Metformin Enhances Excitatory Synaptic Transmission onto Hippocampal CA1 Pyramidal Neurons

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Reagents
2.3. Slice Preparation
2.4. Electrophysiological Recording
2.5. Statistical Analysis
3. Results
3.1. Met Markedly Enhances Glutamatergic Transmission in the CA1 Pyramidal Neurons
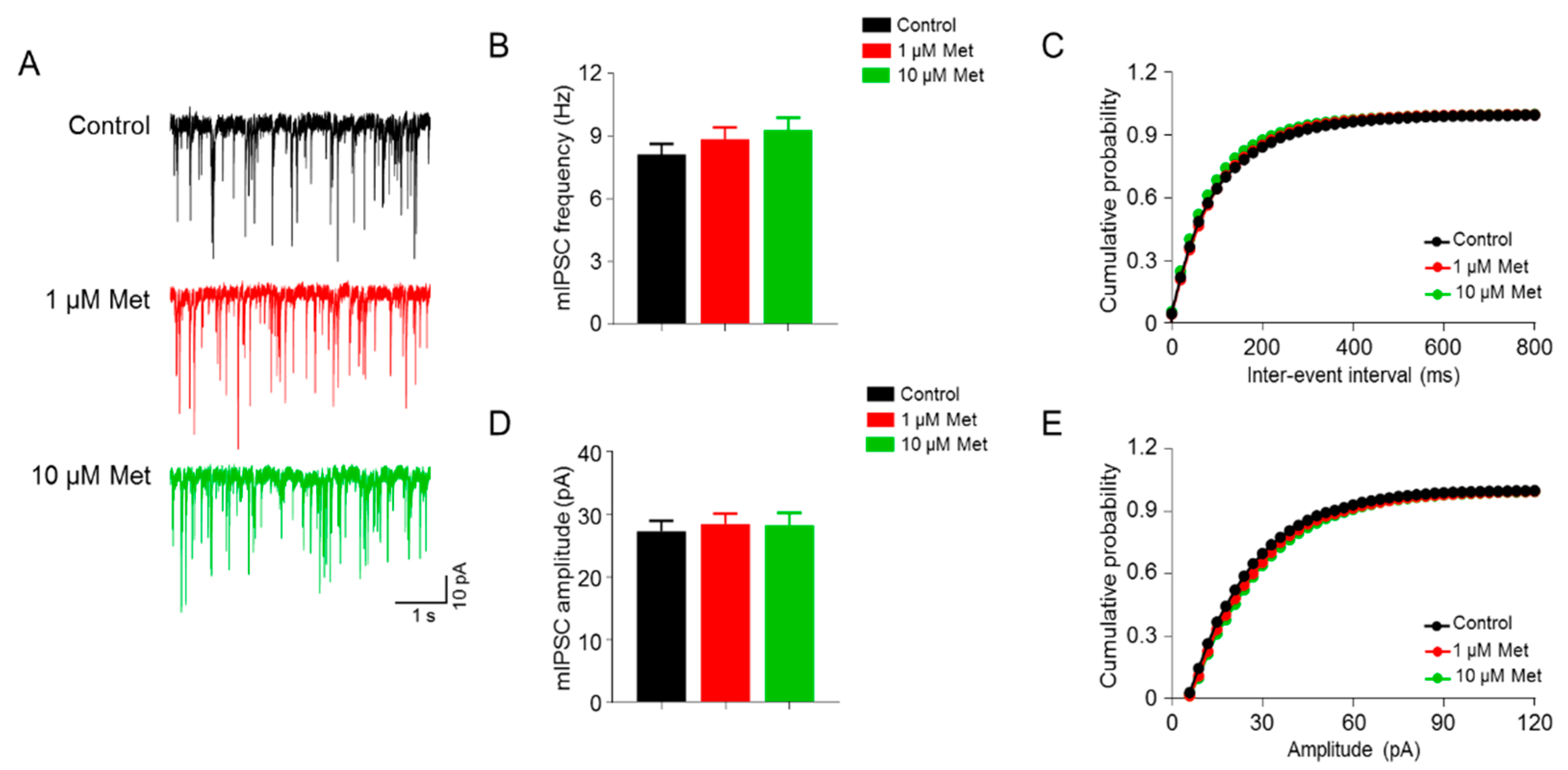
3.2. Met Has No Effect on the GABAergic Transmission in the CA1 Pyramidal Neurons
3.3. Met Increases Glutamate Release from Presynaptic Terminals
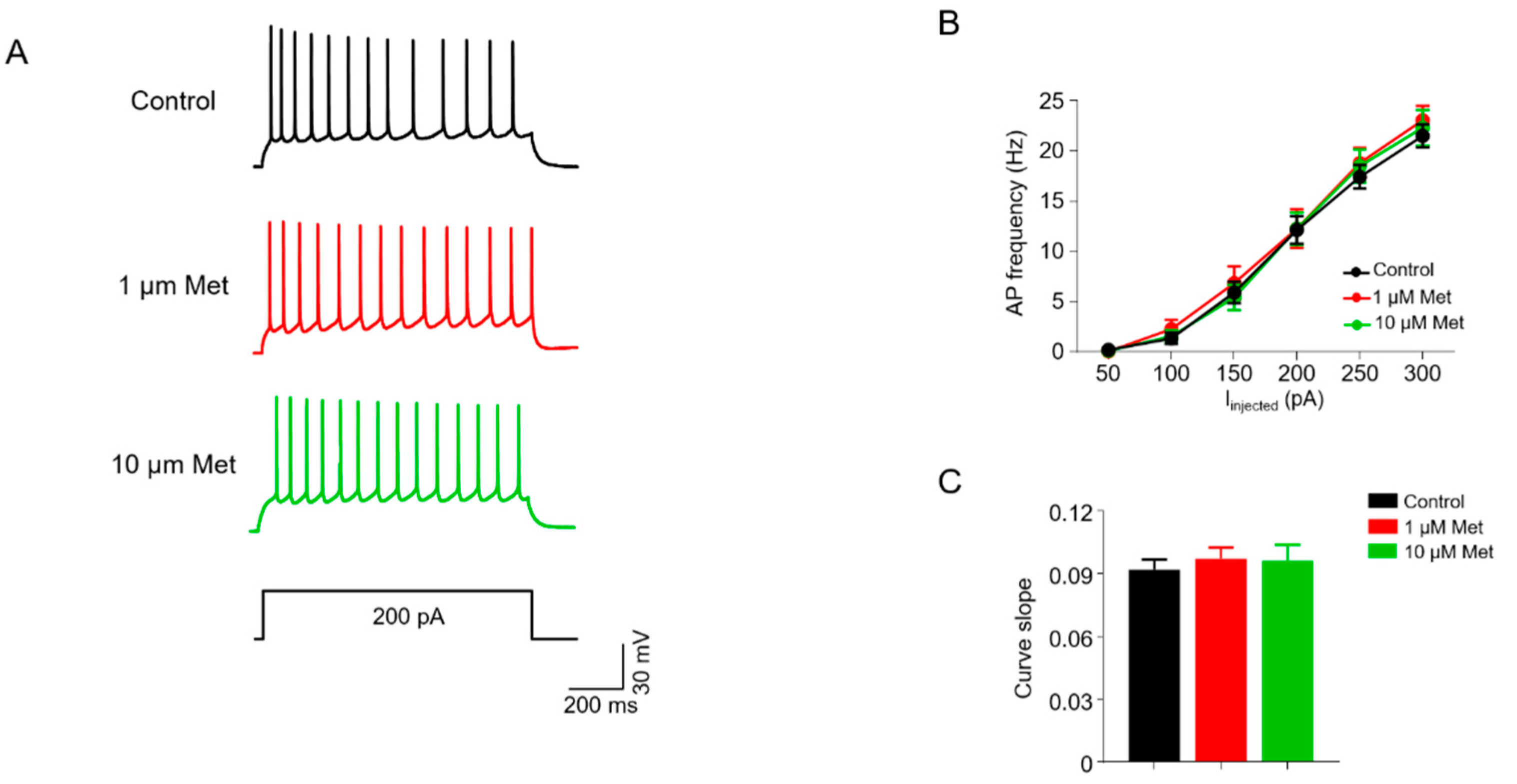
3.4. Met Does Not Alter Intrinsic Excitability in CA1 Pyramidal Neurons
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Availability of Data and Materials
Ethics Approval and Consent to Participate
References
- Pernicova, I.; Korbonits, M. Metformin—Mode of action and clinical implications for diabetes and cancer. Nat. Rev. Endocrinol. 2014, 10, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.S.; Li, M.; Ma, T.; Zong, Y.; Cui, J.; Feng, J.W.; Wu, Y.Q.; Lin, S.Y.; Lin, S.C. Metformin Activates AMPK through the Lysosomal Pathway. Cell Metab. 2016, 24, 521–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Ou, Y.; Li, Y.; Hu, S.; Shao, L.W.; Liu, Y. Metformin extends C. elegans lifespan through lysosomal pathway. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Hebrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Investig. 2010, 120, 2355–2369. [Google Scholar] [CrossRef] [Green Version]
- Gantois, I.; Khoutorsky, A.; Popic, J.; Aguilar-Valles, A.; Freemantle, E.; Cao, R.; Sharma, V.; Pooters, T.; Nagpal, A.; Skalecka, A.; et al. Metformin ameliorates core deficits in a mouse model of fragile X syndrome. Nat. Med. 2017, 23, 674–677. [Google Scholar] [CrossRef]
- Dy, A.B.C.; Tassone, F.; Eldeeb, M.; Salcedo-Arellano, M.J.; Tartaglia, N.; Hagerman, R. Metformin as targeted treatment in fragile X syndrome. Clin. Genet. 2018, 93, 216–222. [Google Scholar] [CrossRef]
- Protic, D.; Aydin, E.Y.; Tassone, F.; Tan, M.M.; Hagerman, R.J.; Schneider, A. Cognitive and behavioral improvement in adults with fragile X syndrome treated with metformin-two cases. Mol. Genet. Genomic. Med. 2019, 7, e00745. [Google Scholar] [CrossRef] [Green Version]
- Biag, H.M.B.; Potter, L.A.; Wilkins, V.; Afzal, S.; Rosvall, A.; Salcedo-Arellano, M.J.; Rajaratnam, A.; Manzano-Nunez, R.; Schneider, A.; Tassone, F.; et al. Metformin treatment in young children with fragile X syndrome. Mol. Genet. Genomic. Med. 2019, 7, e956. [Google Scholar] [CrossRef]
- Ou, Z.; Kong, X.; Sun, X.; He, X.; Zhang, L.; Gong, Z.; Huang, J.; Xu, B.; Long, D.; Li, J.; et al. Metformin treatment prevents amyloid plaque deposition and memory impairment in APP/PS1 mice. Brain Behav. Immun. 2018, 69, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, J.; Guo, F.L.; Gao, X.; Xie, X.; Liu, S.; Yang, X.; Yang, X.; Zhang, L.; Ye, Y.; et al. Metformin Ameliorates Synaptic Defects in a Mouse Model of AD by Inhibiting Cdk5 Activity. Front. Cell. Neurosci. 2020, 14, 170. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.Y.; Huang, S.; Chen, Q.B.; Zhang, D.; Li, W.; Ao, R.; Leung, F.C.; Zhang, Z.; Huang, J.; Tang, Y.; et al. Metformin Ameliorates Abeta Pathology by Insulin-Degrading Enzyme in a Transgenic Mouse Model of Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2020, 2020, 2315106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farr, S.A.; Roesler, E.; Niehoff, M.L.; Roby, D.A.; McKee, A.; Morley, J.E. Metformin Improves Learning and Memory in the SAMP8 Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2019, 68, 1699–1710. [Google Scholar] [CrossRef]
- Patil, S.P.; Jain, P.D.; Ghumatkar, P.J.; Tambe, R.; Sathaye, S. Neuroprotective effect of metformin in MPTP-induced Parkinson’s disease in mice. Neuroscience 2014, 277, 747–754. [Google Scholar] [CrossRef]
- Lu, M.; Su, C.; Qiao, C.; Bian, Y.; Ding, J.; Hu, G. Metformin Prevents Dopaminergic Neuron Death in MPTP/P-Induced Mouse Model of Parkinson’s Disease via Autophagy and Mitochondrial ROS Clearance. Int. J. Neuropsychopharmacol. 2016, 19. [Google Scholar] [CrossRef]
- Arnoux, I.; Willam, M.; Griesche, N.; Krummeich, J.; Watari, H.; Offermann, N.; Weber, S.; Dey, P.N.; Chen, C.W.; Monteiro, O.; et al. Metformin reverses early cortical network dysfunction and behavior changes in Huntington’s disease. Elife 2018, 7. [Google Scholar] [CrossRef]
- Fang, W.; Zhang, J.; Hong, L.; Huang, W.; Dai, X.; Ye, Q.; Chen, X. Metformin ameliorates stress-induced depression-like behaviors via enhancing the expression of BDNF by activating AMPK/CREB-mediated histone acetylation. J. Affect. Disord. 2020, 260, 302–313. [Google Scholar] [CrossRef]
- Liu, W.; Liu, J.; Huang, Z.; Cui, Z.; Li, L.; Liu, W.; Qi, Z. Possible role of GLP-1 in antidepressant effects of metformin and exercise in CUMS mice. J. Affect. Disord. 2019, 246, 486–497. [Google Scholar] [CrossRef]
- Guo, M.; Mi, J.; Jiang, Q.M.; Xu, J.M.; Tang, Y.Y.; Tian, G.; Wang, B. Metformin may produce antidepressant effects through improvement of cognitive function among depressed patients with diabetes mellitus. Clin. Exp. Pharmacol. Physiol. 2014, 41, 650–656. [Google Scholar] [CrossRef]
- Nath, N.; Khan, M.; Paintlia, M.K.; Singh, I.; Hoda, M.N.; Giri, S. Metformin attenuated the autoimmune disease of the central nervous system in animal models of multiple sclerosis. J. Immunol. 2009, 182, 8005–8014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.; Gang, X.; Liu, Y.; Wang, G.; Zhao, X.; Wang, G. Mitochondrial dysfunction plays a key role in the development of neurodegenerative diseases in diabetes. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E750–E764. [Google Scholar] [CrossRef] [PubMed]
- Madeo, F.; Carmona-Gutierrez, D.; Hofer, S.J.; Kroemer, G. Caloric Restriction Mimetics against Age-Associated Disease: Targets, Mechanisms, and Therapeutic Potential. Cell Metab. 2019, 29, 592–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vauzour, D.; Camprubi-Robles, M.; Miquel-Kergoat, S.; Andres-Lacueva, C.; Banati, D.; Barberger-Gateau, P.; Bowman, G.L.; Caberlotto, L.; Clarke, R.; Hogervorst, E.; et al. Nutrition for the ageing brain: Towards evidence for an optimal diet. Ageing Res. Rev. 2017, 35, 222–240. [Google Scholar] [CrossRef] [Green Version]
- Vaiserman, A.M.; Lushchak, O.V.; Koliada, A.K. Anti-aging pharmacology: Promises and pitfalls. Ageing Res. Rev. 2016, 31, 9–35. [Google Scholar] [CrossRef]
- Dong, Z.; Chen, W.; Chen, C.; Wang, H.; Cui, W.; Tan, Z.; Robinson, H.; Gao, N.; Luo, B.; Zhang, L.; et al. CUL3 Deficiency Causes Social Deficits and Anxiety-like Behaviors by Impairing Excitation-Inhibition Balance through the Promotion of Cap-Dependent Translation. Neuron 2020, 105, 475–490. [Google Scholar] [CrossRef]
- Wang, H.; Liu, F.; Chen, W.; Sun, X.; Cui, W.; Dong, Z.; Zhao, K.; Zhang, H.; Li, H.; Xing, G.; et al. Genetic recovery of ErbB4 in adulthood partially restores brain functions in null mice. Proc. Natl. Acad. Sci. USA 2018, 115, 13105–13110. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.H.; Liu, W.Z.; He, Y.; You, W.J.; Zhang, J.Y.; Xu, H.; Tian, X.L.; Li, B.M.; Mei, L.; Holmes, A.; et al. Chronic Stress Causes Projection-Specific Adaptation of Amygdala Neurons via Small-Conductance Calcium-Activated Potassium Channel Downregulation. Biol. Psychiatry 2019, 85, 812–828. [Google Scholar] [CrossRef]
- Sluggett, J.K.; Koponen, M.; Bell, J.S.; Taipale, H.; Tanskanen, A.; Tiihonen, J.; Uusitupa, M.; Tolppanen, A.M.; Hartikainen, S. Metformin and Risk of Alzheimer’s Disease Among Community-Dwelling People with Diabetes: A National Case-Control Study. J. Clin. Endocrinol. Metab. 2020, 105. [Google Scholar] [CrossRef]
- Abdallah, M.S.; Mosalam, E.M.; Zidan, A.A.; Elattar, K.S.; Zaki, S.A.; Ramadan, A.N.; Ebeid, A.M. The Antidiabetic Metformin as an Adjunct to Antidepressants in Patients with Major Depressive Disorder: A Proof-of-Concept, Randomized, Double-Blind, Placebo-Controlled Trial. Neurotherapeutics 2020. [Google Scholar] [CrossRef]
- Ying, M.A.; Maruschak, N.; Mansur, R.; Carvalho, A.F.; Cha, D.S.; McIntyre, R.S. Metformin: Repurposing opportunities for cognitive and mood dysfunction. CNS Neurol. Disord. Drug Targets 2014, 13, 1836–1845. [Google Scholar] [CrossRef] [PubMed]
- Small, S.A.; Schobel, S.A.; Buxton, R.B.; Witter, M.P.; Barnes, C.A. A pathophysiological framework of hippocampal dysfunction in ageing and disease. Nat. Rev. Neurosci. 2011, 12, 585–601. [Google Scholar] [CrossRef] [PubMed]
- Pilipenko, V.; Narbute, K.; Pupure, J.; Langrate, I.K.; Muceniece, R.; Klusa, V. Neuroprotective potential of antihyperglycemic drug metformin in streptozocin-induced rat model of sporadic Alzheimer’s disease. Eur. J. Pharmacol. 2020, 881, 173290. [Google Scholar] [CrossRef]
- Saffari, P.M.; Alijanpour, S.; Takzaree, N.; Sahebgharani, M.; Etemad-Moghadam, S.; Noorbakhsh, F.; Partoazar, A. Metformin loaded phosphatidylserine nanoliposomes improve memory deficit and reduce neuroinflammation in streptozotocin-induced Alzheimer’s disease model. Life Sci. 2020, 255, 117861. [Google Scholar] [CrossRef]
- Liu, B.; Huang, B.; Liu, J.; Shi, J.S. Dendrobium nobile Lindl alkaloid and metformin ameliorate cognitive dysfunction in senescence-accelerated mice via suppression of endoplasmic reticulum stress. Brain Res. 2020, 1741, 146871. [Google Scholar] [CrossRef] [PubMed]
- Yin, D.M.; Chen, Y.J.; Lu, Y.S.; Bean, J.C.; Sathyamurthy, A.; Shen, C.; Liu, X.; Lin, T.W.; Smith, C.A.; Xiong, W.C.; et al. Reversal of behavioral deficits and synaptic dysfunction in mice overexpressing neuregulin 1. Neuron 2013, 78, 644–657. [Google Scholar] [CrossRef] [Green Version]
- Oshima, R.; Yamada, M.; Kurogi, E.; Ogino, Y.; Serizawa, Y.; Tsuda, S.; Ma, X.; Egawa, T.; Hayashi, T. Evidence for organic cation transporter-mediated metformin transport and 5’-adenosine monophosphate-activated protein kinase activation in rat skeletal muscles. Metabolism 2015, 64, 296–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, E.C.; Liang, X.; Yee, S.W.; Geier, E.G.; Stocker, S.L.; Chen, L.; Giacomini, K.M. Targeted disruption of organic cation transporter 3 attenuates the pharmacologic response to metformin. Mol. Pharmacol. 2015, 88, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Segal, E.D.; Yasmeen, A.; Beauchamp, M.C.; Rosenblatt, J.; Pollak, M.; Gotlieb, W.H. Relevance of the OCT1 transporter to the antineoplastic effect of biguanides. Biochem. Biophys. Res. Commun. 2011, 414, 694–699. [Google Scholar] [CrossRef]
- Shang, T.; Uihlein, A.V.; Van Asten, J.; Kalyanaraman, B.; Hillard, C.J. 1-Methyl-4-phenylpyridinium accumulates in cerebellar granule neurons via organic cation transporter 3. J. Neurochem. 2003, 85, 358–367. [Google Scholar] [CrossRef]
- Mayer, F.P.; Schmid, D.; Owens, W.A.; Gould, G.G.; Apuschkin, M.; Kudlacek, O.; Salzer, I.; Boehm, S.; Chiba, P.; Williams, P.H.; et al. An unsuspected role for organic cation transporter 3 in the actions of amphetamine. Neuropsychopharmacology 2018, 43, 2408–2417. [Google Scholar] [CrossRef] [PubMed]
- Gasser, P.J.; Hurley, M.M.; Chan, J.; Pickel, V.M. Organic cation transporter 3 (OCT3) is localized to intracellular and surface membranes in select glial and neuronal cells within the basolateral amygdaloid complex of both rats and mice. Brain Struct. Funct. 2017, 222, 1913–1928. [Google Scholar] [CrossRef] [PubMed]
- Marinangeli, C.; Didier, S.; Ahmed, T.; Caillerez, R.; Domise, M.; Laloux, C.; Begard, S.; Carrier, S.; Colin, M.; Marchetti, P.; et al. AMP-Activated Protein Kinase Is Essential for the Maintenance of Energy Levels during Synaptic Activation. iScience 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Atasoy, D.; Su, H.H.; Sternson, S.M. Hunger states switch a flip-flop memory circuit via a synaptic AMPK-dependent positive feedback loop. Cell 2011, 146, 992–1003. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Luo, Y.; Sun, J.; Ding, Z.; Liu, J.; Yan, W.; Jian, M.; Xue, Y.; Shi, J.; Wang, J.S.; et al. AMPK Signaling in the Dorsal Hippocampus Negatively Regulates Contextual Fear Memory Formation. Neuropsychopharmacology 2016, 41, 1849–1864. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Chaudhari, K.; Shetty, R.; Winters, A.; Gao, X.; Hu, Z.; Ge, W.P.; Sumien, N.; Forster, M.; Liu, R.; et al. Metformin Alters Locomotor and Cognitive Function and Brain Metabolism in Normoglycemic Mice. Aging Dis. 2019, 10, 949–963. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, S.; Moreira, P.I. Antidiabetic drugs for Alzheimer’s and Parkinson’s diseases: Repurposing insulin, metformin, and thiazolidinediones. Int. Rev. Neurobiol. 2020, 155, 37–64. [Google Scholar] [CrossRef]
- Zhu, X.; Shen, J.; Feng, S.; Huang, C.; Liu, Z.; Sun, Y.E.; Liu, H. Metformin improves cognition of aged mice by promoting cerebral angiogenesis and neurogenesis. Aging (Albany NY) 2020, 12. [Google Scholar] [CrossRef]
- Chen, X.; Levy, J.M.; Hou, A.; Winters, C.; Azzam, R.; Sousa, A.A.; Leapman, R.D.; Nicoll, R.A.; Reese, T.S. PSD-95 family MAGUKs are essential for anchoring AMPA and NMDA receptor complexes at the postsynaptic density. Proc. Natl. Acad. Sci. USA 2015, 112, E6983–E6992. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, W.-B.; Chen, J.; Liu, Z.-Y.; Luo, B.; Zhou, T.; Fei, E.-K. Metformin Enhances Excitatory Synaptic Transmission onto Hippocampal CA1 Pyramidal Neurons. Brain Sci. 2020, 10, 706. https://doi.org/10.3390/brainsci10100706
Chen W-B, Chen J, Liu Z-Y, Luo B, Zhou T, Fei E-K. Metformin Enhances Excitatory Synaptic Transmission onto Hippocampal CA1 Pyramidal Neurons. Brain Sciences. 2020; 10(10):706. https://doi.org/10.3390/brainsci10100706
Chicago/Turabian StyleChen, Wen-Bing, Jiang Chen, Zi-Yang Liu, Bin Luo, Tian Zhou, and Er-Kang Fei. 2020. "Metformin Enhances Excitatory Synaptic Transmission onto Hippocampal CA1 Pyramidal Neurons" Brain Sciences 10, no. 10: 706. https://doi.org/10.3390/brainsci10100706
APA StyleChen, W.-B., Chen, J., Liu, Z.-Y., Luo, B., Zhou, T., & Fei, E.-K. (2020). Metformin Enhances Excitatory Synaptic Transmission onto Hippocampal CA1 Pyramidal Neurons. Brain Sciences, 10(10), 706. https://doi.org/10.3390/brainsci10100706