A Way towards Reliable Predictive Methods for the Prediction of Physicochemical Properties of Chemicals Using the Group Contribution and other Methods

Abstract
1. Predictive Methods for Physicochemical Properties of Molecules
1.1. Introduction
1.2. Current Methods
2. Accuracy of Predictive Methods and What Is Required
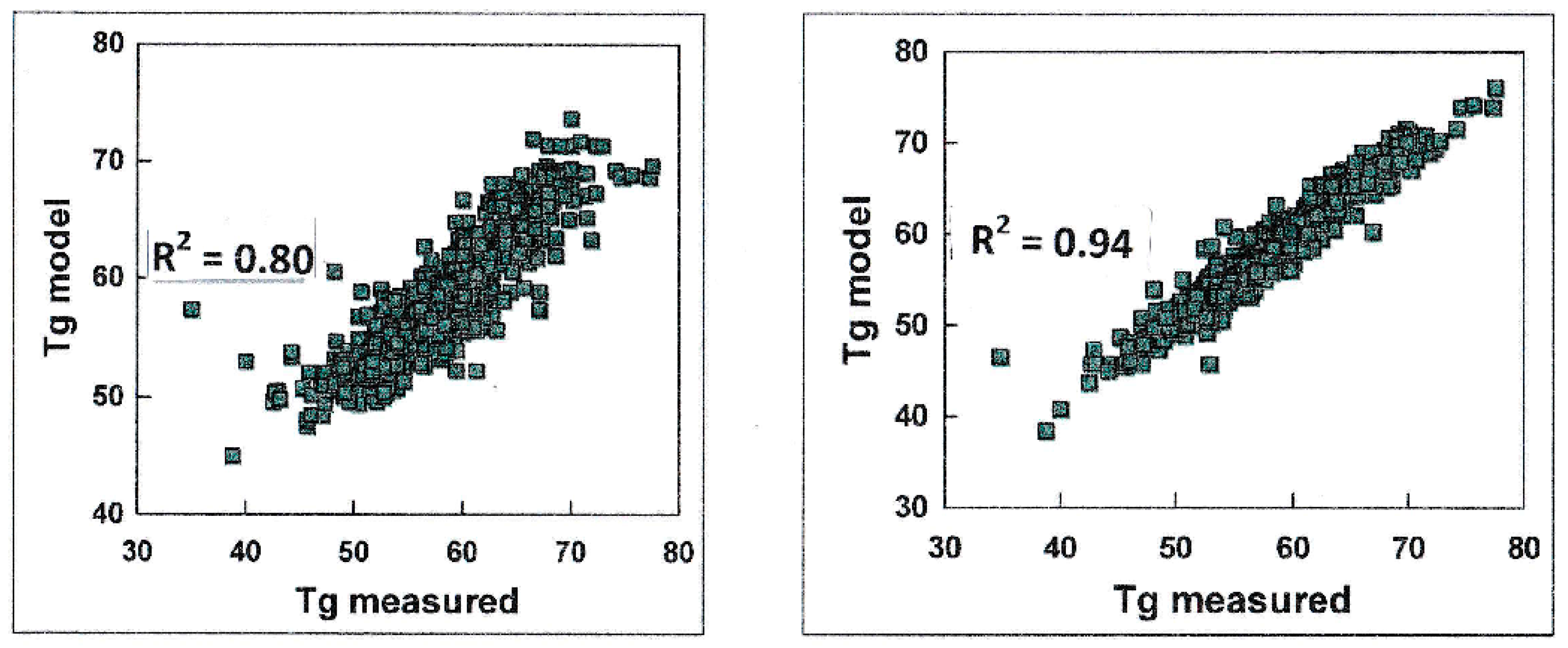
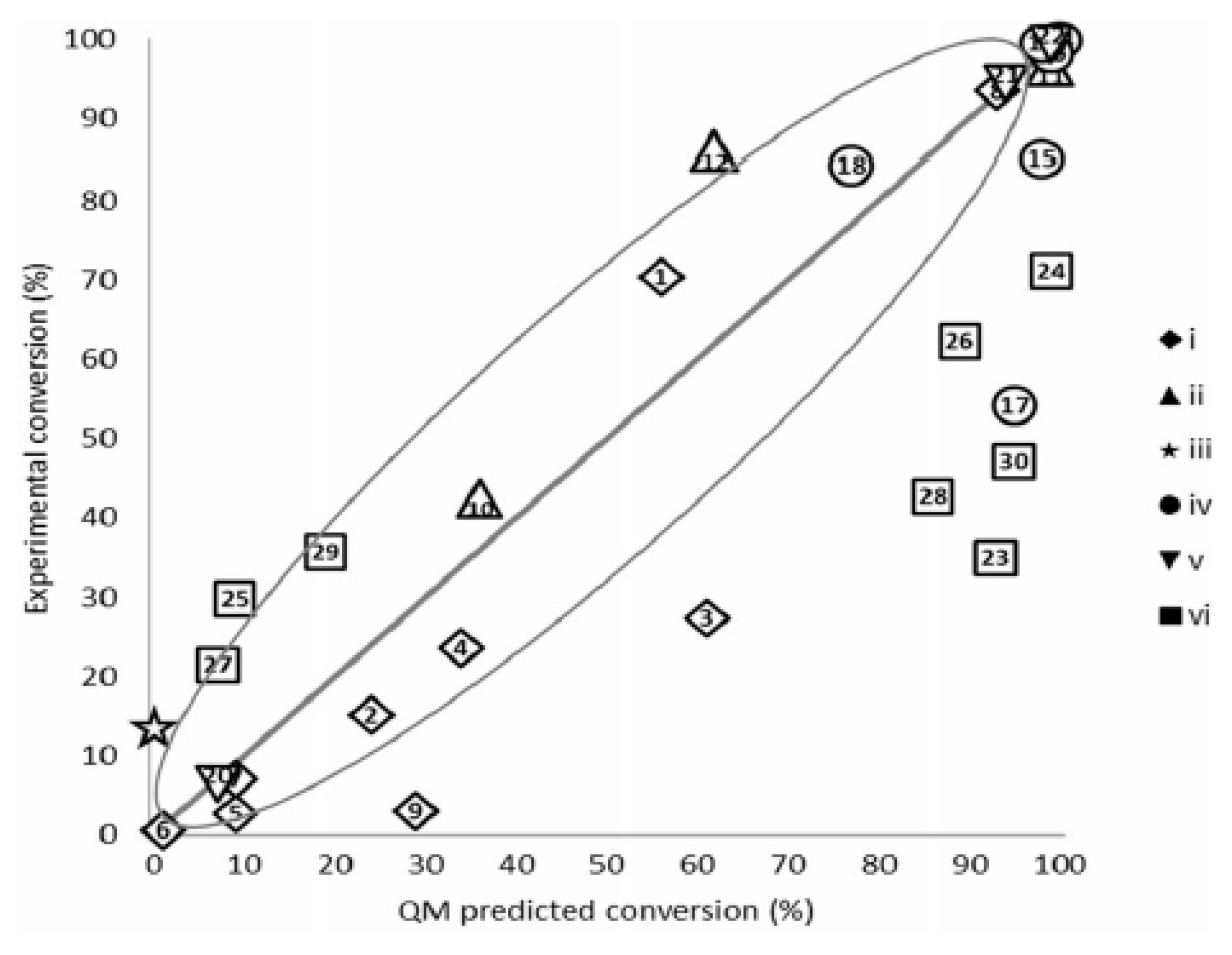
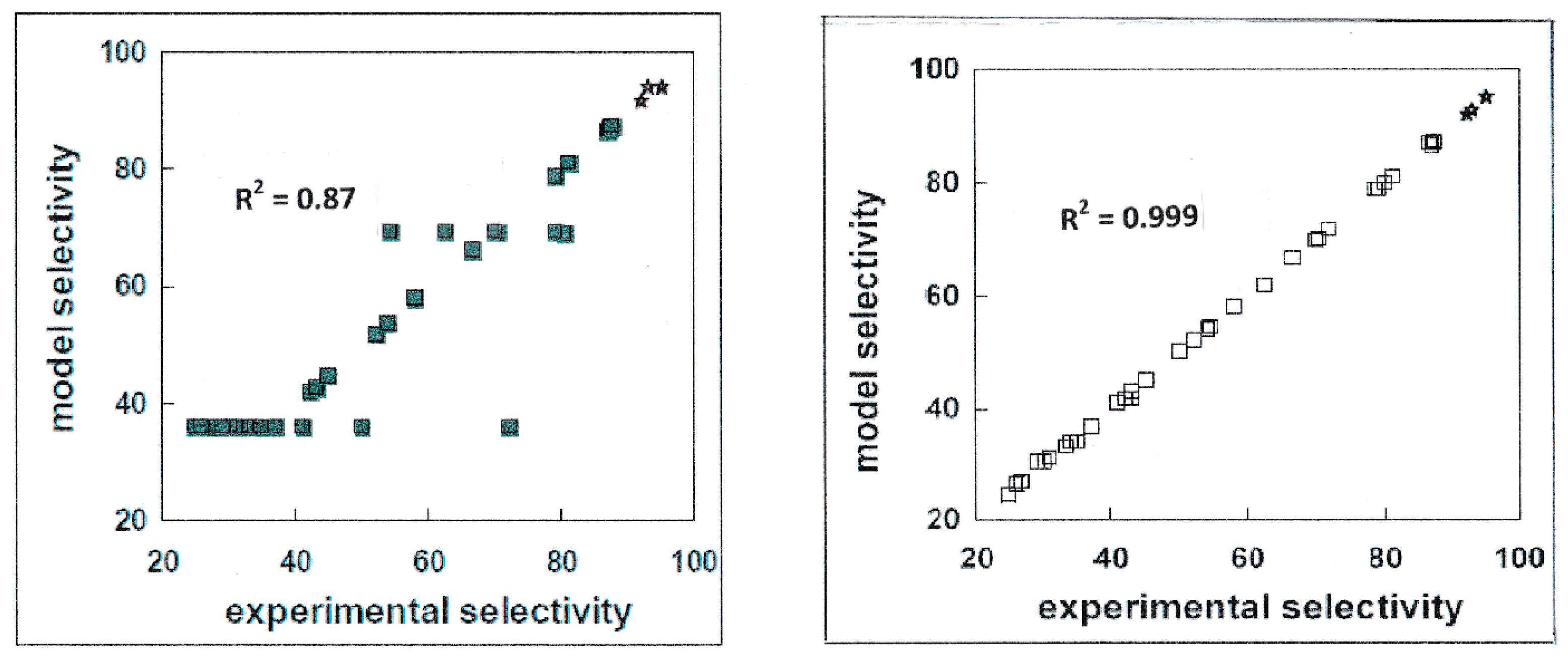
2.1. On the Assessment of the Quality of the Methods Evaluating Physicochemical Properties
2.2. Cases Requiring Quantitative Predictions over the Data Range
2.3. Cases Requiring only Qualitative Predictive Power
2.4. Why do Methods often not Perform up to the Required Level of Accuracy and Reliability?
- (i)
- the preferred or exclusive use of a specific class of methods, for example quantum mechanics or statistical analysis using molecular descriptors
- (ii)
- no optimal parametrization of statistical models or GC methods
- (iii)
- quality of data
- (iv)
- inappropriate account of the physics involved
- (v)
- insufficient transfer of knowledge between different fields of science
2.5. Validation
3. Is There a Way forward towards Reliable Predictive Methods for Physicochemical Property Prediction of Chemicals? How to Arrive at Good Methods for the Prediction of Physicochemical Properties
3.1. Introduction
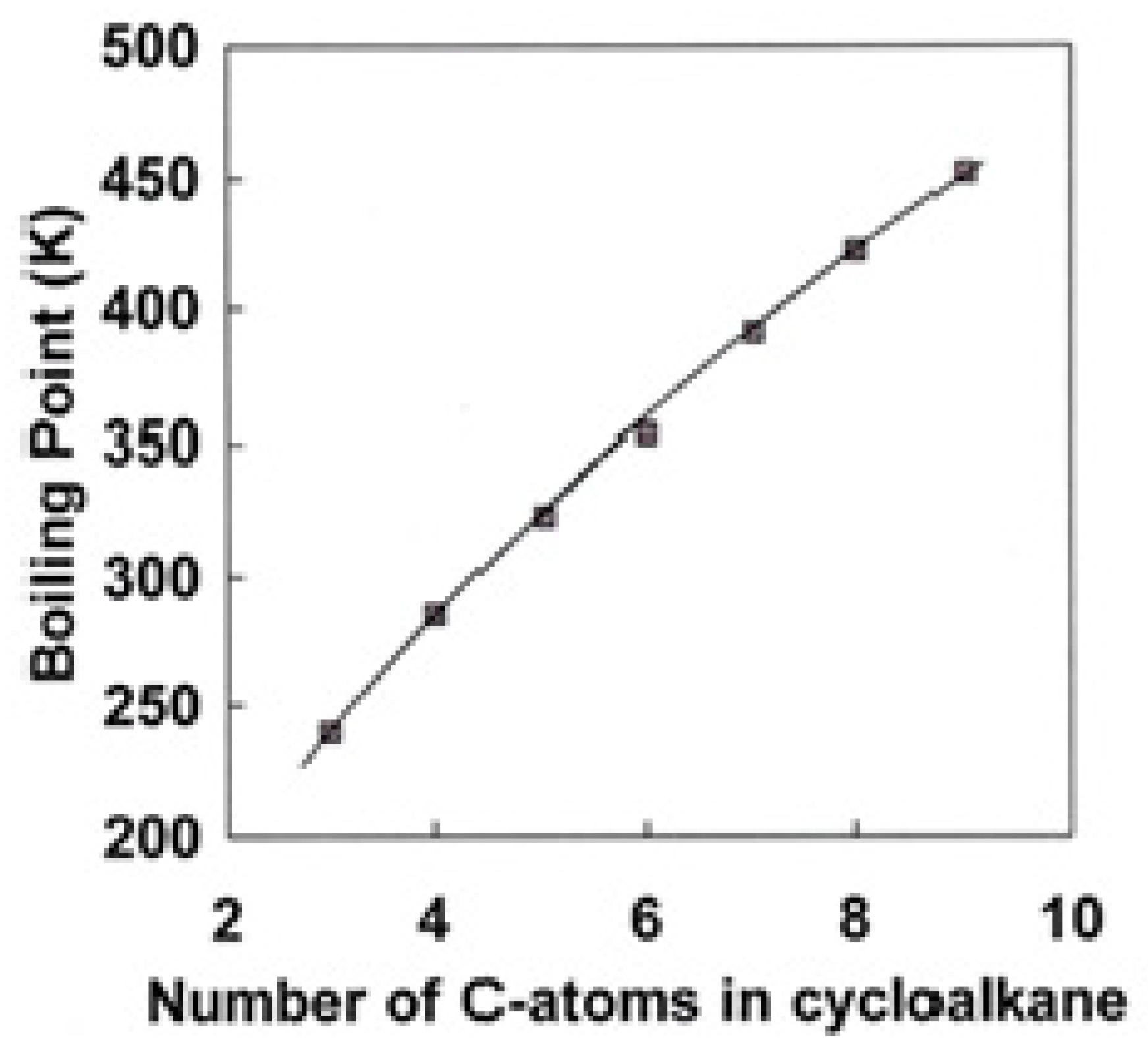
3.2. Organic Molecules: the Virtue of Group Contribution Methods
3.3. QSPR Methods using Molecular Descriptors
4. Conclusions
Conflicts of Interest
References
- Fink, T.; Reymond, J.-L. Virtual exploration of the chemical universe up to 11 atoms of C, N, O, F: Assembly of 26.4 million structures (110.9 million stereoisomers) and analysis for new ring systems, stereochemistry, physicochemical properties, compound classes and drug discovery. J. Chem. Inf. Model. 2007, 47, 342–353. [Google Scholar] [CrossRef]
- Hendriks, E.; Kontogeorgis, G.M.; Dohrn, R.; de Hemptinne, J.-C.; Economou, I.G.; Fele Žilnik, L.; Vesovic, V. Industrial Requirements for Thermodynamics and Transport Properties. Ind. Eng. Chem. Res. 2010, 49, 11131–11141. [Google Scholar] [CrossRef]
- Katritkzy, A.R.; Kuanar, M.; Slavov, S.; Hall, C.D.; Karelson, M.; Kahn, I.; Dobchev, D.A. Quantitative Correlation of Physical and Chemical Properties with Chemical Structure: Utility for Prediction. Chem. Rev. 2010, 110, 5714–5789. [Google Scholar] [CrossRef]
- Nieto-Draghi, C.; Fayet, G.; Creton, B.; Rozanska, X.; Rotureau, P.; De Hemptinne, J.-C.; Ungerer, P.; Rousseau, B.; Adamo, C. A General Guidebook for the Theoretical Prediction of Physicochemical Properties of Chemicals for Regulatory Purposes. Chem. Rev. 2015, 115, 13093–13164. [Google Scholar] [CrossRef] [PubMed]
- Van Speybroeck, V.; Gani, R.; Meier, R.J. The calculation of thermodynamic properties of molecules. Chem. Soc. Rev. 2010, 39, 1764–1779. [Google Scholar] [CrossRef] [PubMed]
- Rozanska, X.; Ungerer, P.; Leblanc, B.; Saxe, P.; Wimmer, E. Automatic and systematic Atomistic Simulations in the MedeA® Software Environment: Application to EU-REACH. Oil Gas Sci. Technol. Rev. IFP Energies Nouv. 2015, 70, 395–403. [Google Scholar] [CrossRef]
- Barrett, R.A.; Meier, R.J. The calculation of molecular entropy using the semiempirical AM1 method. J. Mol. Struct. 1996, 363, 203–209. [Google Scholar] [CrossRef]
- Rozanska, X.; Stewart, J.J.P.; Ungerer, P.; Leblanc, B.; Freeman, C.; Saxe, P.; Wimmer, E. High-Throughput Calculations of Molecular Properties in the MedeA Environment: Accuracy of PM7 in Predicting Vibrational Frequencies, Ideal Gas Entropies, Heat Capacities and Gibbs Free Energies of Organic Molecules. J. Chem. Eng. Data 2014, 59, 3136–3143. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V. (Eds.) Handbook of Molecular Descriptors; Wiley-VCH: Hoboken, NJ, USA, 2000. [Google Scholar]
- Tseng, Y.; Hopfinger, A.J.; Esposito, E.X. The great descriptor melting pot: Mixing descriptors for the common good of QSAR models. J. Comput. Aided Mol. Des. 2012, 26, 39–43. [Google Scholar] [CrossRef]
- Le, T.; Epa, V.C.; Burden, F.R.; Winkler, D.A. Quantitative Structure–Property Relationship Modeling of Diverse Materials Properties. Chem. Rev. 2012, 112, 2889–2919. [Google Scholar] [CrossRef]
- Karelson, M.; Lobanov, V.S.; Katritzky, A.R. Quantum-Chemical Descriptors in QSAR/QSPR Studies. Chem. Rev. 1996, 96, 1027–1044. [Google Scholar] [CrossRef]
- Ren, Y.; Zhao, B.; Chang, Q.; Yao, X. QSPR modeling of nonionic surfactant cloud points: An update. J. Coll. Interf. Sci. 2011, 358, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.C.; Crippen, G.M. Predicting pKa. J. Chem. Inf. Model. 2009, 49, 2013–2033. [Google Scholar] [CrossRef]
- Oprisiu, I.; Marcou, G.; Horvath, D.; Brunel, D.B.; Rivollet, F. Publicly available models to predict normal boiling point of organic compounds. Thermochim. Acta 2013, 553, 60–67. [Google Scholar] [CrossRef]
- Gharagheizi, F.; Mirkhani, S.A.; Ilani-Kashkouli, P.; Mohammad, A.H.; Ramjugernath, D.; Richon, D. Determination of the normal boiling point of chemical compounds using a quantitative structure-property relationship strategy: Application to a very large dataset. Fluid Phase Equilib. 2013, 354, 250–258. [Google Scholar] [CrossRef]
- Piliszek, S.; Wilczyńska-Piliszek, A.J.; Falandysz, J. N-octanol-water partition coefficients (log K(OW)) of 399 congeners of polychlorinated azoxybenzenes (PCAOBs) determined by QSPR- and ANN-based approach. J. Environ. Sci. Health A Tox. Hazard Subst. Environ. Eng. 2011, 46, 1748–1762. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, A.G.; Rothenberg, G. Predictive modeling in homogeneous catalysis: A tutorial. Chem. Soc. Rev. 2010, 39, 1891–1902. [Google Scholar] [CrossRef]
- Joback, K.G.; Reid, R.C. Estimation of Pure-Component Properties from Group-Contributions. Chem. Eng. Commun. 1987, 57, 233–243. [Google Scholar] [CrossRef]
- Benson, S.W.; Cruickshank, F.R.; Golden, D.M.; Haugen, G.R.; O’Neal, E.H.; Rodgers, A.S.; Shaw, R.; Walsh, R. Additivity rules for the estimation of thermochemical properties. Chem. Rev. 1969, 69, 279–324. [Google Scholar] [CrossRef]
- Constantinou, L.; Gani, R. New group contribution method for estimating properties of pure compounds. AIChE J. 1994, 40, 1697–1710. [Google Scholar] [CrossRef]
- Marrero, J.; Gani, R. Group-contribution based estimation of pure component properties. Fluid Phase Equilib. 2001, 183–184, 183–208. [Google Scholar] [CrossRef]
- Bicerano, J. (Ed.) Prediction of Polymer Properties; Marcel Dekker Inc.: New York, NY, USA, 1993. [Google Scholar]
- Kier, L.B.; Hall, H.L. (Eds.) Molecular Connectivity in Structure Activity Analysis; John Wiley & Sons: New York, NY, USA, 1986. [Google Scholar]
- Randic, M. The connectivity index 25 years after. J. Mol. Graph. Model. 2001, 20, 19–35. [Google Scholar] [CrossRef]
- Brown, R.D.J. A quantum-mechanical treatment of aliphatic compounds. Part I. Paraffins. J. Chem Soc. 1953, 2615–2621. [Google Scholar] [CrossRef]
- Constantinou, L.; Prickett, S.E.; Mavrovouniotis, M.L. Estimation of thermodynamic and physical properties of acyclic hydrocarbons using the ABC approach and conjugation operators. Ind. Eng. Chem. Res. 1993, 32, 1734–1746. [Google Scholar] [CrossRef]
- Constantinou, L.; Prickett, S.E.; Mavrovouniotis, M.L. Estimation of Properties of Acyclic Organic Compounds Using Conjugation Operators. Ind. Eng. Chem. Res. 1994, 32, 395–402. [Google Scholar] [CrossRef]
- Katrizky, A.R.; Slavov, S.H.; Dobchev, D.A.; Karelson, M. Rapid QSPR model development technique for prediction of vapor pressure of organic compounds. Comput. Chem. Eng. 2007, 31, 1123–1130. [Google Scholar] [CrossRef]
- Kahrs, O.; Brauner, N.; Cholakov, G.S.; Stateva, R.P.; Marquardt, W.; Shacham, M. Analysis and refinement of the targeted QSPR method. Comput. Chem. Eng. 2008, 32, 1397–1410. [Google Scholar] [CrossRef]
- Ceriani, R.; Gani, R.; Meirelles, A.J.A. Prediction of heat capacities and heats of vaporization of organic liquids by group contribution methods. Fluid Phase Equilib. 2009, 283, 49–55. [Google Scholar] [CrossRef]
- Hukkerikar, A.S.; Meier, R.J.; Sin, G.; Gani, R. A method to estimate the enthalpy of formation of organic compounds with chemical accuracy. Fluid Phase Equilib. 2013, 348, 23–32. [Google Scholar] [CrossRef]
- Hukkerikar, A.S.; Kalakul, S.; Sarup, B.; Young, D.M.; Sin, G.; Gani, R. Estimation of Environment-Related Properties of Chemicals for Design of Sustainable Processes: Development of Group-Contribution+ (GC+) Property Models and Uncertainty Analysis. J. Chem. Inf. Model. 2012, 52, 2823–2839. [Google Scholar] [CrossRef] [PubMed]
- Meier, R.J.; Gundersen, M.T.; Woodley, J.M.; Schürmann, M. A Practical and Fast Method to Predict the Thermodynamic Preference of ω-Transaminase-Based Transformations. Chem. Cat. Chem. 2015, 7, 2594–2597. [Google Scholar] [CrossRef]
- Dearden, J. Expert Systems for Toxicity Prediction. In Situ Toxicology, Chapter 19; Cronin, M., Madden, J., Eds.; The Royal Society of Chemistry: London, UK, 2010. [Google Scholar]
- Thomas, R.S.; Black, M.B.; Li, L.; Healy, E.; Chu, T.M.; Bao, W.; Andersen, M.E.; Wolfinger, R.D. A comprehensive statistical analysis of predicting in vivo hazard using high-throughput in vitro screening. Toxicol. Sci. 2012, 128, 398–417. [Google Scholar] [CrossRef]
- Fourches, D.; Muratov, E.; Tropsha, A. Trust but Verify: On the Importance of Chemical Structure Curation in Cheminformatics and QSAR Modeling Research. J. Chem. Inf. Model. 2010, 50, 1189–1204. [Google Scholar] [CrossRef]
- Renner, R. The KOW Controversy. Doubts about the quality of basic physicochemical data for hydrophobic organic compounds could be undermining many environmental models and assessments. Environ. Sci. Technol. 2002, 36, 411A–413A. [Google Scholar] [CrossRef]
- Atkins, P.W. (Ed.) Physical Chemistry, 2nd ed.; Oxford University Press: Oxford, UK, 1982. [Google Scholar]
- Vansteenkiste, P.; Verstraelen, T.; Van Speybroeck, V.; Waroquier, M. Ab initio calculation of entropy and heat capacity of gas-phase n-alkanes with hetero-elements O and S: Ethers/alcohols and sulfides/thiols. Chem. Phys. 2006, 328, 251–258. [Google Scholar] [CrossRef]
- Böhme, A.; Thaens, D.; Schramm, F.; Paschke, A.; Schüürmann, G. Thiol Reactivity and Its Impact on the Ciliate Toxicity of α,β-Unsaturated Aldehydes, Ketones and Esters. Chem. Res. Toxicol. 2010, 23, 1905–1912. [Google Scholar] [CrossRef] [PubMed]
- Curtiss, L.A.; Raghavachari, K.; Trucks, G.W.; Pople, J.A. Gaussian-2 theory for molecular energies of first- and second-row compounds. J. Chem. Phys. 1991, 94, 7221–7230. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Ceriani, R.; Gani, R.; Liu, Y.A. Prediction of vapor pressure and heats of vaporization of edible oil/fat compounds by group contribution. Fluid Phase Equilib. 2013, 337, 53–59. [Google Scholar] [CrossRef]
- Gasteiger, J.; Zupan, J. Neural Networks in Chemistry. Angew. Chem. Int. Ed. 1993, 32, 503–527. [Google Scholar] [CrossRef]
- Habibi-Yangjeh, A.; Pourbasheer, E.; Danandeh-Jenagharad, M. Prediction of Melting Point for Drug-like Compounds Using Principal Component-Genetic Algorithm-Artificial Neural Network. Bull. Korean Chem. Soc. 2008, 29, 833–841. [Google Scholar] [CrossRef]
- Habibi-Yangjeh, A.; Pourbasheer, E.; Danandeh-Jenagharad, M. Prediction of basicity constants of various pyridines in aqueous solution using a principal component-genetic algorithm-artificial neural networks. Monatsh. Chem. 2008, 139, 1423–1431. [Google Scholar] [CrossRef]
- Smith, J.S.; Isayev, O.; Roitberg, A.E. ANI-1: An extensible neural network potential with DFT accuracy at force field computational cost. Chem. Sci. 2017, 8, 3192–3203. [Google Scholar] [CrossRef] [PubMed]
- Tabares-Mendoza, C.; Guadaramma, P. Predicting the catalytic efficiency by quantum-chemical descriptors: Theoretical study of pincer metallic complexes involved in the catalytic Heck reaction. J. Organometal. Chem. 2006, 691, 2978–2986. [Google Scholar] [CrossRef]
- Jover, J.; Bosque, R.; Martinho Simöes, J.A.; Sales, J. CORAL: QSPRs of enthalpies of formation of organometallic compounds. J. Organometal. Chem. 2008, 693, 1261–1268. [Google Scholar] [CrossRef]
- Fey, N. The contribution of computational studies to organometallic catalysis: Descriptors, mechanisms and models. Dalton Trans. 2010, 39, 296–310. [Google Scholar] [CrossRef] [PubMed]
- Fey, N. Lost in chemical space? Maps to support organometallic catalysis. Chem. Cent. J. 2015, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-Y.; Hsu, M.-T.; Esposito, E.X.; Tseng, Y.J. Oversampling to Overcome Overfitting: Exploring the Relationship between Data Set Composition. Molecular Descriptors and Predictive Modeling Methods. J. Chem. Inf. Model. 2013, 53, 958–971. [Google Scholar] [CrossRef]
- Espinosa, G.; Yaffe, D.; Cohen, Y.; Arenas, A.; Giralt, F. Neural Network Based Quantitative Structural Property Relations (QSPRs) for Predicting Boiling Points of Aliphatic Hydrocarbons. J. Chem. Inf. Comput. Sci. 2000, 40, 859–870. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physical Property | Achieved | Needed |
|---|---|---|
| Heat of formation | 2.5–4 | 4 kJ/mol |
| Liquid heat capacity | >10% | 10% |
| Liquid density | >2% | 2% |
| Vapour pressure | >10% | 10% |
| Normal boiling point | 6 K | 3 K |
| Melting point | 20 K | <10 K |
| Toxicity | typically 70% correct | >95% correct |
| Discipline | r Meaningful if | R2 Meaningful if |
|---|---|---|
| Physics | r < −0.95 or 0.95 < r | 0.9 < R2 |
| Chemistry | r < −0.9 or 0.9 < r | 0.8 < R2 |
| Biology | r < −0.7 or 0.7 < r | 0.5 < R2 |
| Social Sciences | r < −0.6 or 0.6 < r | 0.35 < R2 |
| Tool | Concordance (%) = Agreement with Experiment |
|---|---|
| TOPKAT | 58 |
| DEREK | 59 |
| CASE | 49 |
| COMPACT | 54 |
| Human Experts | 75 |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meier, R.J. A Way towards Reliable Predictive Methods for the Prediction of Physicochemical Properties of Chemicals Using the Group Contribution and other Methods. Appl. Sci. 2019, 9, 1700. https://doi.org/10.3390/app9081700
Meier RJ. A Way towards Reliable Predictive Methods for the Prediction of Physicochemical Properties of Chemicals Using the Group Contribution and other Methods. Applied Sciences. 2019; 9(8):1700. https://doi.org/10.3390/app9081700
Chicago/Turabian StyleMeier, Robert J. 2019. "A Way towards Reliable Predictive Methods for the Prediction of Physicochemical Properties of Chemicals Using the Group Contribution and other Methods" Applied Sciences 9, no. 8: 1700. https://doi.org/10.3390/app9081700
APA StyleMeier, R. J. (2019). A Way towards Reliable Predictive Methods for the Prediction of Physicochemical Properties of Chemicals Using the Group Contribution and other Methods. Applied Sciences, 9(8), 1700. https://doi.org/10.3390/app9081700