Synthesis and Investigation of Thermal Properties of Highly Pure Carboxylic Fatty Esters to Be Used as PCM

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis Development
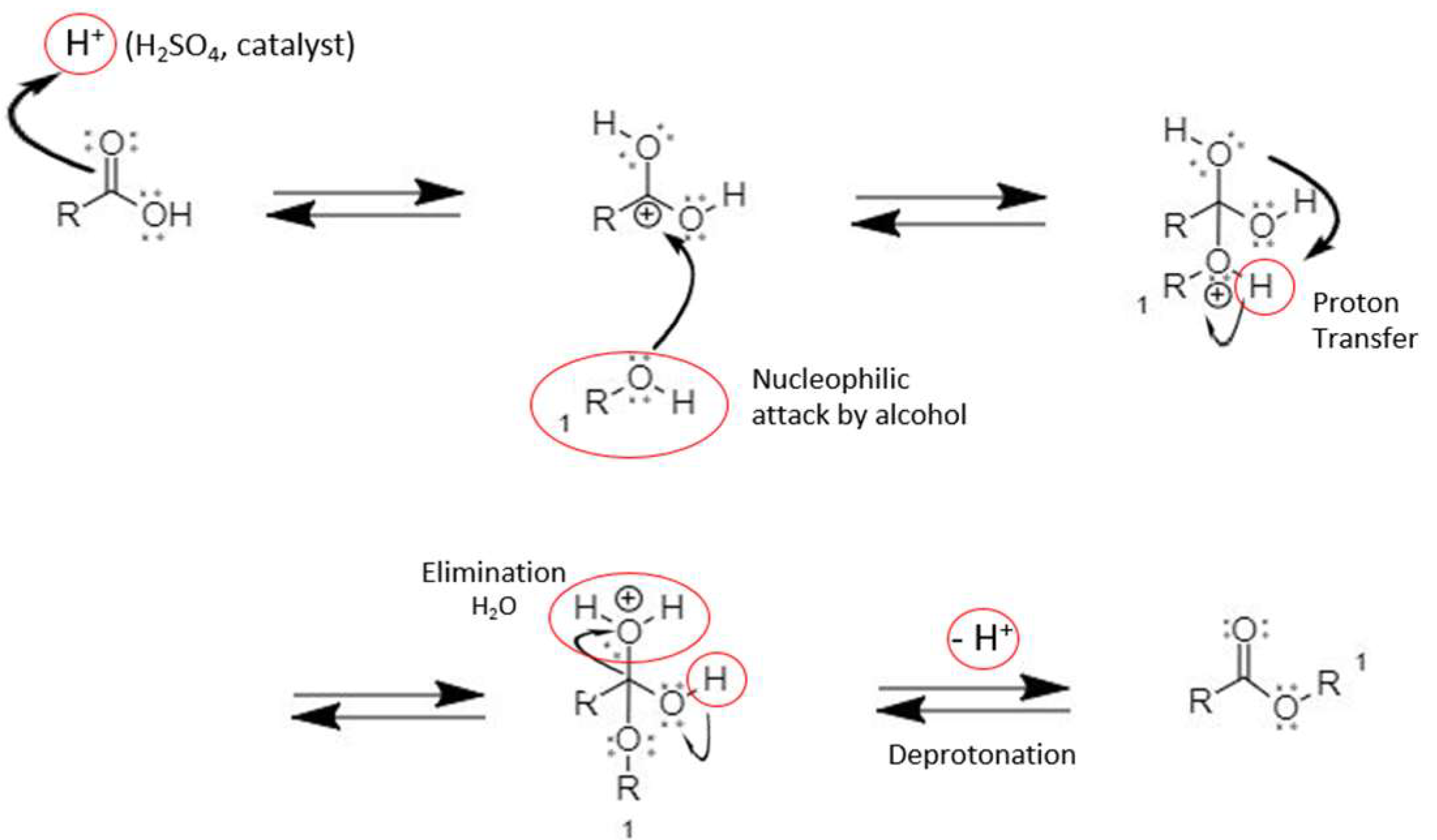
2.1.1. Synthesis Mechanism and Instrumentation
2.1.2. Purification
2.2. Characterization
2.2.1. Differential Scanning Calorimetry (DSC)
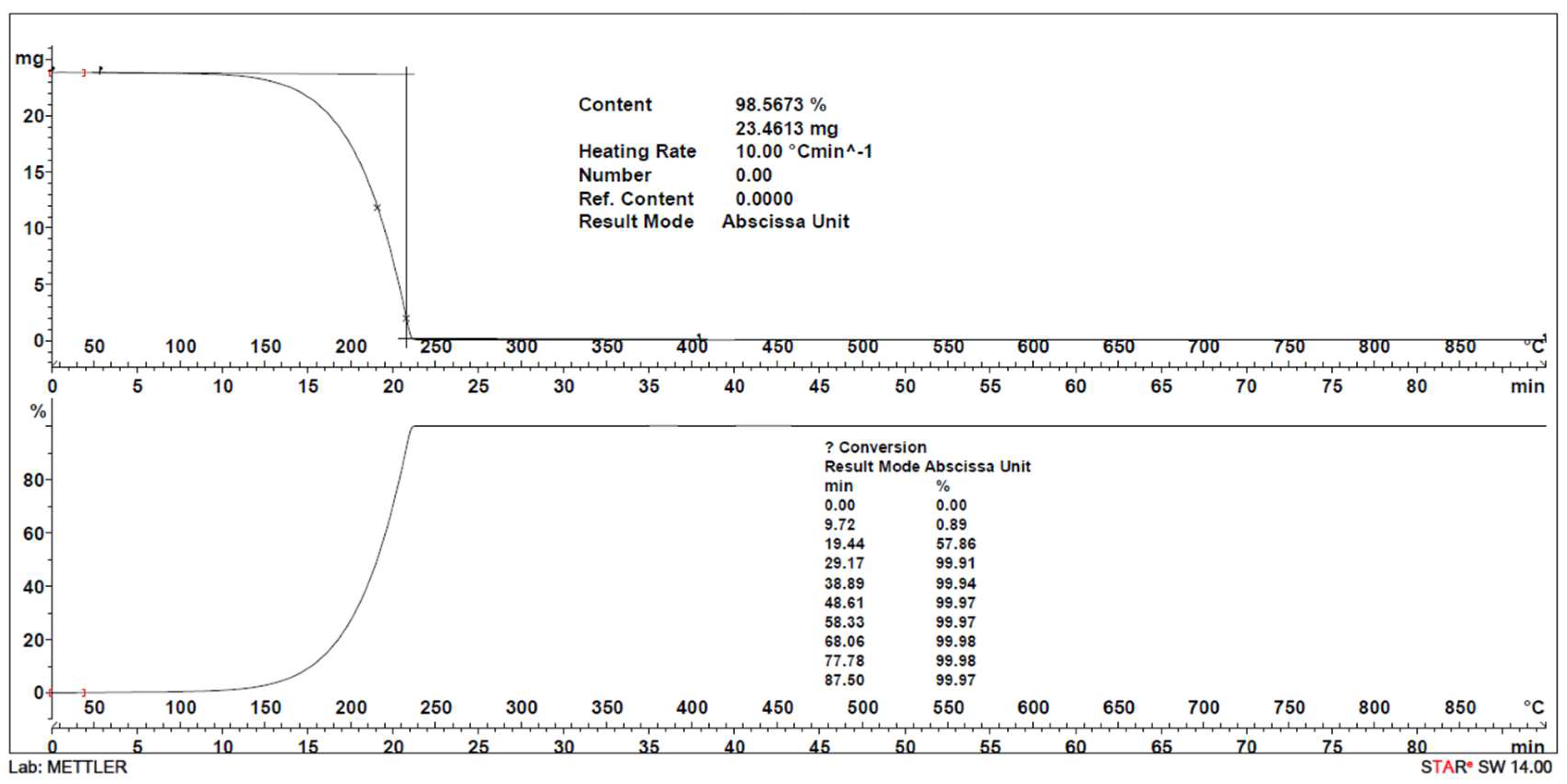
2.2.2. Thermal Gravimetric Analysis (TGA)
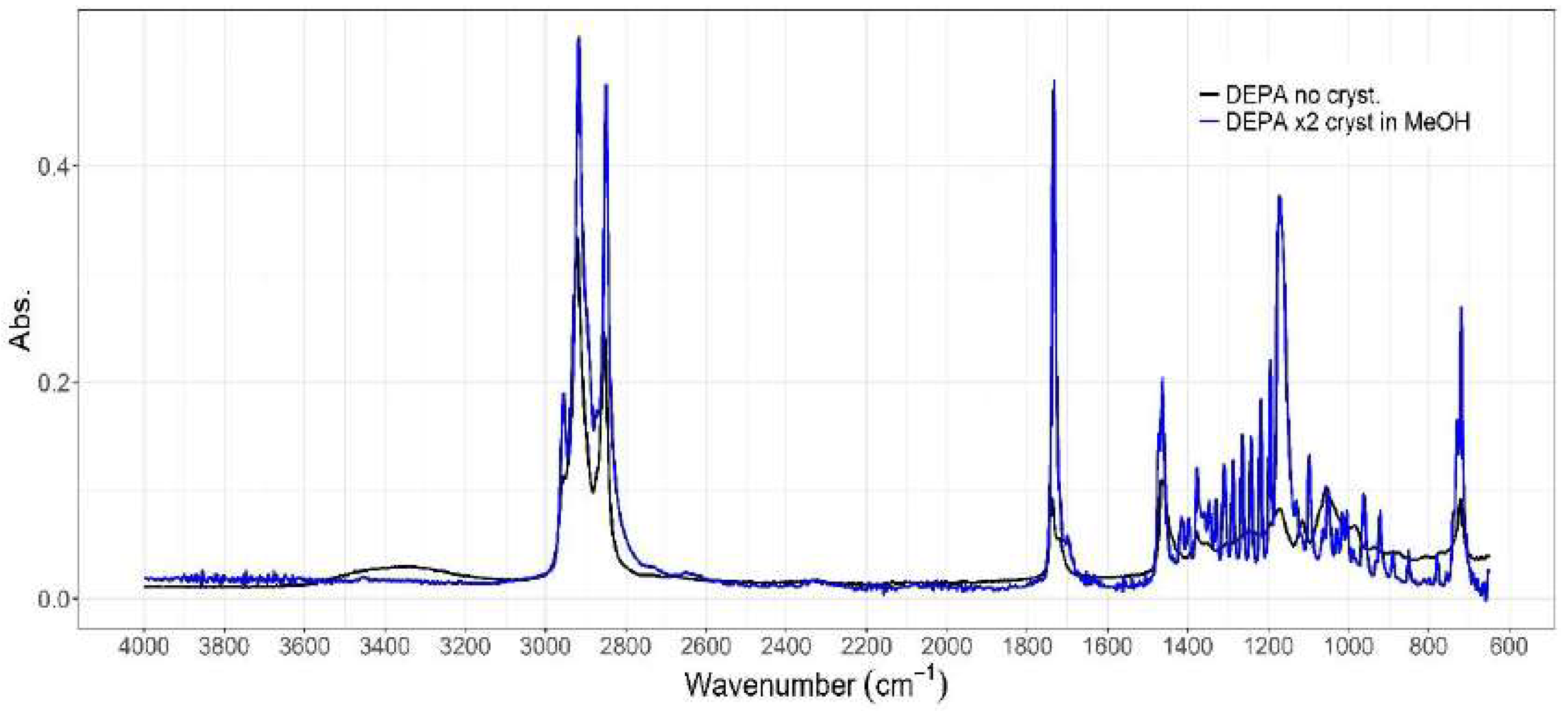
2.2.3. Attenuated Total Reflectance Infrared Spectroscopy (ATR-IR)
2.2.4. Gas Chromatography Coupled with Mass Spectroscopy (GC-MS)
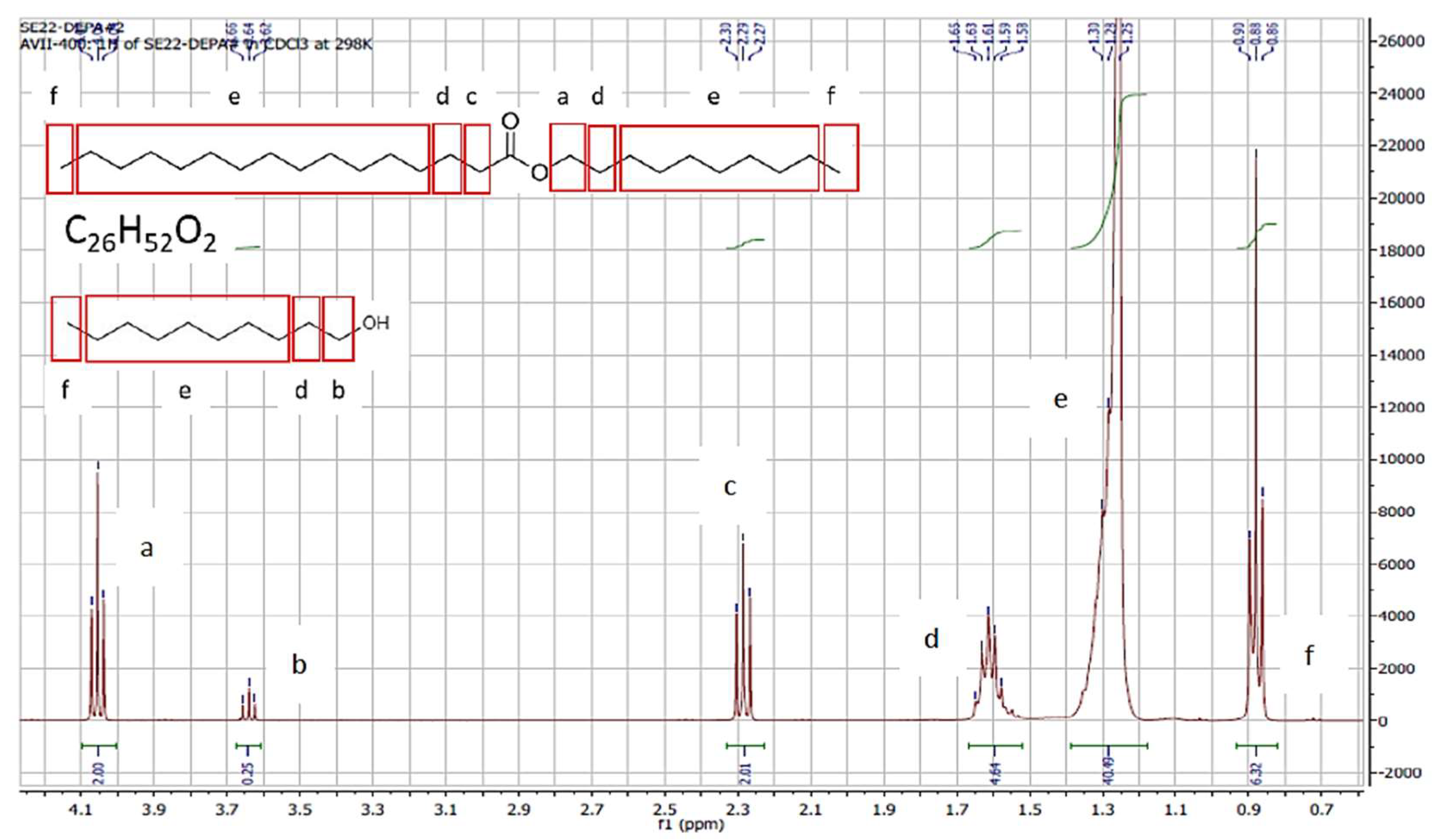
2.2.5. Nuclear Magnetic Resonance (NMR)
3. Results and Discussion
3.1. Synthesis Development and Optimization
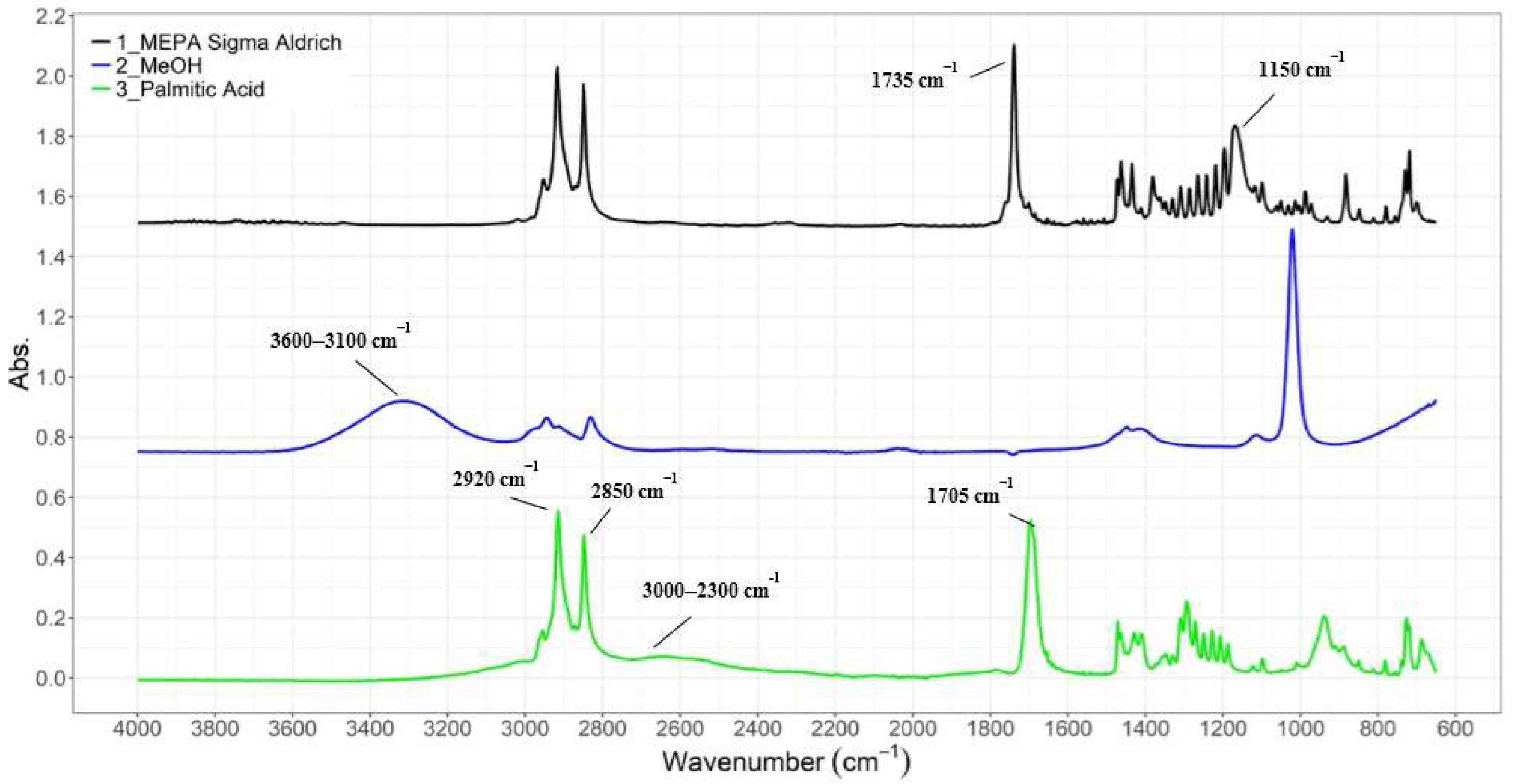
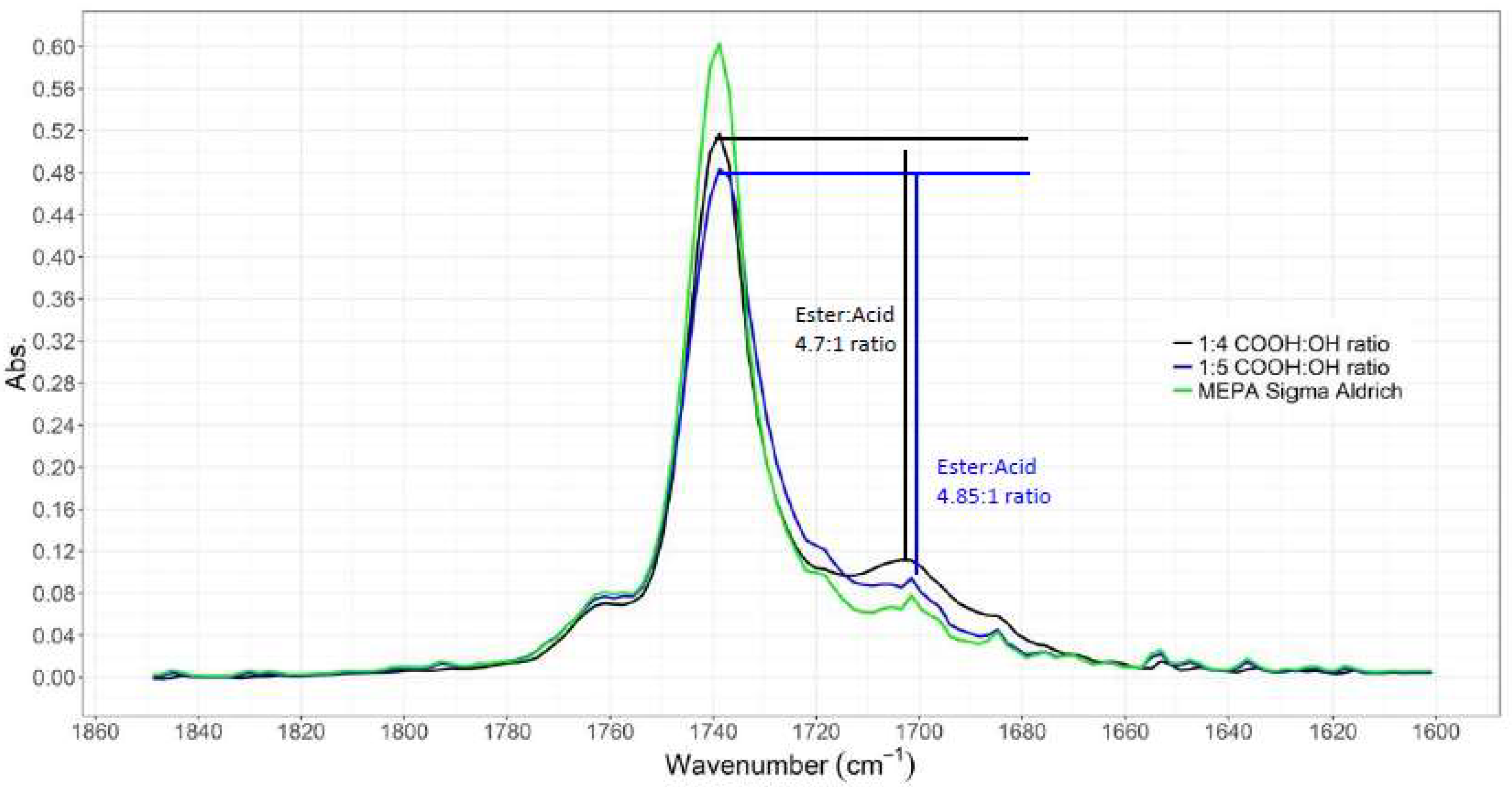
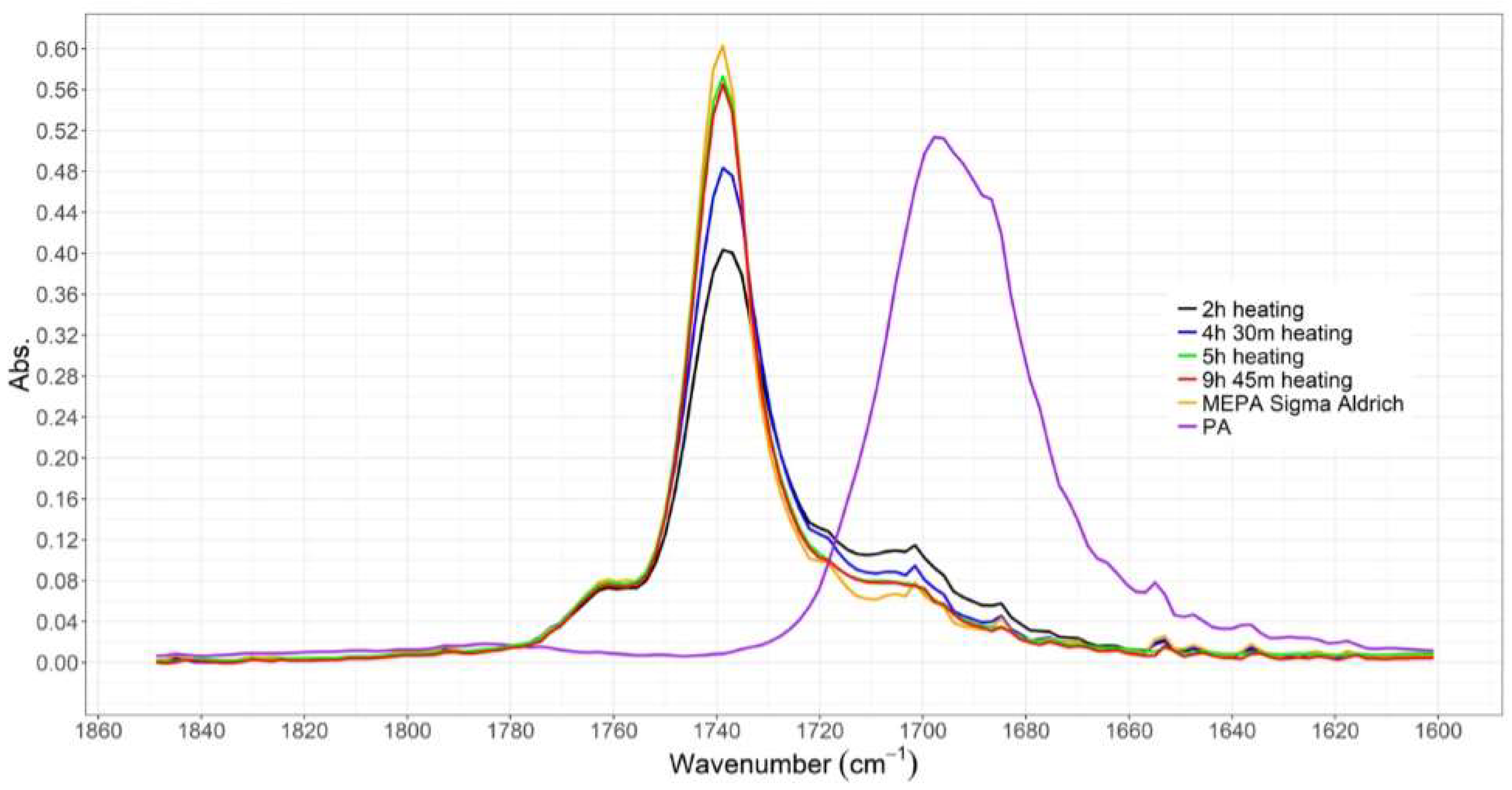
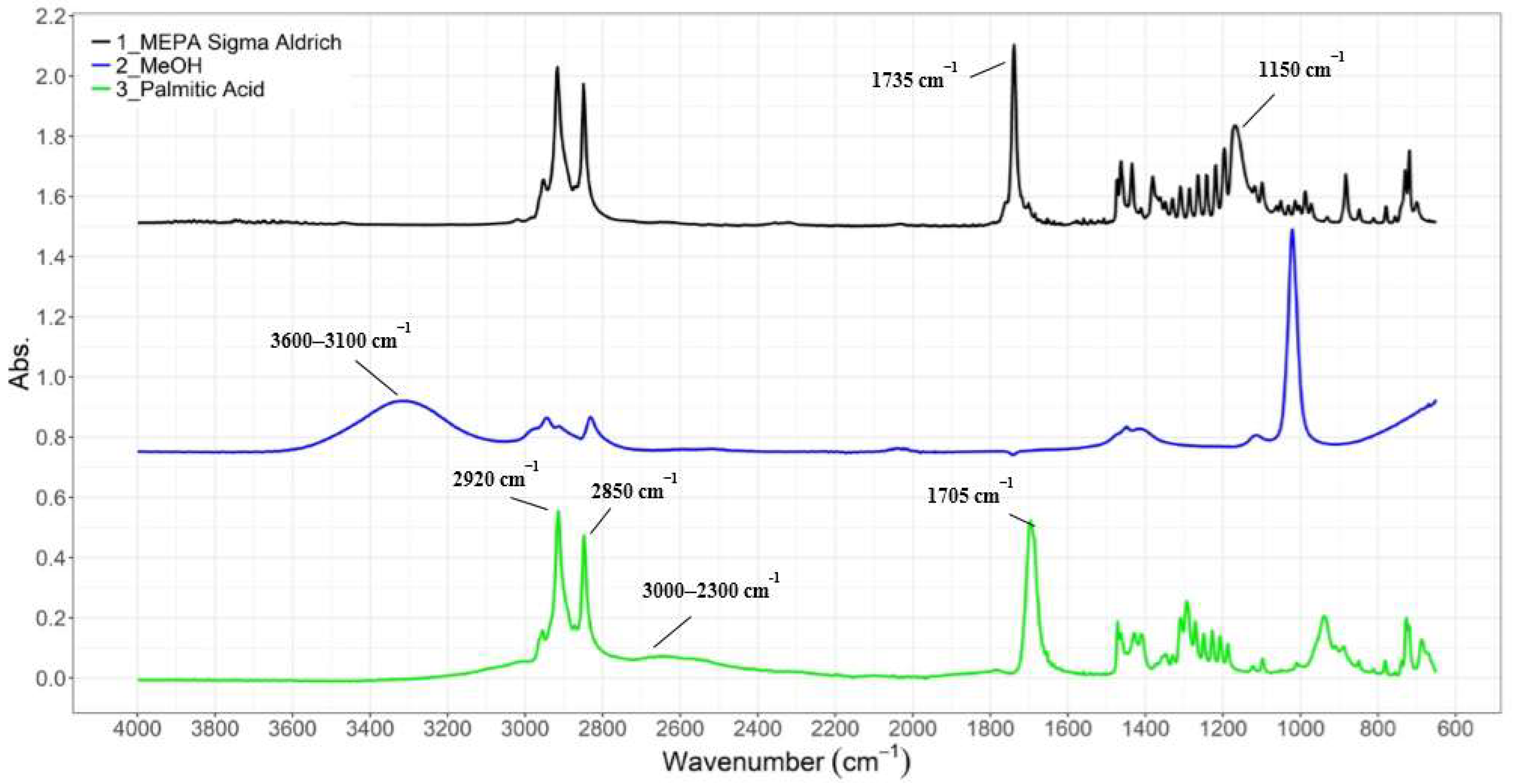
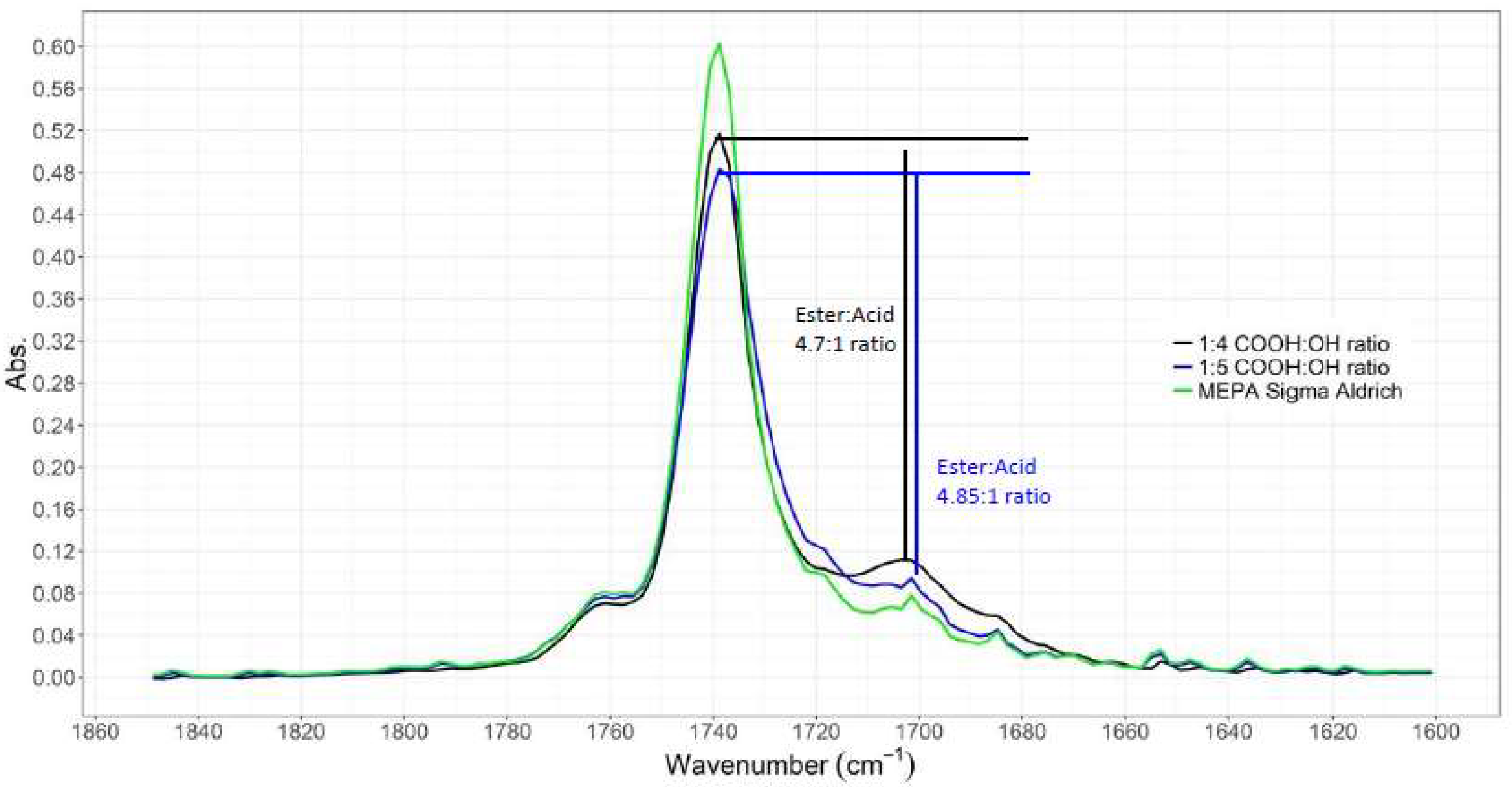
3.1.1. ATR-IR Analysis
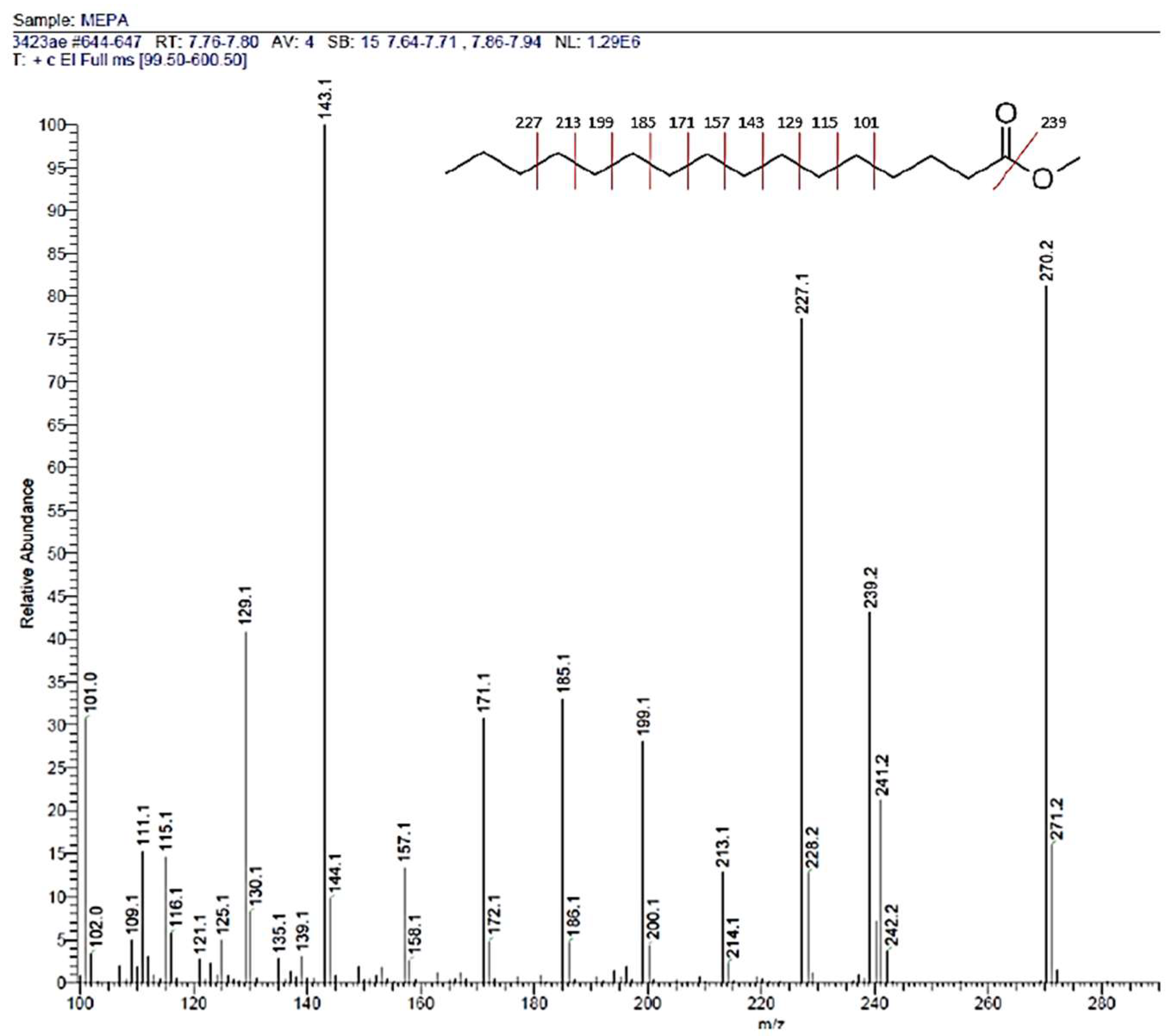
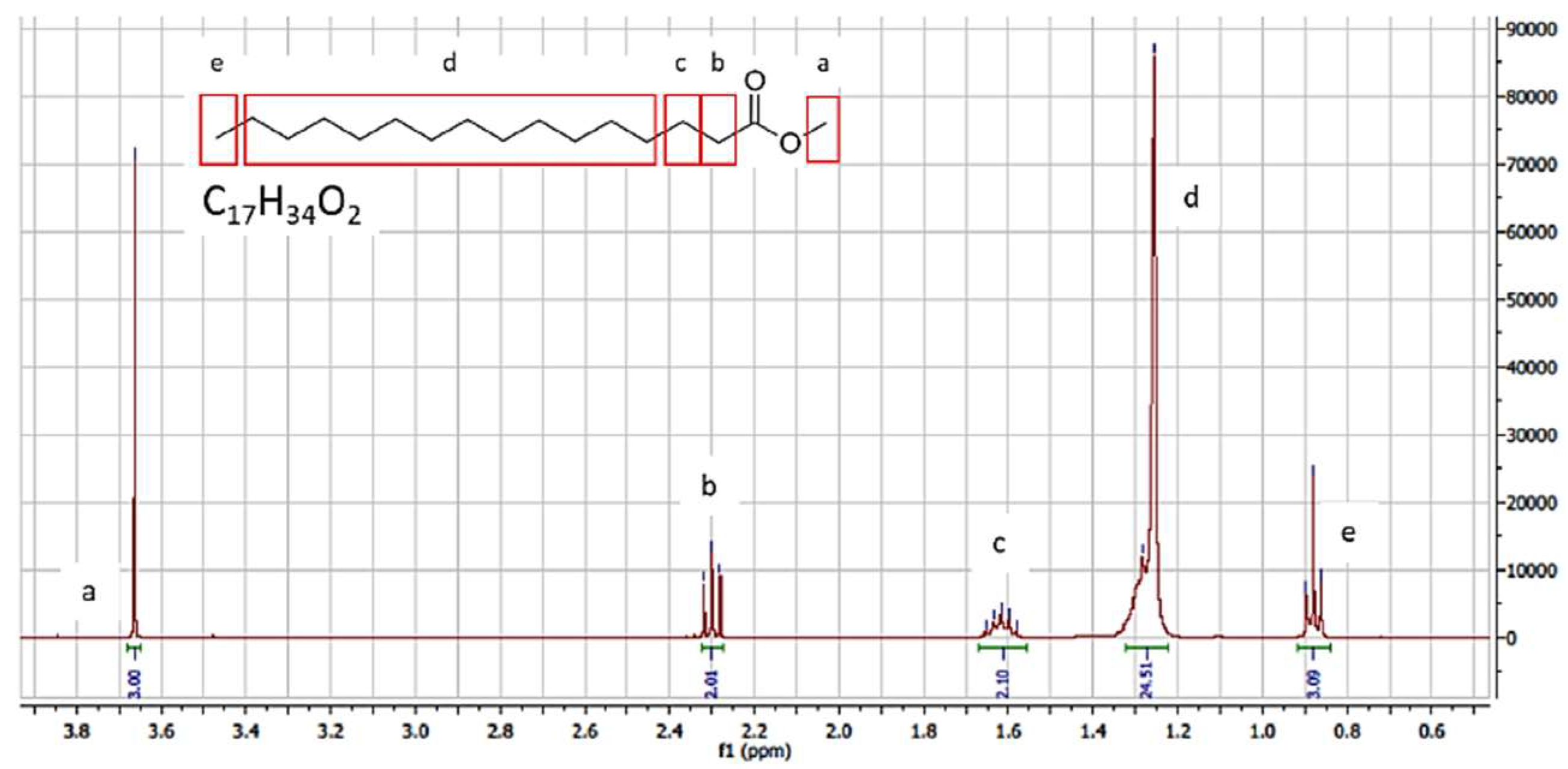
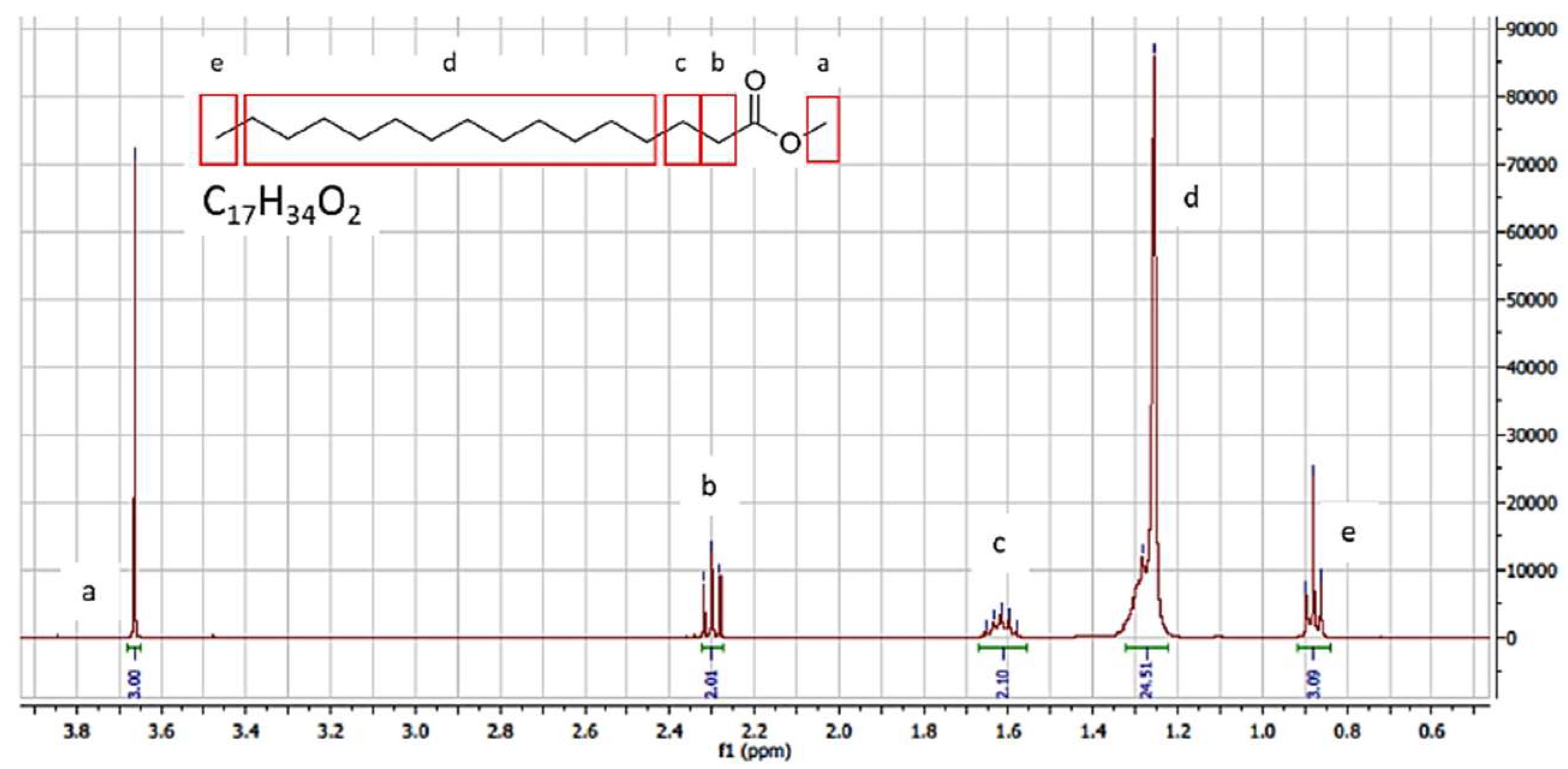
3.1.2. GC-MS and NMR Analysis
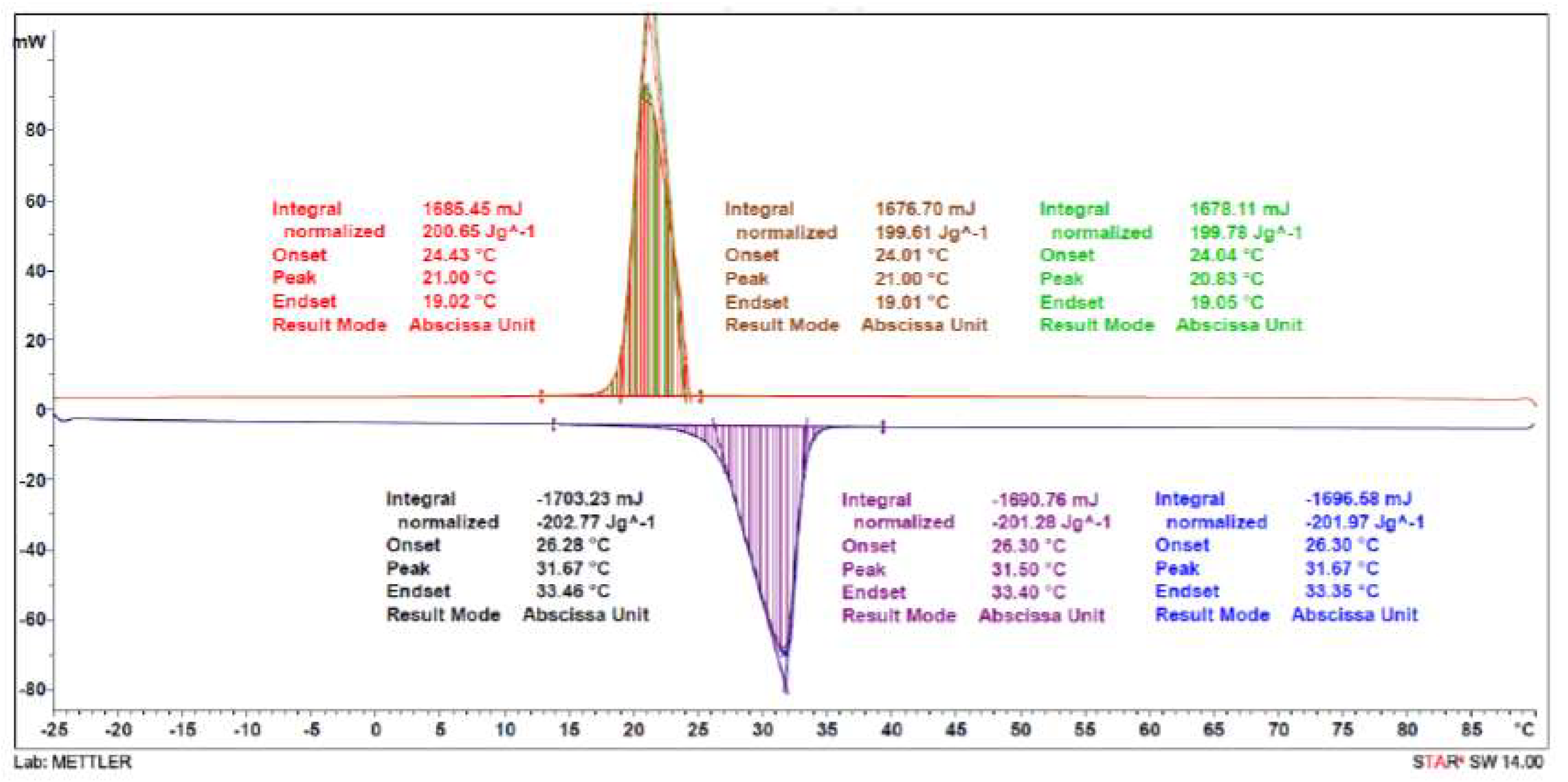
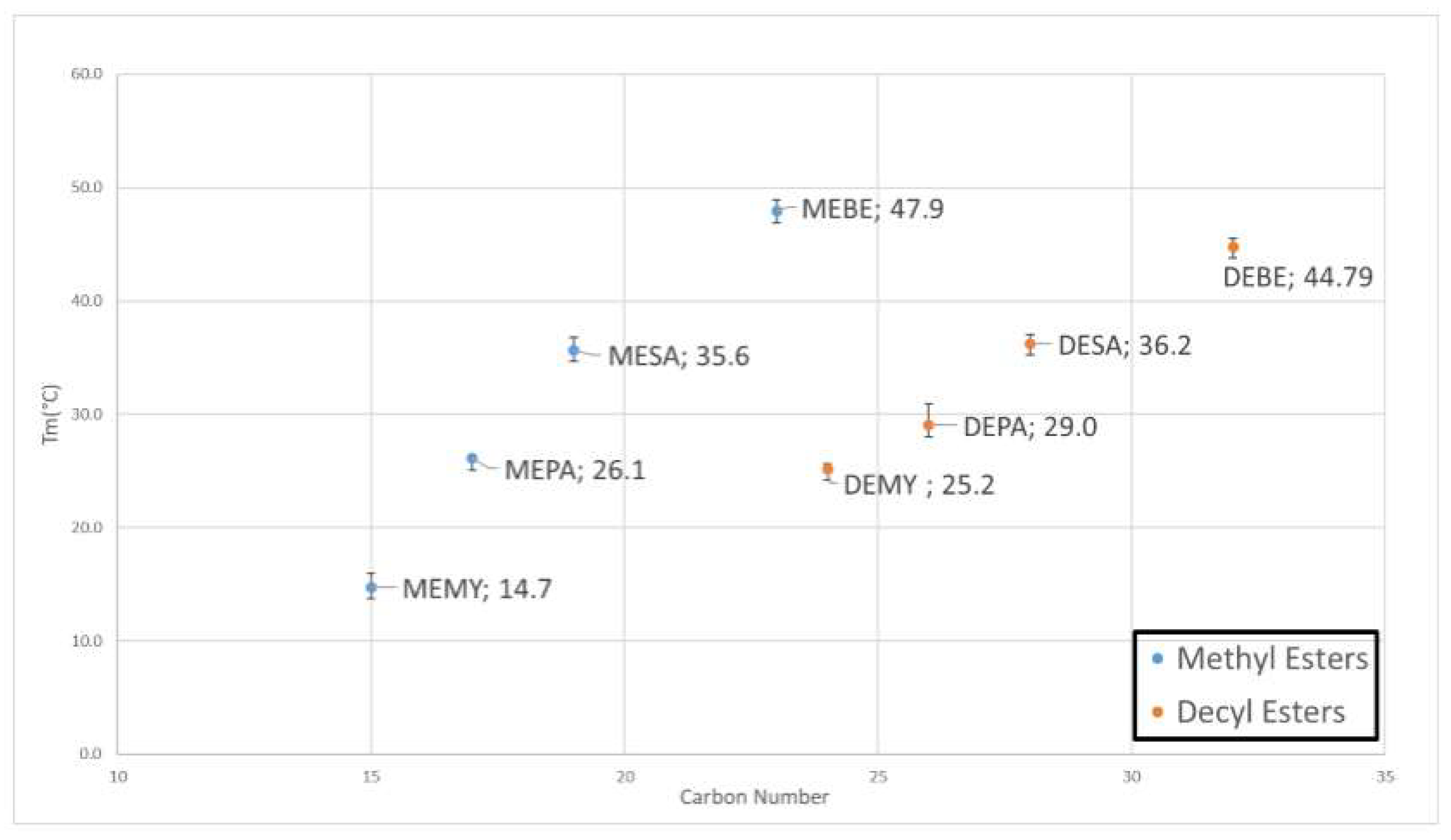
3.1.3. DSC and TGA Analysis
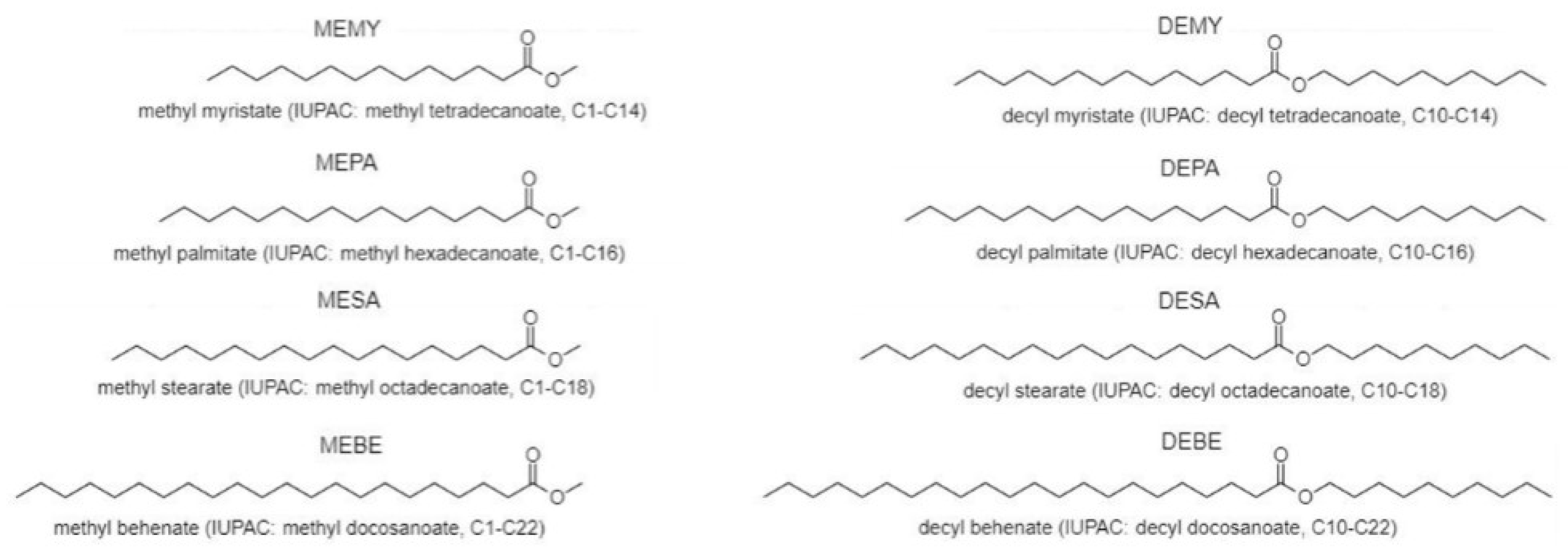
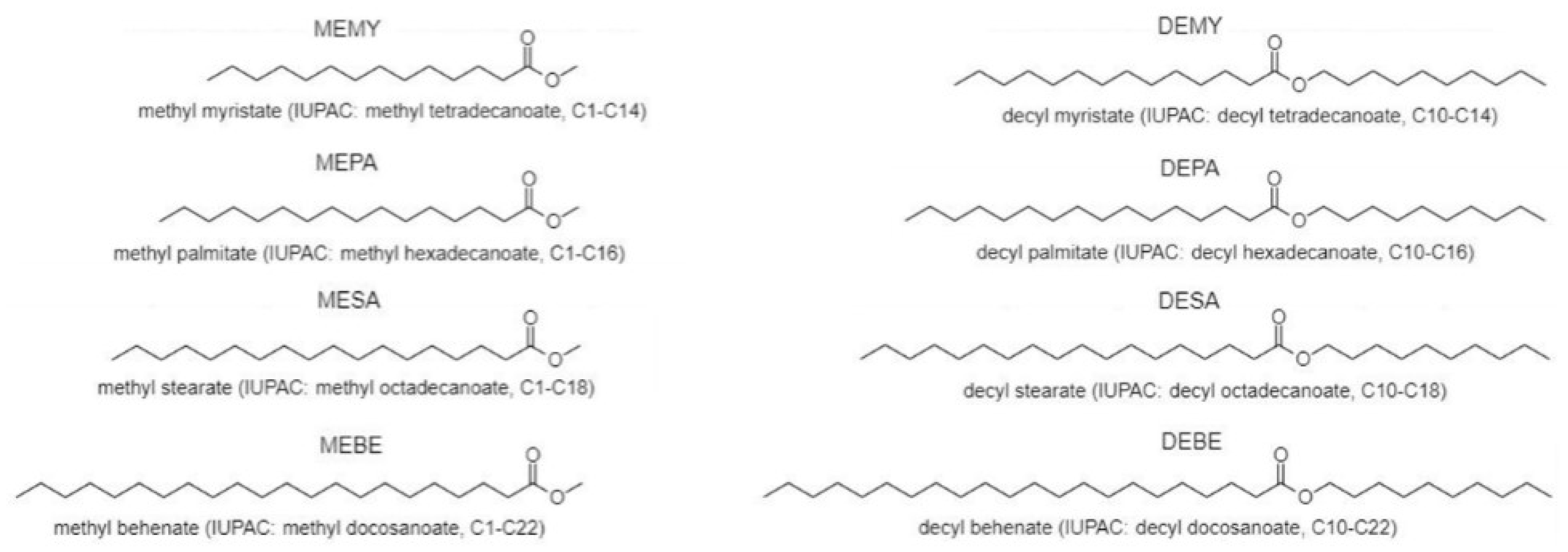
3.2. Synthesis of Methyl and Decyl Esters
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
List of Abbreviations
| ATR-IR | Attenuated Total Reflectance InfraRed Spectroscopy |
| BE | Behenic Acid |
| DEBE | Decyl Behenate (C10-C22) |
| DEMY | Decyl Myristate (C10-C14) |
| DEPA | Decyl Palmitate (C10-C16) |
| DESA | Decyl Stearate (C10-C18) |
| DSC | Differential Scanning Calorimetry |
| Et2O | Diethyl Ether |
| EtOAc | Ethyl Acetate |
| GC-MS | Gas-Chromatography coupled with Mass Spectrometry |
| H2SO4 | Sulfuric Acid |
| IEA | International Energy Agency |
| LHS | Latent Heat Storage |
| MEBE | Methyl Behenate (C1-C22) |
| MEMY | Methyl Myristate (C1-C14) |
| MeOH | Methanol |
| MEPA | Methyl Palmitate (C1-C16) |
| MESA | Methyl Stearate (C1-C18) |
| MY | Myristic Acid |
| Na2SO4 | Sodium Sulfate |
| NMR | Nuclear Magnetic Resonance |
| PA | Palmitic Acid |
| PCM | Phase Change Material |
| SA | Stearic Acid |
| SHS | Sensible Heat Storage |
| Tc | Crystallization Temperature |
| TCS | Thermochemical Storage Material |
| TGA | Thermogravimetric Analysis |
| Tm | Melting Temperature |
| ΔH | Enthalpy of fusion |
Appendix A
References
- International Energy Agency (IEA). Key World Energy Statistics. 2017. Available online: https://www.iea.org/publications/freepublications/publication/KeyWorld2017.pdf (accessed on 19 May 2018).
- Pielichowska, K.; Pielichowski, K. Phase change materials for thermal energy storage. Prog. Mater. Sci. 2014, 65, 67–123. [Google Scholar] [CrossRef]
- Nkwetta, D.N.; Haghighat, F. Thermal energy storage with phase change material—A state-of-the art review. Sustain. Cities Soc. 2013, 10, 87–100. [Google Scholar] [CrossRef]
- Sharma, A.; Tyagi, V.V.; Chen, C. Experimental study for the melting of PCM inside concentric horizontal cylindrical annuli. In Proceedings of the 6th International Conference on Thermal Energy Storage, Espoo, Finland, 15–17 August 1994; pp. 503–511. [Google Scholar]
- Sarbu, I.; Sebarchievici, C. A Comprehensive Review of Thermal Energy Storage. Sustainability 2018, 10, 191–223. [Google Scholar] [CrossRef]
- Mehling, H.; Cabeza, L.F. Heat and Cold Storage with PCM. An up to Date Introduction into Basics and Applications; Springer: Berlin, Germany, 2008; ISBN 978-3-540-68556-2. [Google Scholar]
- Liu, M.; Saman, W.; Bruno, F. Review on storage materials and thermal performance enhancement techniques for high temperature phase change thermal storage systems. Renew. Sustain. Energy Rev. 2012, 16, 2118–2132. [Google Scholar] [CrossRef]
- Abhat, A. Low Temperature Latent Heat Thermal Energy Storage: Heat Storage Materials. Sol. Energy 1983, 30, 313–332. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhang, N.; Tao, W.; Cao, X.; He, Y. Fatty Acids as phase change materials: A review. Renew. Sustain. Energy Rev. 2014, 29, 482–498. [Google Scholar] [CrossRef]
- Cabeza, F.L.; Castell, A.; Barreneche, C.; de Gracia, A.; Fernandez, A.I. Materials used as PCM in thermal energy storage in buildings: A review. Renew. Sustain. Energy Rev. 2011, 15, 1675–1695. [Google Scholar] [CrossRef]
- Menoufli, K.; Castell, A.; Farid, M.M.; Boer, D.; Cabeza, F.L. Life cycle assessment of experimental cubicles including PCM manufactured from natural resources (Esters): A theoretical study. Energy Convers. Manag. 2003, 44, 2277–2287. [Google Scholar] [CrossRef]
- Stamatiou, A.; Obermeyer, M.; Fischer, L.J.; Schuetz, P.; Wortlischek, J. Investigation of unbranched, saturated, Carboxylic Esters as phase change materials. Renew. Energy 2017, 108, 401–409. [Google Scholar] [CrossRef]
- Aydin, A.A.; Okutan, H. High-chain Fatty Acid Esters of myristic Alcohol with even carbon number: Novel organic phase change materials for thermal energy storage-1. Sol. Energy Mater. Sol. Cells 2011, 95, 2752–2762. [Google Scholar] [CrossRef]
- Aydin, A.A. DiEsters of high-chain diCarboxylic Acids with 1-tetradecanol as novel organic phase change materials for thermal energy storage. Sol. Energy Mater. Sol. Cells 2012, 104, 102–108. [Google Scholar] [CrossRef]
- Carnelley, T. Chemical Symmetry, or the Influence of Atomic Arrangement on the Physical Properties of Compounds. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1882, 12, 112–130. [Google Scholar] [CrossRef]
- Boese, R.; Kirchner, M.T.; Dunitz, J.D.; Filippin, G.; Gavezzotti, A. Solid-State Behaviour of the Dichlorobenzenes: Actual, Semi-Virtual and Virtual Christallography. Helv. Chim. Acta 2001, 84, 1561–1577. [Google Scholar] [CrossRef]
- Noël, J.A.; Samer, K.; White, M.A. Molecular Structure and Melting: Implications for Phase Change Materials. Can. J. Chem. 2017, 1–8. [Google Scholar] [CrossRef]
- Yang, K.; Cai, Z.; Jaiswal, A.; Tyagi, M.; Moore, J.S. Dynamic Odd-Even Effect in Liquid n-Alkanes near their Melting Points. Angew. Chem. Int. Ed. 2006, 55, 14090–14095. [Google Scholar] [CrossRef] [PubMed]
- Sari, A.; Biçer, A.; Karaipekli, A. Synthesis, characterization, thermal properties of a series of stearic Acid Esters as novel solid-liquid phase change materials. Mater. Lett. 2009, 63, 1213–1216. [Google Scholar] [CrossRef]
- Becker, H.G.O.; Berger, W.; Domschke, G.; Fanghänel, E.; Faust, J.; Fischer, M.; Gentz, F.; Gewald, K.; Gluch, R.; Mayer R Müller, K. Organikum, Organisch-Chemisches Grundpraktikum, 2nd ed.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2004; pp. 226–233. ISBN 3-527-31148-3. [Google Scholar]
- Buddrus, J.; Schmidt, B. Grundlagen der Organischen Chemie, 5th ed.; Walter de Gruyter GmbH: München, Germany, 2015; pp. 154–196. ISBN 978-3-11-030559-3. [Google Scholar]
- Cassel, R.B. TA Instruments Application Note. Available online: http://www.tainstruments.com/pdf/literature/TA297.pdf (accessed on 04 May 2018).
- Spectra Analysis Application Note 005. Available online: http://www.spectra-analysis.com (accessed on 19 April 2018).
- Jabbar, A.; Ali, A.; Tawab, A.; Iqbal, M. Fatty Acid Profiling of Lipid A Isolated from Indigenous Salmonella Typhi Strain by Gas Chromatography Mass Spectrometry. J. Chem. Soc. Pak. 2014, 36, 140–149. [Google Scholar]
- Du, K.; Calautit, J.; Wang, Z.; Wu, Y.; Liu, H. A review of the applications of phase change materials in cooling, heating and power generation in different temperature ranges. Appl. Energy 2018, 220, 242–273. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Carbon Number | MW (g/mol) | Purity | Tc (onset, °C) | Tm (onset, °C) | Supercooling (°C) | ΔH (J/g) | ΔH (KJ/mol) | Tdegradation (Start, °C) | Tdegradation (End, °C) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| MEMY (C1-C14) | C15H30O2 | 15 | 242.4 | 98% | 13.5 ± 1.2 | 14.7 ± 1.9 | 1.2 | 190 ± 2 | 46.1 | 87 ± 12 | 233 ± 12 |
| MEPA (C1-C16) | C17H34O2 | 17 | 270.5 | 98% | 24.2 ± 0.2 | 26.3 ± 0.0 | 2.1 | 201 ± 1 | 54.4 | 119 ± 10 | 272 ± 4 |
| MESA (C1-C18) | C19H38O2 | 19 | 298.5 | 98% | 31.9 ± 1.1 | 35.6 ± 1.0 | 3.7 | 204 ± 1 | 60.9 | 117 ± 7 | 287 ± 6 |
| MEBE (C1-C22) | C23H46O2 | 23 | 354.6 | 98% | 41.5 ± 1.0 | 47.9 ± 0.7 | 6.4 | 200 ± 5 | 70.9 | 155 ± 7 | 305 ±20 |
| DEMY (C10-C14) | C24H48O2 | 24 | 368.7 | 98% | 22.9 ± 0.2 | 25.2 ± 0.4 | 2.3 | 196 ± 3 | 72.3 | 130 ± 5 | 318 ± 11 |
| DEPA (C10-C16) | C26H52O2 | 26 | 396.7 | 89% | 28.9 ± 1.2 | 29.0 ± 1.9 | 0.1 | 193 ± 6 | 76.6 | 145 ± 7 | 310 ± 14 |
| DESA (C10-C18) | C28H56O2 | 28 | 424.8 | 98% | 33.8 ± 1.3 | 36.2 ± 0.8 | 2.4 | 192 ± 6 | 81.6 | 205 ± 20 | 350 ± 14 |
| DEBE (C10-C22) | C32H64O2 | 32 | 480.9 | 95% | 44.7 ± 0.6 | 44.8 ± 0.7 | 0.1 | 193 ± 9 | 92.8 | 200 ± 20 | 375 ± 21 |
| Carbon Number | Tc (Onset, °C) | Tm (Onset, °C) | |
|---|---|---|---|
| MEMY (C1-C14) | 15 | 13.5 ± 1.2 | 14.7 ± 1.9 |
| DEMY (C10-C14) | 24 | 22.9 ± 0.2 | 25.2 ± 0.4 |
| MEPA (C1-C16) | 17 | 24.2 ± 0.2 | 26.3 ± 0.0 |
| DEPA (C10-C16) | 26 | 28.9 ± 1.2 | 29.0 ± 1.9 |
| MESA (C1-C18) | 19 | 31.9 ± 1.1 | 35.6 ± 1.0 |
| DESA (C10-C18) | 28 | 33.8 ± 1.3 | 36.2 ± 0.8 |
| MEBE (C1-C22) | 23 | 41.5 ± 1.0 | 47.9 ± 0.7 |
| DEBE (C10-C22) | 32 | 44.7 ± 0.6 | 44.8 ± 0.7 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravotti, R.; Fellmann, O.; Lardon, N.; Fischer, L.J.; Stamatiou, A.; Worlitschek, J. Synthesis and Investigation of Thermal Properties of Highly Pure Carboxylic Fatty Esters to Be Used as PCM. Appl. Sci. 2018, 8, 1069. https://doi.org/10.3390/app8071069
Ravotti R, Fellmann O, Lardon N, Fischer LJ, Stamatiou A, Worlitschek J. Synthesis and Investigation of Thermal Properties of Highly Pure Carboxylic Fatty Esters to Be Used as PCM. Applied Sciences. 2018; 8(7):1069. https://doi.org/10.3390/app8071069
Chicago/Turabian StyleRavotti, Rebecca, Oliver Fellmann, Nicolas Lardon, Ludger J. Fischer, Anastasia Stamatiou, and Jörg Worlitschek. 2018. "Synthesis and Investigation of Thermal Properties of Highly Pure Carboxylic Fatty Esters to Be Used as PCM" Applied Sciences 8, no. 7: 1069. https://doi.org/10.3390/app8071069
APA StyleRavotti, R., Fellmann, O., Lardon, N., Fischer, L. J., Stamatiou, A., & Worlitschek, J. (2018). Synthesis and Investigation of Thermal Properties of Highly Pure Carboxylic Fatty Esters to Be Used as PCM. Applied Sciences, 8(7), 1069. https://doi.org/10.3390/app8071069