Distinct Bacterial Consortia Established in ETBE-Degrading Enrichments from a Polluted Aquifer


 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Site Characteristics and Water Sample Collection
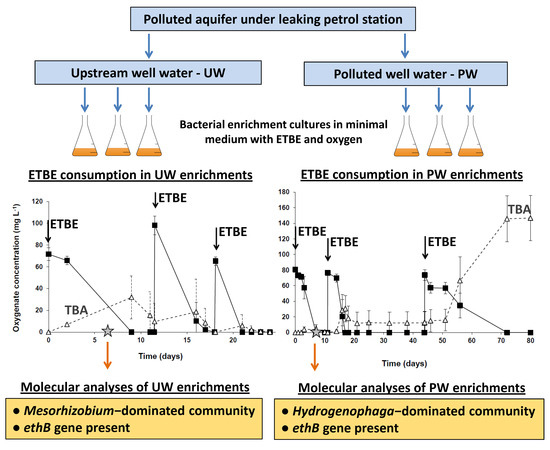
2.2. Enrichment Cultures and Their Biodegradation Potential
2.3. DNA Extraction
2.4. PCR Amplification of 16S rRNA (rrs) Genes and ethB Genes
2.5. Sequencing and Sequence Analysis
2.6. Definition of New Probes for 16S rRNA Gene-Based Taxonomical Microarray
2.7. In Vitro Transcription, Microarray Hybridization, Scanning and Image Analysis
2.8. Microarray Data Filtration and Statistical Analysis
3. Results
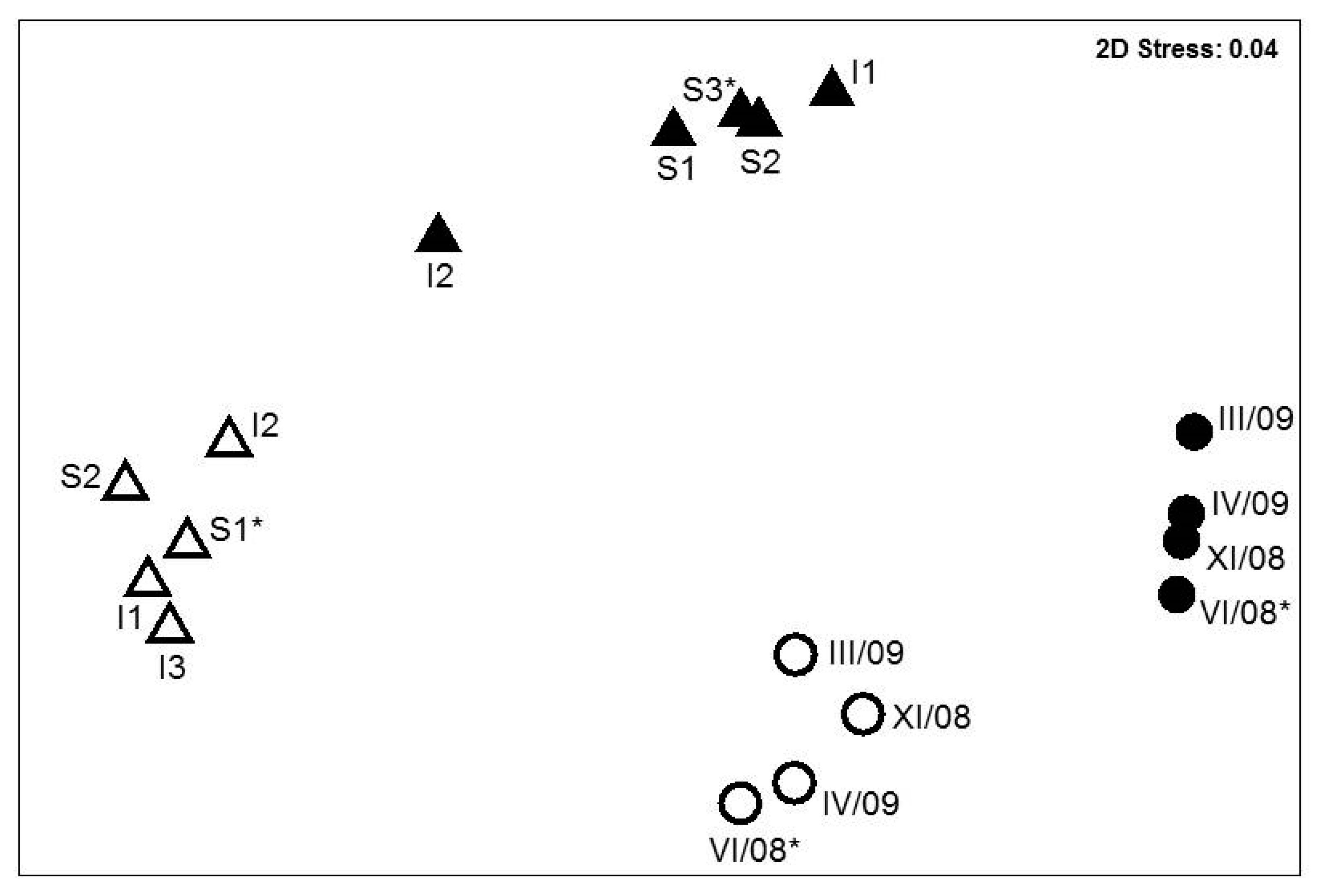
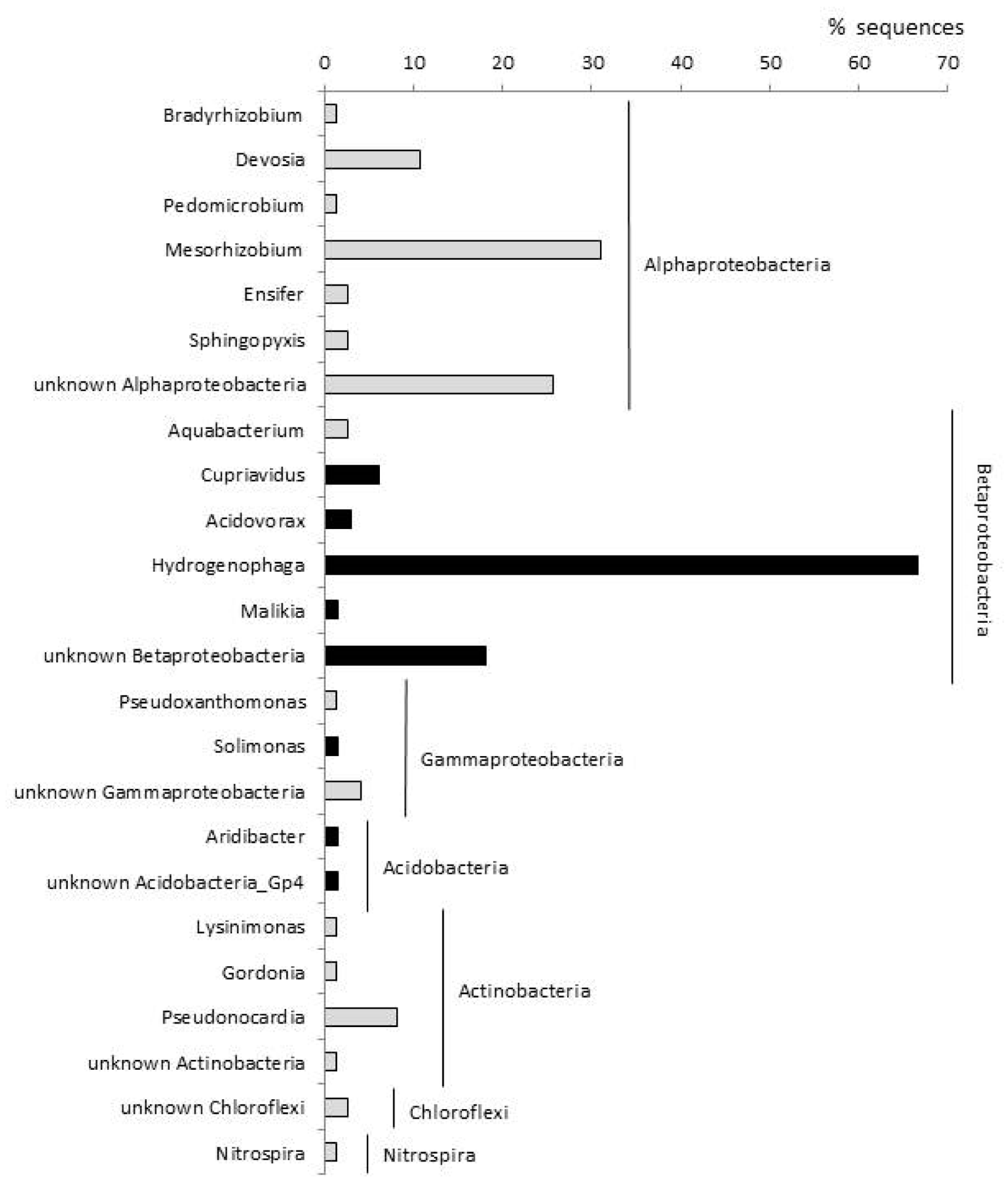
3.1. Bacterial Communities in Polluted and Upstream Well Water
3.2. ETBE Biodegradation Potential in the ETBE Enrichments from the Upstream and the Polluted Well
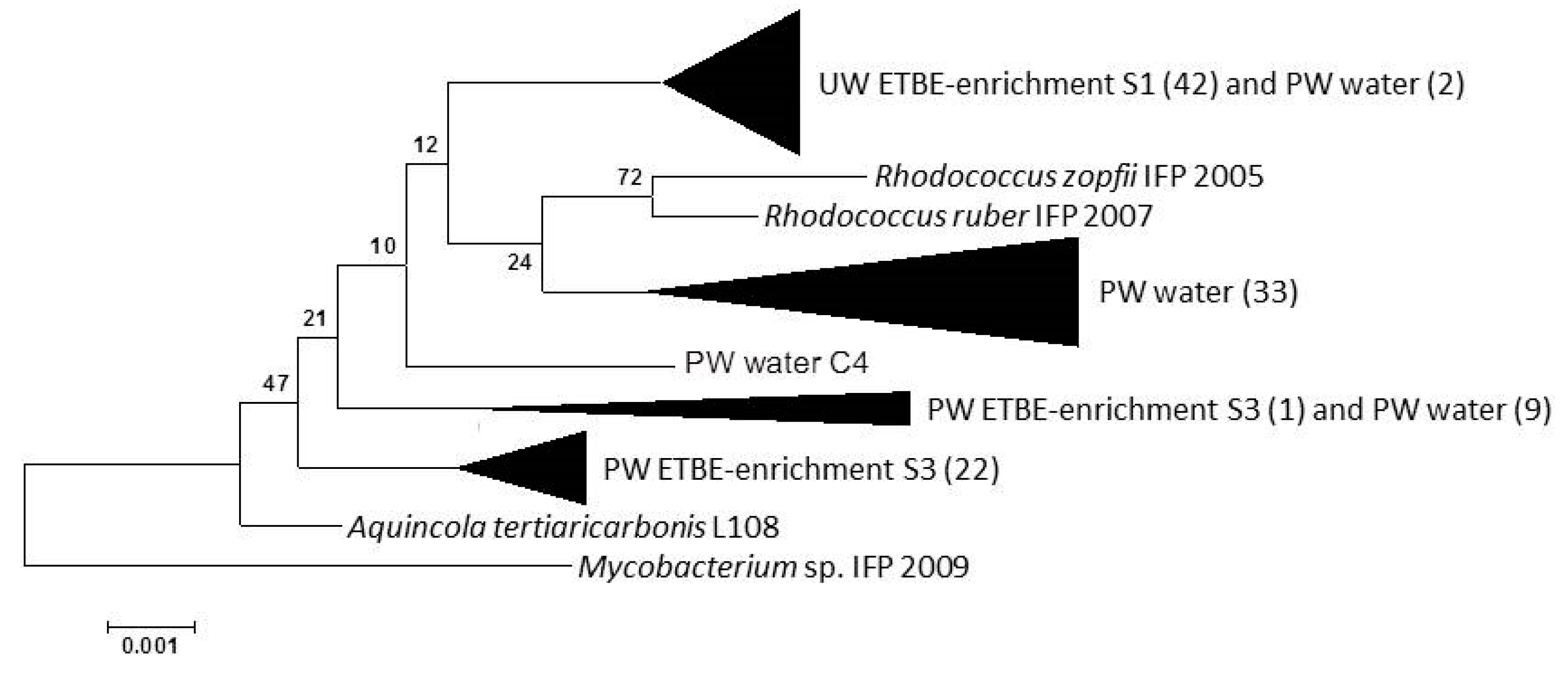
3.3. Bacterial Communities in the ETBE-Enrichment Cultures
3.4. Genetic Diversity of the ethB Gene
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lopes Ferreira, N.; Malandain, C.; Fayolle-Guichard, F. Enzymes and genes involved in the aerobic biodegradation of methyl tert-butyl ether (MTBE). Appl. Microbiol. Biot. 2006, 72, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Rosell i Linares, M. Vigilencia Ambiental de Metil Tert-Butil eer (MTBE) un Additiu de la Gasolina en Aigües i Sols. Ph.D. Thesis, Université de Barcelone Faculté de Chimie, Barcelona, Spain, July 2006. [Google Scholar]
- Hyman, M. Biodegradation of gasoline ether oxygenates. Curr. Opin. Biotechnol. 2013, 24, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Fayolle-Guichard, F.; Durand, J.; Cheucle, M.; Rosell, M.; Michelland, R.J.; Tracol, J.P.; Le Roux, F.; Grundmann, G.; Atteia, O.; Richnow, H.H.; et al. Study of an aquifer contaminated by ethyl tert-butyl ether (ETBE): Site characterization and on-site bioremediation. J. Hazard. Mater. 2012, 201, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Fayolle, F.; Hernandez, G.; Le Roux, F.; Vandecasteele, J.P. Isolation of two aerobic strains that degrade efficiently ethyl t-butyl ether (ETBE). Biotechnol. Lett. 1998, 20, 283–286. [Google Scholar] [CrossRef]
- Kharoune, M.; Kharoune, L.; Lebeault, J.M.; Pauss, A. Isolation and characterization of two aerobic bacterial strains that completely degrade ethyl tert-butyl ether (ETBE). Appl. Microbiol. Biot. 2001, 55, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Béguin, P.; Chauvaux, S.; Miras, I.; Francois, A.; Fayolle, F.; Monot, F. Genes involved in the degradation of ether fuels by bacteria of the Mycobacterium/Rhodococcus group. Oil Gas Sci. Technol. 2003, 58, 489–495. [Google Scholar] [CrossRef]
- Müller, R.H.; Rohwerder, T.; Harms, H. Degradation of fuel oxygenates and their main intermediates by Aquincola tertiaricarbonis L108. Microbiology 2008, 154, 1414–1421. [Google Scholar] [CrossRef] [PubMed]
- Auffret, M.; Labbé, D.; Thouand, G.; Greer, C.W.; Fayolle-Guichard, F. Degradation of a mixture of hydrocarbons, gasoline, and diesel oil additives by Rhodococcus aetherivorans and Rhodococcus wratislaviensis. Appl. Environ. Microbiol. 2009, 75, 7774–7782. [Google Scholar] [CrossRef]
- McKelvie, J.R.; Hyman, M.R.; Elsner, M.; Smith, C.; Aslett, D.M.; Lacrampe-Couloume, G.; Lollar, B.S. Isotopic fractionation of methyl tert-butyl ether suggests different initial reaction mechanisms during aerobic biodegradation. Environ. Sci. Technol. 2009, 43, 2793–2799. [Google Scholar] [CrossRef]
- Le Digabel, Y.; Demanèche, S.; Benoit, Y.; Vogel, T.M.; Fayolle-Guichard, F. Ethyl tert-butyl ether (ETBE) biodegradation by a syntrophic association of Rhodococcus sp. IFP 2042 and Bradyrhizobium sp. IFP 2049 isolated from a polluted aquifer. Appl. Microbiol. Biot. 2013, 97, 10531–10539. [Google Scholar] [CrossRef]
- Le Digabel, Y.; Demanèche, S.; Benoit, Y.; Fayolle-Guichard, F.; Vogel, T.M. Ethyl tert-butyl ether (ETBE)-degrading microbial communities in enrichments from polluted environments. J. Hazard. Mater. 2014, 279, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Bombach, P.; Nägele, N.; Rosell, M.; Richnow, H.H.; Fischer, A. Evaluation of ethyl tert-butyl ether biodegradation in a contaminated aquifer by compound-specific isotope analysis and in situ microcosms. J. Hazard. Mater. 2015, 286, 100–106. [Google Scholar] [CrossRef]
- Van der Waals, M.J.; Pijls, C.; Sinke, A.J.; Langenhoff, A.A.; Smidt, H.; Gerritse, J. Anaerobic degradation of a mixture of MtBE, EtBE, TBA, and benzene under different redox conditions. Appl. Microbiol. Biot. 2018, 102, 3387–3397. [Google Scholar] [CrossRef]
- Chauvaux, S.; Chevalier, F.; Le Dantec, C.; Fayolle, F.; Miras, I.; Kunst, F.; Beguin, P. Cloning of a genetically unstable cytochrome P-450 gene cluster involved in degradation of the pollutant ethyl tert-butyl ether by Rhodococcus ruber. J. Bacteriol. 2001, 183, 6551–6557. [Google Scholar] [CrossRef]
- Malandain, C.; Fayolle-Guichard, F.; Vogel, T.M. Cytochromes P450-mediated degradation of fuel oxygenates by environmental isolates. FEMS Microbiol. Ecol. 2010, 72, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Mikesková, H.; Novotný, Č.; Svobodová, K. Interspecific interactions in mixed microbial cultures in a biodegradation perspective. Appl. Microbiol. Biot. 2012, 95, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.C.; Schirmer, M.; Weiss, H.; Haderlein, S.B. Microbial degradation of methyl tert-butyl ether and tert-butyl alcohol in the subsurface. J. Contam. Hydrol. 2004, 70, 173–203. [Google Scholar] [CrossRef] [PubMed]
- Babé, A.; Labbé, D.; Monot, F.; Greer, C.; Fayolle-Guichard, F. Biodegradability of Oxygenates by Microflora from MTBE-Contaminated Sites: New Molecular Tools. In Fuel Oxygenates. The Handbook of Environmental Chemistry; Barceló, D., Ed.; Springer: Berlin/Heidelberg, Germany, 2007. [Google Scholar]
- Lin, C.W.; Tsai, S.L.; Hou, S.N. Effects of environmental settings on MTBE removal for a mixed culture and its monoculture isolation. Appl. Microbiol. Biot. 2007, 74, 194–201. [Google Scholar] [CrossRef]
- Kharoune, M.; Kharoune, L.; Lebeault, J.M.; Pauss, A. Aerobic degradation of ethyl-tert-butyl ether by a microbial consortium: Selection and evaluation of biodegradation ability. Environ. Toxicol. Chem. 2002, 21, 2052–2058. [Google Scholar] [CrossRef]
- Van der Waals, M.J.; Plugge, C.; Meima-Franke, M.; de Waard, P.; Bodelier, P.L.; Smidt, H.; Gerritse, J. Ethyl tert-butyl ether (EtBE) degradation by an algal-bacterial culture obtained from contaminated groundwater. Water Res. 2019, 148, 314–323. [Google Scholar] [CrossRef]
- Piveteau, P.; Fayolle, F.; Vandecasteele, J.P.; Monot, F. Biodegradation of tert-butyl alcohol and related xenobiotics by a methylotrophic bacterial isolate. Appl. Microbiol. Biot. 2001, 55, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Pospiech, A.; Neumann, B. A versatile quick-prep of genomic DNA from gram-positive bacteria. Trends Genet. 1995, 66, 217–218. [Google Scholar] [CrossRef]
- Bruce, K.D.; Hiorns, W.D.; Hobman, J.L.; Osborn, A.M.; Strike, P.; Ritchie, D.A. Amplification of DNA from native populations of soil bacteria by using the polymerase chain-reaction. Appl. Environ. Microbiol. 1992, 58, 3413–3416. [Google Scholar] [PubMed]
- Sanguin, H.; Herrera, A.; Oger-Desfeux, C.; Dechesne, A.; Simonet, P.; Navarro, E.; Vogel, T.M.; Moënne-Loccoz, Y.; Nesme, X.; Grundmann, G.L. Development and validation of a prototype 16S rRNA-based taxonomic microarray for Alphaproteobacteria. Environ. Microbiol. 2006, 8, 289–307. [Google Scholar] [CrossRef]
- Huber, T.; Faulkner, G.; Hugenholtz, P. Bellerophon: A program to detect chimeric sequences in multiple sequence alignments. Bioinformatics 2004, 20, 2317–2319. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Tamura, K.; Dudley, J.; Nei, M.; Kumar, S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) Software Version 4.0. Mol. Biol. Evol. 2007, 24, 1596–1599. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Pommier, T.; Canback, B.; Lundberg, P.; Hagström, A.; Tunlid, A. RAMI: A tool for identification and characterization of phylogenetic clusters in microbial communities. Bioinformatics 2009, 25, 736–742. [Google Scholar] [CrossRef]
- Kyselková, M.; Kopecký, J.; Frapolli, M.; Défago, G.; Ságová-Marečková, M.; Grundmann, G.L.; Moënne-Loccoz, Y. Comparison of rhizobacterial community composition in soil suppressive or conducive to tobacco black root rot disease. ISME J. 2009, 3, 1127–1138. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, W.; Strunk, O.; Westram, R.; Richter, L.; Meier, H.; Yadhukumar Buchner, A.; Lai, T.; Steppi, S.; Jobb, G.; Förster, W.; et al. ARB: A software environment for sequence data. Nucl. Acids Res. 2004, 32, 1363–1371. [Google Scholar] [CrossRef]
- Sanguin, H.; Remenant, B.; Dechesne, A.; Thioulouse, J.; Vogel, T.M.; Nesme, X.; Moënne-Loccoz, Y.; Grundmann, G.L. Potential of a 16S rRNA-based taxonomic microarray for analyzing the rhizosphere effects of maize on Agrobacterium spp. and bacterial communities. Appl. Environ. Microbiol. 2006, 72, 4302–4312. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Gloeckner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucl. Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef] [PubMed]
- Stralis-Pavese, N.; Sessitsch, A.; Weilharter, A.; Reichenauer, T.; Riesing, J.; Csontos, J.; Murrell, J.C.; Bodrossy, L. Optimization of diagnostic microarray for application in analysing landfill methanotroph communities under different plant covers. Environ. Microbiol. 2004, 6, 347–363. [Google Scholar] [CrossRef] [PubMed]
- Bouffaud, M.L.; Kyselková, M.; Gouesnard, B.; Grundmann, G.; Muller, D.; Moënne-Loccoz, Y. Is diversification history of maize influencing selection of soil bacteria by roots? Mol. Ecol. 2012, 21, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Clarke, K.R.; Gorley, R.N. PRIMER V6: User Manual/Tutorial, PRIMER-E Plymouth; Plymouth Marine Laboratory: Plymouth, UK, 2006. [Google Scholar]
- Clarke, K.R. Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 1993, 18, 117–143. [Google Scholar] [CrossRef]
- Fahy, A.; McGenity, T.J.; Timmis, K.N.; Ball, A.S. Heterogeneous aerobic benzene-degrading communities in oxygen-depleted groundwaters. FEMS Microbiol. Ecol. 2006, 58, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Kyselková, M.; Kopecký, J.; Ságová-Marečková, M.; Grundmann, G.L.; Moënne-Loccoz, Y. Oligonucleotide microarray methodology for taxonomic and functional monitoring of microbial community composition. Plant Soil Environ. 2009, 55, 379–388. [Google Scholar] [CrossRef]
- Hendrickx, B.; Dejonghe, W.; Boënne, M.; Brennerova, M.; Cernik, M.; Lederer, T.; Bucheli-Witschel, M.; Bastiaens, L.; Verstraete, W.; Top, E.M.; et al. Dynamics of an oligotrophic bacterial aquifer community during contact with a groundwater plume contaminated with benzene toluene ethylbenzene and xylenes: An in situ mesocosm study. Appl. Environ. Microbiol. 2005, 71, 3815–3825. [Google Scholar] [CrossRef]
- Fahy, A.; Lethbridge, G.; Earle, R.; Ball, A.S.; Timmis, K.N.; McGenity, T.J. Effects of long-term benzene pollution on bacterial diversity and community structure in groundwater. Environ. Microbiol. 2005, 7, 1192–1199. [Google Scholar] [CrossRef] [PubMed]
- Bundy, J.G.; Paton, G.I.; Campbell, C.D. Microbial communities in different soil types do not converge after diesel contamination. J. Appl. Microbiol. 2002, 92, 276–288. [Google Scholar] [CrossRef] [PubMed]
- Auffret, M.D.; Yergeau, E.; Labbé, D.; Fayolle-Guichard, F.; Greer, C.W. Importance of Rhodococcus strains in a bacterial consortium degrading a mixture of hydrocarbons, gasoline, and diesel oil additives revealed by metatranscriptomic analysis. Appl. Microbiol. Biotechnol. 2015, 99, 2419–2430. [Google Scholar] [CrossRef] [PubMed]
- Hatzinger, P.B.; McClay, K.; Vainberg, S.; Tugusheva, M.; Condee, C.W.; Steffan, R.J. Biodegradation of methyl tert-butyl ether by a pure bacterial culture. Appl. Environ. Microbiol. 2001, 67, 5601–5607. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Raynal, M.; Pruden, A. Aerobic MTBE biodegradation in the presence of BTEX by two consortia under batch and semi-batch conditions. Biodegradation 2008, 19, 269–282. [Google Scholar] [CrossRef]
- Bastida, F.; Rosell, M.; Franchini, A.G.; Seifert, J.; Finsterbusch, S.; Jehmlich, N.; Jechalke, S.; von Bergen, M.; Richnow, H.H. Elucidating MTBE degradation in a mixed consortium using a multidisciplinary approach. FEMS Microbiol. Ecol. 2010, 73, 370–384. [Google Scholar] [CrossRef] [PubMed]
- Schuster, J.; Purswani, J.; Breuer, U.; Pozo, C.; Harms, H.; Mï, R.H.; Rohwerder, T. Constitutive expression of the cytochrome P450 EthABCD monooxygenase system enables degradation of synthetic dialkyl ethers in Aquincola tertiaricarbonis L108. Appl. Environ. Microbiol. 2013, 79, 2321–2327. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ETBE (mg/L) | MTBE (mg/L) | TBA (mg/L) | BTEX (mg/L) | TOC a (mg/L) | Nitrates (mg/L) | Sulfates (mg/L) | Oxygen (mg/L) | |
|---|---|---|---|---|---|---|---|---|
| PW | 160.8 | 2.5 | 1.9 | 4.4 | 204 | ND b | 7 | 0.33 |
| UW | ND | ND | ND | ND | ND | 3 | 25 | 2.54 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kyselková, M.; Falcão Salles, J.; Dumestre, A.; Benoit, Y.; Grundmann, G.L. Distinct Bacterial Consortia Established in ETBE-Degrading Enrichments from a Polluted Aquifer. Appl. Sci. 2019, 9, 4247. https://doi.org/10.3390/app9204247
Kyselková M, Falcão Salles J, Dumestre A, Benoit Y, Grundmann GL. Distinct Bacterial Consortia Established in ETBE-Degrading Enrichments from a Polluted Aquifer. Applied Sciences. 2019; 9(20):4247. https://doi.org/10.3390/app9204247
Chicago/Turabian StyleKyselková, Martina, Joana Falcão Salles, Alain Dumestre, Yves Benoit, and Geneviève L. Grundmann. 2019. "Distinct Bacterial Consortia Established in ETBE-Degrading Enrichments from a Polluted Aquifer" Applied Sciences 9, no. 20: 4247. https://doi.org/10.3390/app9204247
APA StyleKyselková, M., Falcão Salles, J., Dumestre, A., Benoit, Y., & Grundmann, G. L. (2019). Distinct Bacterial Consortia Established in ETBE-Degrading Enrichments from a Polluted Aquifer. Applied Sciences, 9(20), 4247. https://doi.org/10.3390/app9204247