hATTR Pathology: Nerve Biopsy Results from Italian Referral Centers

 ,
,  , , , ,
, , , ,  , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Nerve Biopsy
2.3. Statistical Analysis
2.4. Ethics
3. Results
3.1. Clinical Results
3.2. Pathological Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Plante-Bordeneuve, V.; Said, G. Familial amyloid polyneuropathy. Lancet Neurol. 2011, 10, 1086–1097. [Google Scholar] [CrossRef]
- Ruberg, F.L.; Berk, J.L. Transthyretin (TTR) cardiac amyloidosis. Circulation 2012, 126, 1286–1300. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Kincaid, J.C. The molecular biology and clinical features of amyloid neuropathy. Muscle Nerve 2007, 36, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Luigetti, M.; Conte, A.; Del Grande, A.; Bisogni, G.; Madia, F.; Lo Monaco, M.; Laurenti, L.; Obici, L.; Merlini, G.; Sabatelli, M. TTR-related amyloid neuropathy: Clinical, electrophysiological and pathological findings in 15 unrelated patients. Neurol. Sci. 2013, 34, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Mazzeo, A.; Stancanelli, C.; Di Leo, R.; Gentile, L.; Di Bella, G.; Minutoli, F.; Baldari, S.; Vita, G. Transthyretin-related familial amyloidotic polyneuropathy: Description of a cohort of patients with Leu64 mutation and late onset. J. Peripher. Nerv. Syst. 2012, 17, 385–390. [Google Scholar] [CrossRef]
- Russo, M.; Obici, L.; Bartolomei, I.; Cappelli, F.; Luigetti, M.; Fenu, S.; Cavallaro, T.; Chiappini, M.G.; Gemelli, C.; Pradotto, L.G.; et al. ATTRv amyloidosis Italian Registry: Clinical and epidemiological data. Amyloid 2020, 1–7. [Google Scholar] [CrossRef]
- Koike, H.; Misu, K.; Ikeda, S.; Ando, Y.; Nakazato, M.; Ando, E.; Yamamoto, M.; Hattori, N.; Sobue, G. Study Group for Hereditary Neuropathy in, J. Type I (transthyretin Met30) familial amyloid polyneuropathy in Japan: Early- vs late-onset form. Arch. Neurol. 2002, 59, 1771–1776. [Google Scholar] [CrossRef]
- Adams, D.; Ando, Y.; Beirao, J.M.; Coelho, T.; Gertz, M.A.; Gillmore, J.D.; Hawkins, P.N.; Lousada, I.; Suhr, O.B.; Merlini, G. Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J. Neurol. 2020, 1–14. [Google Scholar] [CrossRef]
- Westermark, P. Diagnosing amyloidosis. Scand. J. Rheumatol. 1995, 24, 327–329. [Google Scholar] [CrossRef]
- Leung, N.; Nasr, S.H.; Sethi, S. How I treat amyloidosis: The importance of accurate diagnosis and amyloid typing. Blood 2012, 120, 3206–3213. [Google Scholar] [CrossRef]
- Planté-Bordeneuve, V.; Ferreira, A.; Lalu, T.; Zaros, C.; Lacroix, C.; Adams, D.; Said, G. Diagnostic pitfalls in sporadic transthyretin familial amyloid polyneuropathy (TTR-FAP). Neurology 2007, 69, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Luigetti, M.; Romano, A.; Di Paolantonio, A.; Bisogni, G.; Sabatelli, M. Diagnosis and Treatment of Hereditary Transthyretin Amyloidosis (hATTR) Polyneuropathy: Current Perspectives on Improving Patient Care. Ther. Clin. Risk Manag. 2020, 16, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Do Amaral, B.; Coelho, T.; Sousa, A.; Guimarães, A. Usefulness of labial salivary gland biopsy in familial amyloid polyneuropathy Portuguese type. Amyloid 2009, 16, 232–238. [Google Scholar] [CrossRef] [PubMed]
- De Paula, F.; Teshima, T.H.N.; Hsieh, R.; Souza, M.M.; Nico, M.M.S.; Lourenco, S.V. Overview of Human Salivary Glands: Highlights of Morphology and Developing Processes. Anat. Rec. (Hoboken) 2017, 300, 1180–1188. [Google Scholar] [CrossRef]
- Cappellari, M.; Cavallaro, T.; Ferrarini, M.; Cabrini, I.; Taioli, F.; Ferrari, S.; Merlini, G.; Obici, L.; Briani, C.; Fabrizi, G.M. Variable presentations of TTR-related familial amyloid polyneuropathy in seventeen patients. J. Peripher. Nerv. Syst. 2011, 16, 119–129. [Google Scholar] [CrossRef]
- Koike, H.; Hashimoto, R.; Tomita, M.; Kawagashira, Y.; Iijima, M.; Tanaka, F.; Sobue, G. Diagnosis of sporadic transthyretin Val30Met familial amyloid polyneuropathy: A practical analysis. Amyloid 2011, 18, 53–62. [Google Scholar] [CrossRef]
- Mazzeo, A.; Russo, M.; Di Bella, G.; Minutoli, F.; Stancenelli, C.; Gentile, L.; Toscano, A.; Vita, G. TTR-FAP: A single-center experience in Sicily, an Italian endemic area. Orphanet J. Rare Dis. 2015, 10, O1. [Google Scholar] [CrossRef]
- Živkovic, S.A.; Mnatsakanova, D.; Lacomis, D. Phenotypes of Late-Onset Transthyretin Amyloid Neuropathy: A Diagnostic Challenge. J. Clin. Neuromuscul. Dis. 2019, 21, 1–6. [Google Scholar] [CrossRef]
- Suhr, O.; Danielsson, A.; Holmgren, G.; Steen, L. Malnutrition and gastrointestinal dysfunction as prognostic factors for survival in familial amyloidotic polyneuropathy. J. Intern. Med. 1994, 235, 479–485. [Google Scholar] [CrossRef]
- Luigetti, M.; Di Paolantonio, A.; Bisogni, G.; Romano, A.; Conte, A.; Barbato, F.; Del Grande, A.; Madia, F.; Rossini, P.M.; Lauretti, L.; et al. Sural nerve biopsy in peripheral neuropathies: 30-year experience from a single center. Neurol. Sci. 2020, 41, 341–346. [Google Scholar] [CrossRef]
- Cortese, A.; Vegezzi, E.; Lozza, A.; Alfonsi, E.; Montini, A.; Moglia, A.; Merlini, G.; Obici, L. Diagnostic challenges in hereditary transthyretin amyloidosis with polyneuropathy: Avoiding misdiagnosis of a treatable hereditary neuropathy. J. Neurol. Neurosurg. Psychiatry 2017, 88, 457–458. [Google Scholar] [CrossRef]
- Behse, F.; Buchthal, F.; Carlsen, F. Nerve biopsy and conduction studies in diabetic neuropathy. J. Neurol. Neurosurg Psychiatry 1977, 40, 1072–1082. [Google Scholar] [CrossRef]
- Dyck, P.J.; Lais, A.; Karnes, J.L.; O’Brien, P.; Rizza, R. Fiber loss is primary and multifocal in sural nerves in diabetic polyneuropathy. Ann. Neurol. 1986, 19, 425–439. [Google Scholar] [CrossRef]
- Murakami, T.; Sunada, Y. Transthyretin Amyloid Neuropathy: The Schwann Cell Hypothesis. Adv. Exp. Med. Biol. 2019, 1190, 371–378. [Google Scholar] [CrossRef]
- Koike, H.; Nishi, R.; Ikeda, S.; Kawagashira, Y.; Iijima, M.; Sakurai, T.; Shimohata, T.; Katsuno, M.; Sobue, G. The morphology of amyloid fibrils and their impact on tissue damage in hereditary transthyretin amyloidosis: An ultrastructural study. J. Neurol. Sci. 2018, 394, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.L.; Gandhi, S. Crucial role of protein oligomerization in the pathogenesis of Alzheimer’s and Parkinson’s diseases. FEBS J. 2018, 285, 3631–3644. [Google Scholar] [CrossRef] [PubMed]
- European Federation of Neurological Societies/Peripheral Nerve Society Guideline on Management of Multifocal Motor Neuropathy. Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society—First revision. J. Peripher. Nerv. Syst. 2010, 15, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Said, G.; Planté-Bordeneuve, V. Familial amyloid polyneuropathy: A clinico-pathologic study. J Neurol Sci. 2009, 284, 149–154. [Google Scholar] [CrossRef]
- Gabriel, C.M.; Gregson, N.A.; Wood, N.W.; Hughes, R.A. Immunological study of hereditary motor and sensory neuropathy type 1a (HMSN1a). J. Neurol. Neurosurg. Psychiatry 2002, 72, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, L.; Malik, O.; Kenton, A.R.; Sharp, D.; Muddle, J.R.; Davis, M.B.; Winer, J.B.; Orrell, R.W.; King, R.H.M. Coexistent hereditary and inflammatory neuropathy. Brain 2004, 127, 193–202. [Google Scholar] [CrossRef]
- Duchesne, M.; Mathis, S.; Richard, L.; Magdelaine, C.; Corcia, P.; Nouioua, S.; Tazir, M.; Magy, L.; Vallat, J.-M. Nerve Biopsy Is Still Useful in Some Inherited Neuropathies. J. Neuropathol. Exp. Neurol. 2017, 77, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.; Coelho, T.; Rodrigues, A.; Felgueiras, H.; Oliveira, P.; Guimarães, A.; Melo-Pires, M.; Taipa, R. Clinicopathological correlations of sural nerve biopsies in TTR Val30Met familial amyloid polyneuropathy. Brain Commun. 2019, 1. [Google Scholar] [CrossRef] [PubMed]
- Luigetti, M.; Bisogni, G.; Romano, A.; Di Paolantonio, A.; Barbato, F.; Primicerio, G.; Rossini, P.M.; Servidei, S.; Sabatelli, M. Sudoscan in the evaluation and follow-up of patients and carriers with TTR mutations: Experience from an Italian Centre. Amyloid 2018, 25, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Ebenezer, G.J.; Liu, Y.; Judge, D.P.; Cunningham, K.; Truelove, S.; Carter, N.D.; Sebastian, B.; Byrnes, K.; Polydefkis, M. Cutaneous nerve biomarkers in transthyretin familial amyloid polyneuropathy. Ann. Neurol. 2017, 82, 44–56. [Google Scholar] [CrossRef]
- Chao, C.C.; Hsueh, H.W.; Kan, H.W.; Liao, C.H.; Jiang, H.H.; Chiang, H.; Lin, W.M.; Yeh, T.Y.; Lin, Y.H.; Cheng, Y.Y.; et al. Skin nerve pathology: Biomarkers of premanifest and manifest amyloid neuropathy. Ann. Neurol. 2019, 85, 560–573. [Google Scholar] [CrossRef]
- Musumeci, M.B.; Cappelli, F.; Russo, D.; Tini, G.; Canepa, M.; Milandri, A.; Bonfiglioli, R.; Di Bella, G.; My, F.; Luigetti, M.; et al. Low Sensitivity of Bone Scintigraphy in Detecting Phe64Leu Mutation-Related Transthyretin Cardiac Amyloidosis. JACC Cardiovasc. Imaging 2020, 13, 1314–1321. [Google Scholar] [CrossRef]









{kind=link}
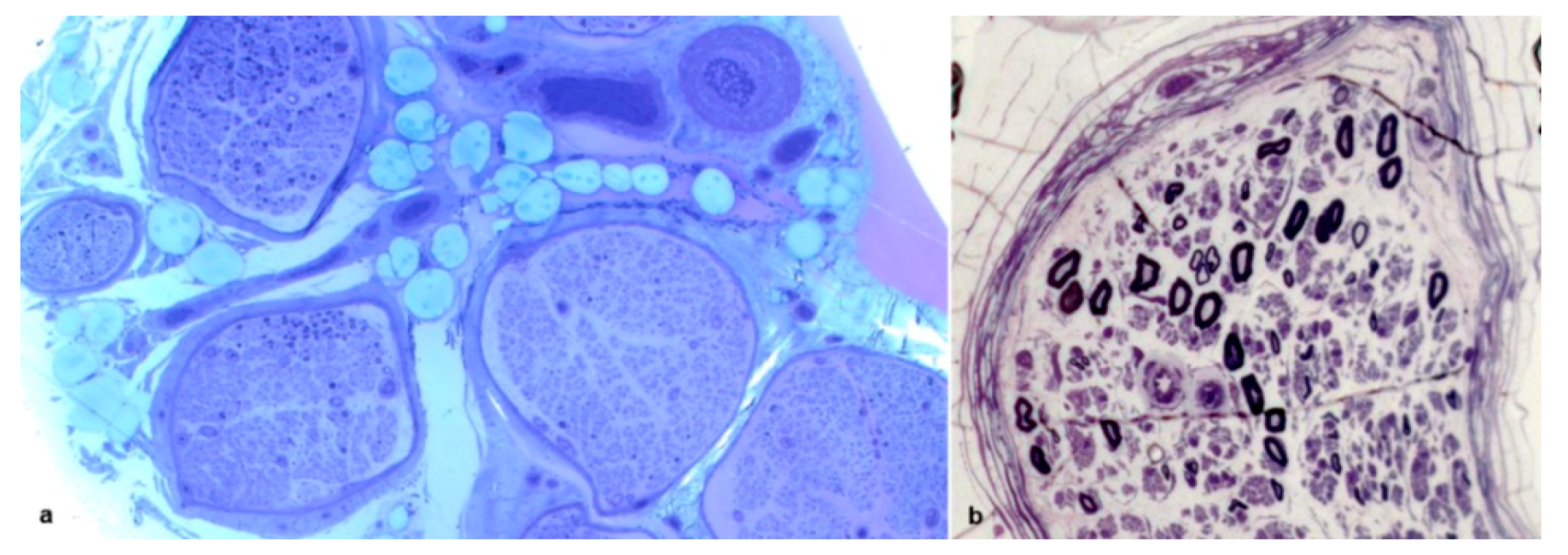{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All Patients | Phe64Leu | Val30Met | Glu89Gln | Tyr68Phe | |
|---|---|---|---|---|---|
| Number of patients | 67 | 26 | 24 | 6 | 3 |
| M/F | 54/13 | 22/4 | 20/4 | 5/1 | 3/0 |
| Age at onset | 64.1 | 68.7 | 67.3 | 48.2 | 63.3 |
| Age at biopsy | 67.0 | 71.1 | 69.6 | 55.3 | 64.7 |
| Disease duration at biopsy (years) | 2.9 | 2.4 | 2.3 | 7.1 | 3.4 |
| Origin | |||||
| Northern Italy | 13/67 (19.4%) | 1/26 (3.8%) | 8/24 (33.3%) | 0/6 (0.0%) | 2/3 (66.7%) |
| Central Italy | 19/67 (28.3%) | 2/26 (7.6%) | 15/24 (62.5%) | 0/6 (0.0%) | 0/3 (0.0%) |
| Southern Italy | 33/67 (49.3%) | 23/26 (88.6%) | 0/24 (0.0%) | 6/6 (100%) | 1/3 (33.3%) |
| Other countries | 2/67 (3.0%) | 0/26 (0.0%) | 1/24 (4.2%) | 0/6 (0.0%) | 0/3 (0.0%) |
| Polyneuropathy | |||||
| Axonal | 54/67 (80.6%) | 23/26 (88.5%) | 21/24 (87.5%) | 4/6 (66.7%) | 1/3 (33.3%) |
| Mixed | 11/67 (16.4%) | 3/26 (11.5%) | 2/24 (8.3%) | 2/6 (33.3%) | 1/3 (33.3%) |
| Demyelinating | 2/67 (3.0%) | 0/26 (0.0%) | 1/24 (4.2%) | 0/6 (0.0%) | 1/3 (33.3%) |
| Cardiomyopathy | 47/67 (70.2%) | 14/26 (53.9%) | 20/24 (83.4%) | 5/6 (83.3%) | 1/3 (33.3%) |
| Autonomic disturbances | 43/67 (64.2%) | 21/26 (80.8%) | 10/24 (41.7%) | 5/6 (83.3%) | 2/3 (66.7%) |
| Weight loss | 34/67 (50.8%) | 17/26 (65.4%) | 10/24 (41.7%) | 2/6 (33.3%) | 1/3 (33.3%) |
| Congo red positive abdominal fat needle aspiration | 8/20 (40.0%) | 1/6 (16.6%) | 6/11 (54.5%) | - | 1/1 (100%) |
| Number of biopsies | 69 | 27 | 24 | 6 | 3 |
| Congo red positive nerve biopsy | 50/69 (72.5%) | 13/27 (48.2%) | 21/24 (87.5%) | 5/6 (83.3%) | 2/3 (66.7%) |
| Axonal loss | |||||
| Slight | 4/69 (5.8%) | 0/27 (0.0%) | 0/24 (0.0%) | 0/6 (0.0%) | 1/3 (33.3%) |
| Moderate | 15/69 (21.7%) | 1/27 (3.7%) | 7/24 (29.2%) | 2/6 (33.3%) | 2/3 * (66.7%) |
| Severe | 42/69 (60.9%) | 24/27 (88.9%) | 13/24 (54.2%) | 4/6 (66.7%) | 0/3 (0.0%) |
| Total | 8/69 (11.6%) | 2/27 (7.4%) | 4/24 (16.7%) | 0/6 (0.0%) | 0/3 (0.0%) |
| Myelin abnormalities | 10/69 (14.5%) | 4/27 (14.8%) | 2/24 (8.3%) | 1/6 (16.7%) | 2/3 (66.7%) |
| Inflammatory infiltrates | 4/69 (5.8%) | 1/27 (3.7%) | 0/24 (0.0%) | 0/6 (0.0%) | 0/3 (0.0%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luigetti, M.; Romozzi, M.; Bisogni, G.; Cardellini, D.; Cavallaro, T.; Di Paolantonio, A.; Fabrizi, G.M.; Fenu, S.; Gentile, L.; Grandis, M.; et al. hATTR Pathology: Nerve Biopsy Results from Italian Referral Centers. Brain Sci. 2020, 10, 780. https://doi.org/10.3390/brainsci10110780
Luigetti M, Romozzi M, Bisogni G, Cardellini D, Cavallaro T, Di Paolantonio A, Fabrizi GM, Fenu S, Gentile L, Grandis M, et al. hATTR Pathology: Nerve Biopsy Results from Italian Referral Centers. Brain Sciences. 2020; 10(11):780. https://doi.org/10.3390/brainsci10110780
Chicago/Turabian StyleLuigetti, Marco, Marina Romozzi, Giulia Bisogni, Davide Cardellini, Tiziana Cavallaro, Andrea Di Paolantonio, Gian Maria Fabrizi, Silvia Fenu, Luca Gentile, Marina Grandis, and et al. 2020. "hATTR Pathology: Nerve Biopsy Results from Italian Referral Centers" Brain Sciences 10, no. 11: 780. https://doi.org/10.3390/brainsci10110780
APA StyleLuigetti, M., Romozzi, M., Bisogni, G., Cardellini, D., Cavallaro, T., Di Paolantonio, A., Fabrizi, G. M., Fenu, S., Gentile, L., Grandis, M., Marucci, G., Massucco, S., Mazzeo, A., Pareyson, D., Romano, A., Russo, M., Schenone, A., Tagliapietra, M., Tozza, S., ... Sabatelli, M. (2020). hATTR Pathology: Nerve Biopsy Results from Italian Referral Centers. Brain Sciences, 10(11), 780. https://doi.org/10.3390/brainsci10110780