestin | F: CAGTACTCGGAATGCAGCAA | 98 |
| R: CTTCTGTGTCCAGACCACTTT | ||
| GFAP | F: CACCCTGCATCTCCAACTAAC | 109 |
| R: GGAAGAAAGAGGAAAGACAGGG | ||
| GAPDH | F: ACTCCCATTCTTCCACCTTTG | 105 |
| R: CCCTGTTGCTGTAGCCATATT |
| EGF-R | FGFR-2 | FGFR-8 | VEGF-R | |
|---|---|---|---|---|
| EGF | 2.2 | 2.5 | 0.7 | 0.2 |
| FGF2 | 2.7 | 1.3 | 0.9 | 0.7 |
| FGF8 | 9.3 | 2 | 1.3 | 0.4 |
| VEGF | 18.9 | 5.2 | 6 | 2.8 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peña, J.S.; Vazquez, M. VEGF Upregulates EGFR Expression to Stimulate Chemotactic Behaviors in the rMC-1 Model of Müller Glia. Brain Sci. 2020, 10, 330. https://doi.org/10.3390/brainsci10060330
Peña JS, Vazquez M. VEGF Upregulates EGFR Expression to Stimulate Chemotactic Behaviors in the rMC-1 Model of Müller Glia. Brain Sciences. 2020; 10(6):330. https://doi.org/10.3390/brainsci10060330
Chicago/Turabian StylePeña, Juan S., and Maribel Vazquez. 2020. "VEGF Upregulates EGFR Expression to Stimulate Chemotactic Behaviors in the rMC-1 Model of Müller Glia" Brain Sciences 10, no. 6: 330. https://doi.org/10.3390/brainsci10060330
APA StylePeña, J. S., & Vazquez, M. (2020). VEGF Upregulates EGFR Expression to Stimulate Chemotactic Behaviors in the rMC-1 Model of Müller Glia. Brain Sciences, 10(6), 330. https://doi.org/10.3390/brainsci10060330