Efficient Photoelectrochemical Water Splitting Reaction using Electrodeposited Co3Se4 Catalyst



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Electrodeposition of Co3Se4
2.3. Photoelectrochemical Measurements
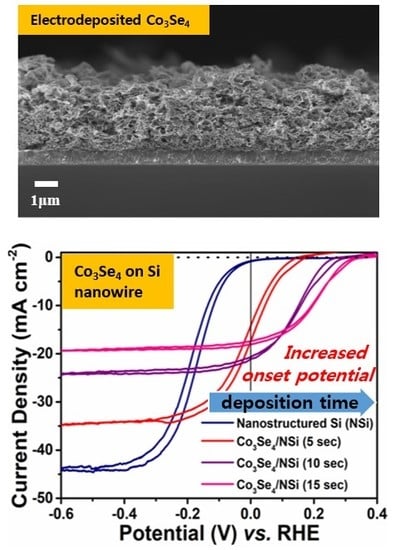
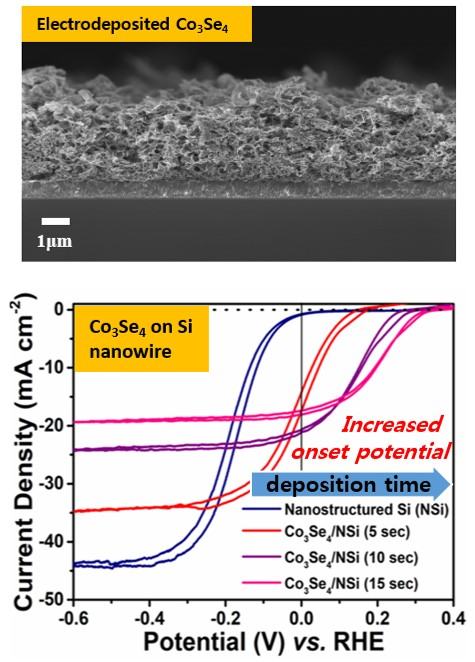
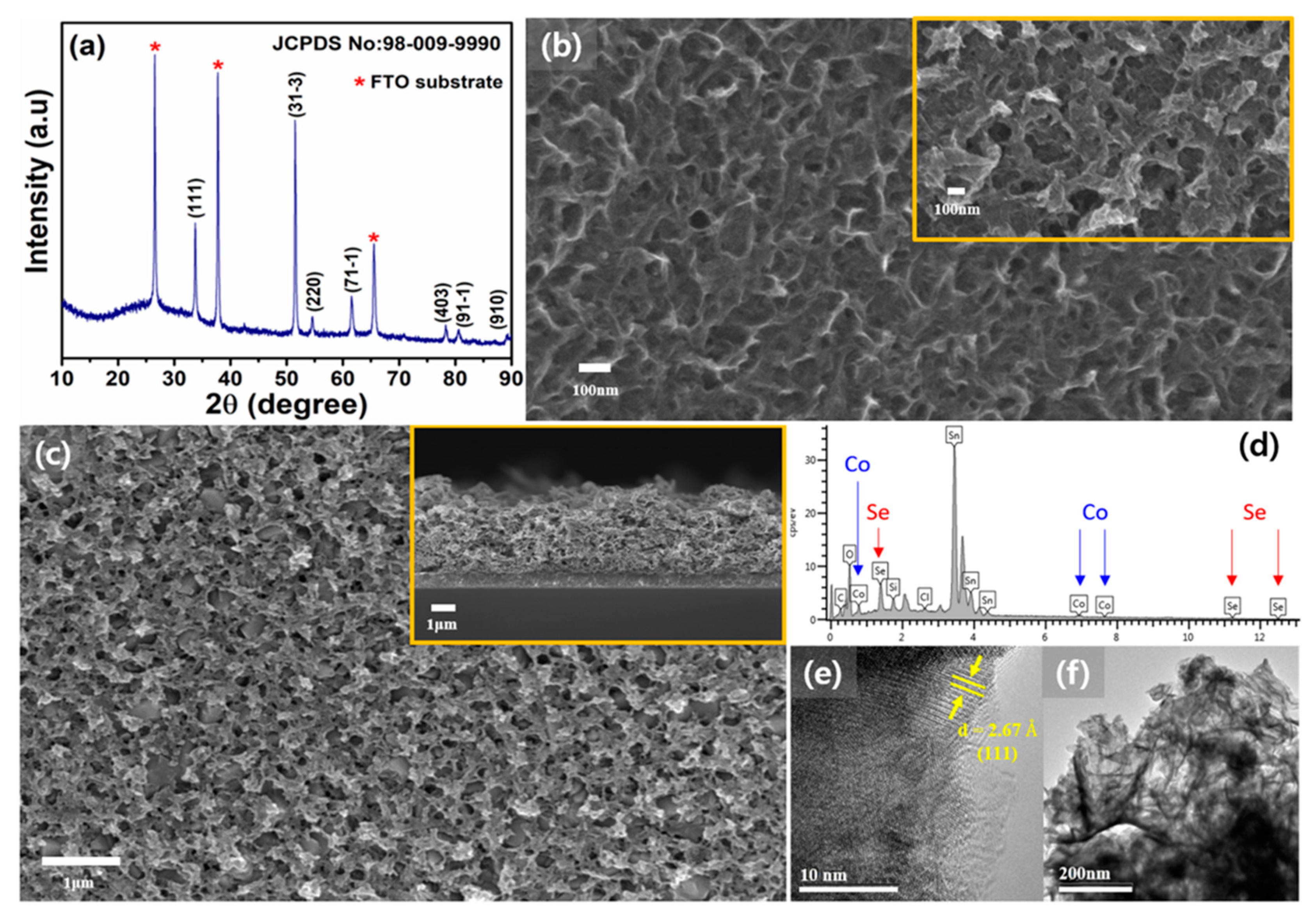
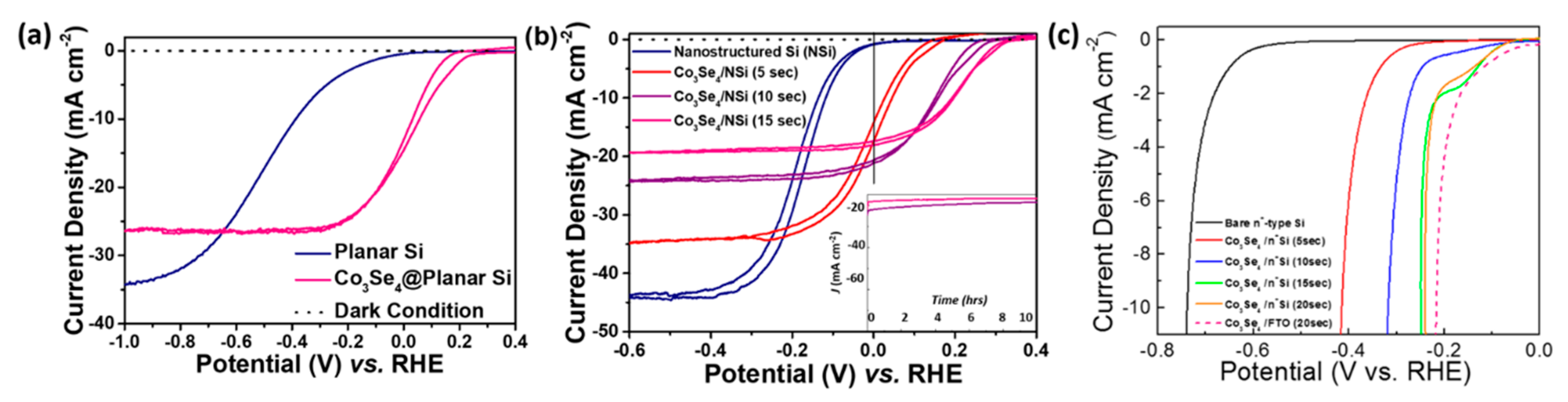
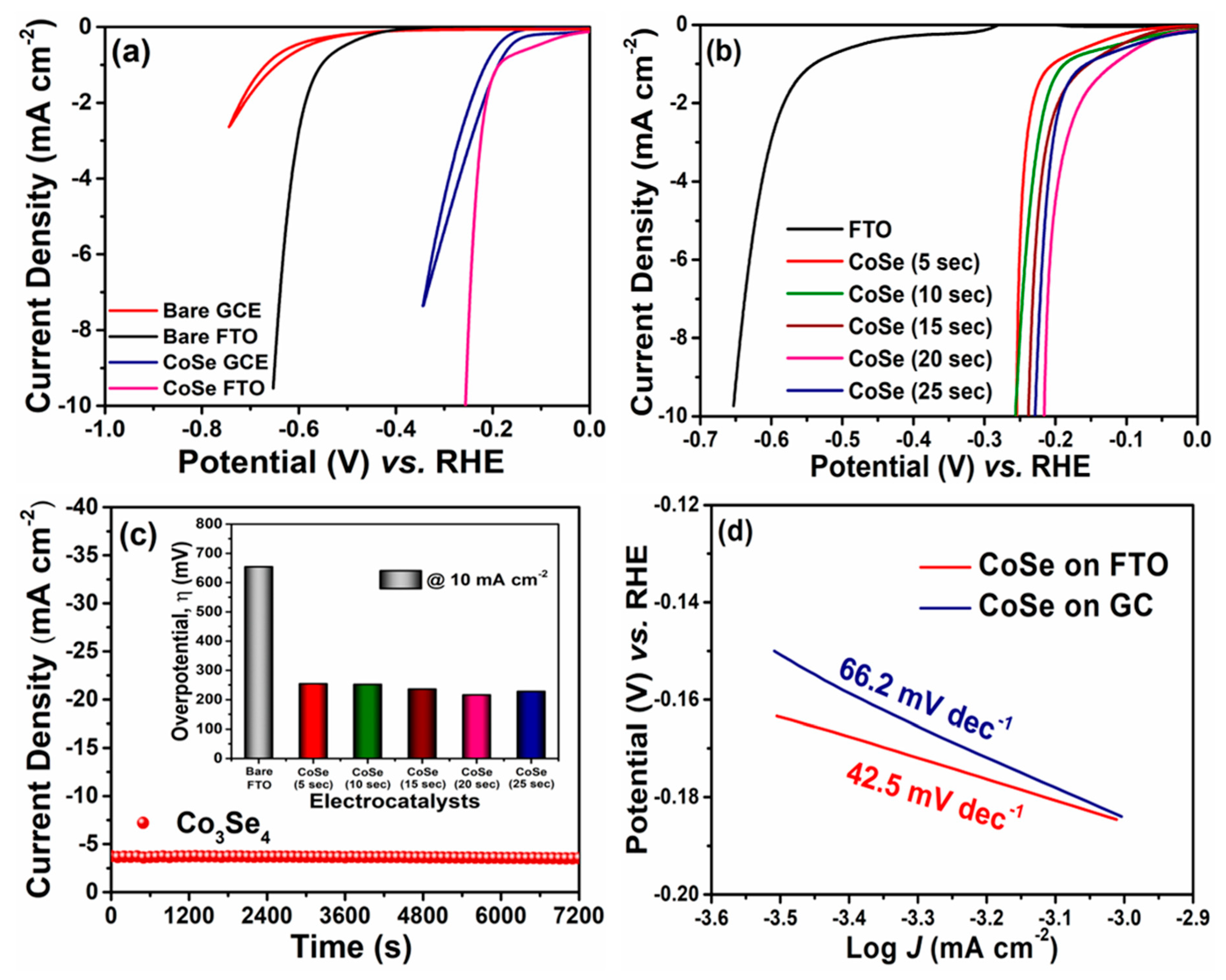
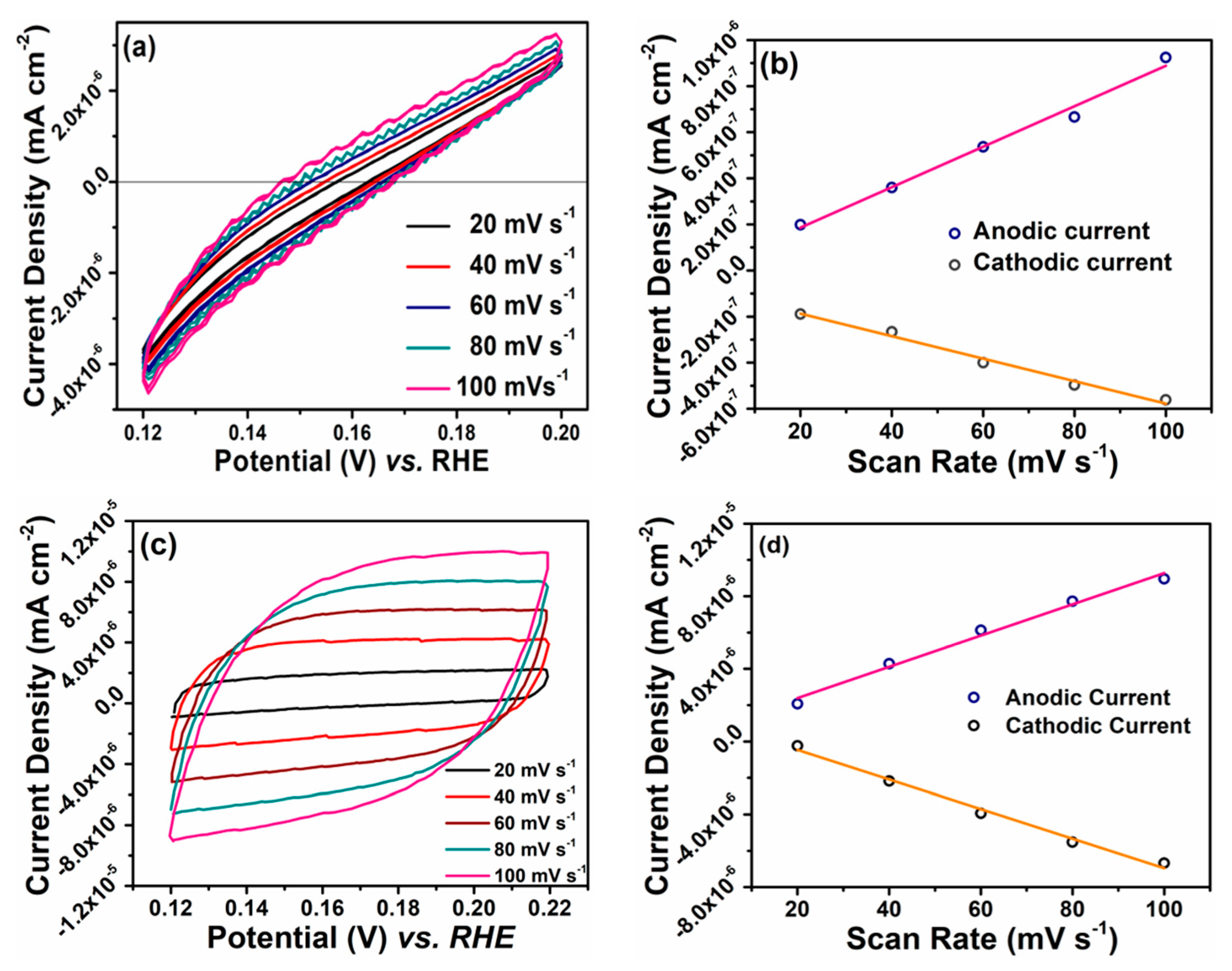
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Barber, J. Photosynthetic energy conversion: Natural and artificial. Chem. Soc. Rev. 2009, 38, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.G.; Warren, E.L.; McKone, J.R.; Boettcher, S.W.; Mi, Q.; Santori, E.A.; Lewis, N.S. Solar Water Splitting Cells. Chem. Rev. 2010, 110, 6446–6473. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Zhang, Y. Noble metal-free hydrogen evolution catalysts for water splitting. Chem. Soc. Rev. 2015, 44, 5148–5180. [Google Scholar] [CrossRef]
- Zhang, H.; Ding, Q.; He, D.; Liu, H.; Liu, W.; Li, Z.; Yang, B.; Zhang, X.; Lei, L.; Jin, S. A p-Si/NiCoSex core/shell nanopillar array photocathode for enhanced photoelectrochemical hydrogen production. Energy Environ. Sci. 2016, 9, 3113–3119. [Google Scholar] [CrossRef]
- Benck, J.D.; Lee, S.C.; Fong, K.D.; Kibsgaard, J.; Sinclair, R.; Jaramillo, T.F. Designing Active and Stable Silicon Photocathodes for Solar Hydrogen Production Using Molybdenum Sulfide Nanomaterials. Adv. Energy Mater. 2014, 4, 1400739. [Google Scholar] [CrossRef]
- Züttel, A.; Remhof, A.; Borgschulte, A.; Friedrichs, O. Hydrogen: The future energy carrier. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2010, 368, 3329–3342. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.A. Sustainable Hydrogen Production. Science 2004, 305, 972–974. [Google Scholar] [CrossRef]
- Fujishima, A.; Honda, K. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef]
- McKone, J.R.; Lewis, N.S.; Gray, H.B. Will Solar-Driven Water-Splitting Devices See the Light of Day? Chem. Mater. 2014, 26, 407–414. [Google Scholar] [CrossRef]
- Chen, X.; Liu, L.; Yu, P.Y.; Mao, S.S. Increasing Solar Absorption for Photocatalysis with Black Hydrogenated Titanium Dioxide Nanocrystals. Science 2011, 331, 746–750. [Google Scholar] [CrossRef]
- Warren, S.C.; Voïtchovsky, K.; Dotan, H.; Leroy, C.M.; Cornuz, M.; Stellacci, F.; Hébert, C.; Rothschild, A.; Grätzel, M. Identifying champion nanostructures for solar water-splitting. Nat. Mater. 2013, 12, 842. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Zhang, Q.; Su, Z.; Zhao, Z.; Wang, Y.; Li, Y.; Lu, X.; Wei, D.; Feng, G.; Yu, Q.; et al. Efficient solar water-splitting using a nanocrystalline CoO photocatalyst. Nat. Nanotechnol. 2013, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Takei, K.; Zhang, J.; Kapadia, R.; Zheng, M.; Chen, Y.-Z.; Nah, J.; Matthews, T.S.; Chueh, Y.-L.; Ager, J.W.; et al. p-Type InP Nanopillar Photocathodes for Efficient Solar-Driven Hydrogen Production. Angew. Chem. Int. Ed. 2012, 51, 10760–10764. [Google Scholar] [CrossRef] [PubMed]
- Kenney, M.J.; Gong, M.; Li, Y.; Wu, J.Z.; Feng, J.; Lanza, M.; Dai, H. High-Performance Silicon Photoanodes Passivated with Ultrathin Nickel Films for Water Oxidation. Science 2013, 342, 836–840. [Google Scholar] [CrossRef] [PubMed]
- Kye, J.; Shin, M.; Lim, B.; Jang, J.-W.; Oh, I.; Hwang, S. Platinum Monolayer Electrocatalyst on Gold Nanostructures on Silicon for Photoelectrochemical Hydrogen Evolution. ACS Nano 2013, 7, 6017–6023. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Pang, X.; Shen, S.; Qian, X.; Cheung, J.S.; Wang, D. Metal Oxide Composite Enabled Nanotextured Si Photoanode for Efficient Solar Driven Water Oxidation. Nano Lett. 2013, 13, 2064–2072. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.M.; Winkler, M.T.; Choi, H.J.; Simmons, C.B.; Needleman, D.B.; Buonassisi, T. Crystalline silicon photovoltaics: A cost analysis framework for determining technology pathways to reach baseload electricity costs. Energy Environ. Sci. 2012, 5, 5874–5883. [Google Scholar] [CrossRef]
- Oh, I.; Kye, J.; Hwang, S. Enhanced Photoelectrochemical Hydrogen Production from Silicon Nanowire Array Photocathode. Nano Lett. 2011, 12, 298–302. [Google Scholar] [CrossRef]
- Wang, X.; Peng, K.-Q.; Pan, X.-J.; Chen, X.; Yang, Y.; Li, L.; Meng, X.-M.; Zhang, W.-J.; Lee, S.-T. High-Performance Silicon Nanowire Array Photoelectrochemical Solar Cells through Surface Passivation and Modification. Angew. Chem. Int. Ed. 2011, 50, 9861–9865. [Google Scholar] [CrossRef]
- Strandwitz, N.C.; Comstock, D.J.; Grimm, R.L.; Nichols-Nielander, A.C.; Elam, J.; Lewis, N.S. Photoelectrochemical Behavior of n-type Si(100) Electrodes Coated with Thin Films of Manganese Oxide Grown by Atomic Layer Deposition. J. Phys. Chem. C 2013, 117, 4931–4936. [Google Scholar] [CrossRef]
- Lana-Villarreal, T.; Straboni, A.; Pichon, L.; Alonso-Vante, N. Photoelectrochemical characterization of p-type silicon electrodes covered with tunnelling nitride dielectric films. Thin Solid Films 2007, 515, 7376–7381. [Google Scholar] [CrossRef]
- Peng, K.-Q.; Wang, X.; Wu, X.-L.; Lee, S.-T. Platinum Nanoparticle Decorated Silicon Nanowires for Efficient Solar Energy Conversion. Nano Lett. 2009, 9, 3704–3709. [Google Scholar] [CrossRef] [PubMed]
- Kemppainen, E.; Bodin, A.; Sebok, B.; Pedersen, T.; Seger, B.; Mei, B.; Bae, D.; Vesborg, P.C.K.; Halme, J.; Hansen, O.; et al. Scalability and feasibility of photoelectrochemical H2 evolution: The ultimate limit of Pt nanoparticle as an HER catalyst. Energy Environ. Sci. 2015, 8, 2991–2999. [Google Scholar] [CrossRef]
- Faber, M.S.; Lukowski, M.A.; Ding, Q.; Kaiser, N.S.; Jin, S. Earth-Abundant Metal Pyrites (FeS2, CoS2, NiS2, and Their Alloys) for Highly Efficient Hydrogen Evolution and Polysulfide Reduction Electrocatalysis. J. Phys. Chem. C 2014, 118, 21347–21356. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Wang, H.; Lu, Z.; Cui, Y. CoSe2 Nanoparticles Grown on Carbon Fiber Paper: An Efficient and Stable Electrocatalyst for Hydrogen Evolution Reaction. J. Am. Chem. Soc. 2014, 136, 4897–4900. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, Y.; Zhang, G.; Xu, T.; Wan, P.; Sun, X. A metallic CoS2 nanopyramid array grown on 3D carbon fiber paper as an excellent electrocatalyst for hydrogen evolution. J. Mater. Chem. A 2015, 3, 6306–6310. [Google Scholar] [CrossRef]
- Gao, M.-R.; Liang, J.-X.; Zheng, Y.-R.; Xu, Y.-F.; Jiang, J.; Gao, Q.; Li, J.; Yu, S.-H. An efficient molybdenum disulfide/cobalt diselenide hybrid catalyst for electrochemical hydrogen generation. Nat. Commun. 2015, 6, 5982. [Google Scholar] [CrossRef]
- Zheng, Y.-R.; Gao, M.-R.; Gao, Q.; Li, H.-H.; Xu, J.; Wu, Z.-Y.; Yu, S.-H. An Efficient CeO2/CoSe2 Nanobelt Composite for Electrochemical Water Oxidation. Small 2015, 11, 182–188. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, B.; Wu, X.; Li, Z.; Lei, L.; Zhang, X. Polymorphic CoSe2 with Mixed Orthorhombic and Cubic Phases for Highly Efficient Hydrogen Evolution Reaction. ACS Appl. Mater. Interfaces 2015, 7, 1772–1779. [Google Scholar] [CrossRef]
- Huang, Z.; Chen, Z.; Chen, Z.; Lv, C.; Meng, H.; Zhang, C. Ni12P5 Nanoparticles as an Efficient Catalyst for Hydrogen Generation via Electrolysis and Photoelectrolysis. ACS Nano 2014, 8, 8121–8129. [Google Scholar] [CrossRef]
- Bao, X.-Q.; Fatima Cerqueira, M.; Alpuim, P.; Liu, L. Silicon nanowire arrays coupled with cobalt phosphide spheres as low-cost photocathodes for efficient solar hydrogen evolution. Chem. Commun. 2015, 51, 10742–10745. [Google Scholar] [CrossRef] [PubMed]
- Masud, J.; Swesi, A.T.; Liyanage, W.P.R.; Nath, M. Cobalt Selenide Nanostructures: An Efficient Bifunctional Catalyst with High Current Density at Low Coverage. ACS Appl. Mater. Interfaces 2016, 8, 17292–17302. [Google Scholar] [CrossRef] [PubMed]
- Roske, C.W.; Popczun, E.J.; Seger, B.; Read, C.G.; Pedersen, T.; Hansen, O.; Vesborg, P.C.; Brunschwig, B.S.; Schaak, R.E.; Chorkendorff, I. Comparison of the performance of cop-coated and pt-coated radial junction n+ p-silicon microwire-array photocathodes for the sunlight-driven reduction of water to H2 (g). J. Phys. Chem. Lett. 2015, 6, 1679–1683. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.; Bard, L.R.F. Electrochemical Methods: Fundamentals and Applications; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2001; p. 864. [Google Scholar]
- Morita, M.; Ohmi, T.; Hasegawa, E.; Kawakami, M.; Ohwada, M. Growth of native oxide on a silicon surface. J. Appl. Phys. 1990, 68, 1272–1281. [Google Scholar] [CrossRef]
- Aswal, D.; Lenfant, S.; Guerin, D.; Yakhmi, J.; Vuillaume, D. Self assembled monolayers on silicon for molecular electronics. Anal. Chim. Acta 2006, 568, 84–108. [Google Scholar] [CrossRef] [PubMed]
- Takahagi, T.; Nagai, I.; Ishitani, A.; Kuroda, H.; Nagasawa, Y. The formation of hydrogen passivated silicon single-crystal surfaces using ultraviolet cleaning and HF etching. J. Appl. Phys. 1988, 64, 3516–3521. [Google Scholar] [CrossRef]
- Chen, Y.W.; Prange, J.D.; Dühnen, S.; Park, Y.; Gunji, M.; Chidsey, C.E.D.; McIntyre, P.C. Atomic layer-deposited tunnel oxide stabilizes silicon photoanodes for water oxidation. Nat. Mater. 2011, 10, 539–544. [Google Scholar] [CrossRef]
- Randin, J.P.; Yeager, E. Differential Capacitance Study of Stress-Annealed Pyrolytic Graphite Electrodes. J. Electrochem. Soc. 1971, 118, 711–714. [Google Scholar] [CrossRef]
- Trasatti, S.; Petrii, O.A. Real surface area measurements in electrochemistry. J. Electroanal. Chem. 1992, 327, 353–376. [Google Scholar] [CrossRef]
- McCrory, C.C.L.; Jung, S.; Peters, J.C.; Jaramillo, T.F. Benchmarking Heterogeneous Electrocatalysts for the Oxygen Evolution Reaction. J. Am. Chem. Soc. 2013, 135, 16977–16987. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Si Photocathode | Efficiency (%) |
|---|---|
| Planar Si | 0.02 |
| Co3Se4 on planar Si | 0.20 |
| Nanostructured Si | 0.04 |
| Co3Se4 on nanostructured Si (5 s) | 0.47 |
| Co3Se4 on nanostructured Si (10 s) | 1.72 |
| Co3Se4 on nanostructured Si (15 s) | 2.71 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sim, Y.; John, J.; Surendran, S.; Moon, B.; Sim, U. Efficient Photoelectrochemical Water Splitting Reaction using Electrodeposited Co3Se4 Catalyst. Appl. Sci. 2019, 9, 16. https://doi.org/10.3390/app9010016
Sim Y, John J, Surendran S, Moon B, Sim U. Efficient Photoelectrochemical Water Splitting Reaction using Electrodeposited Co3Se4 Catalyst. Applied Sciences. 2019; 9(1):16. https://doi.org/10.3390/app9010016
Chicago/Turabian StyleSim, Yelyn, Jude John, Subramani Surendran, Byeolee Moon, and Uk Sim. 2019. "Efficient Photoelectrochemical Water Splitting Reaction using Electrodeposited Co3Se4 Catalyst" Applied Sciences 9, no. 1: 16. https://doi.org/10.3390/app9010016
APA StyleSim, Y., John, J., Surendran, S., Moon, B., & Sim, U. (2019). Efficient Photoelectrochemical Water Splitting Reaction using Electrodeposited Co3Se4 Catalyst. Applied Sciences, 9(1), 16. https://doi.org/10.3390/app9010016