Two-Stage Strategy for CO Removal from H2-Rich Streams over (Nano-) CuO/CeO2 Structured Catalyst at Low Temperature




Abstract
1. Introduction
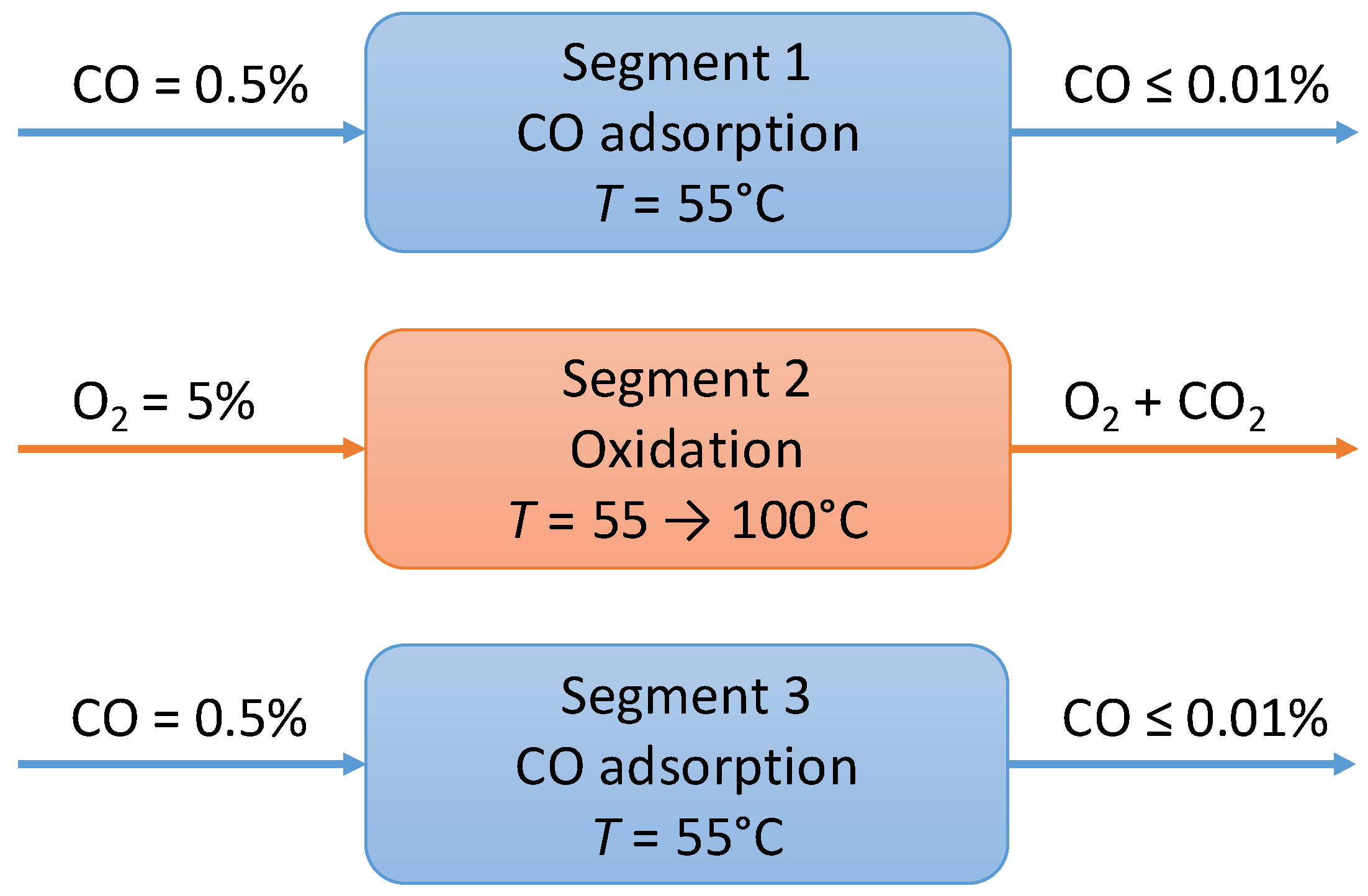
Description of the Process
- CO adsorption: During this phase, CO is adsorbed over the catalyst surface at very low temperature (T1 < 100 °C) at which H2 does not interact with the catalytic sites;
- CO oxidation: CO trapped over the catalyst surface is oxidized and the CO2 formed is desorbed.
2. Materials and Methods
2.1. Catalyst Preparation
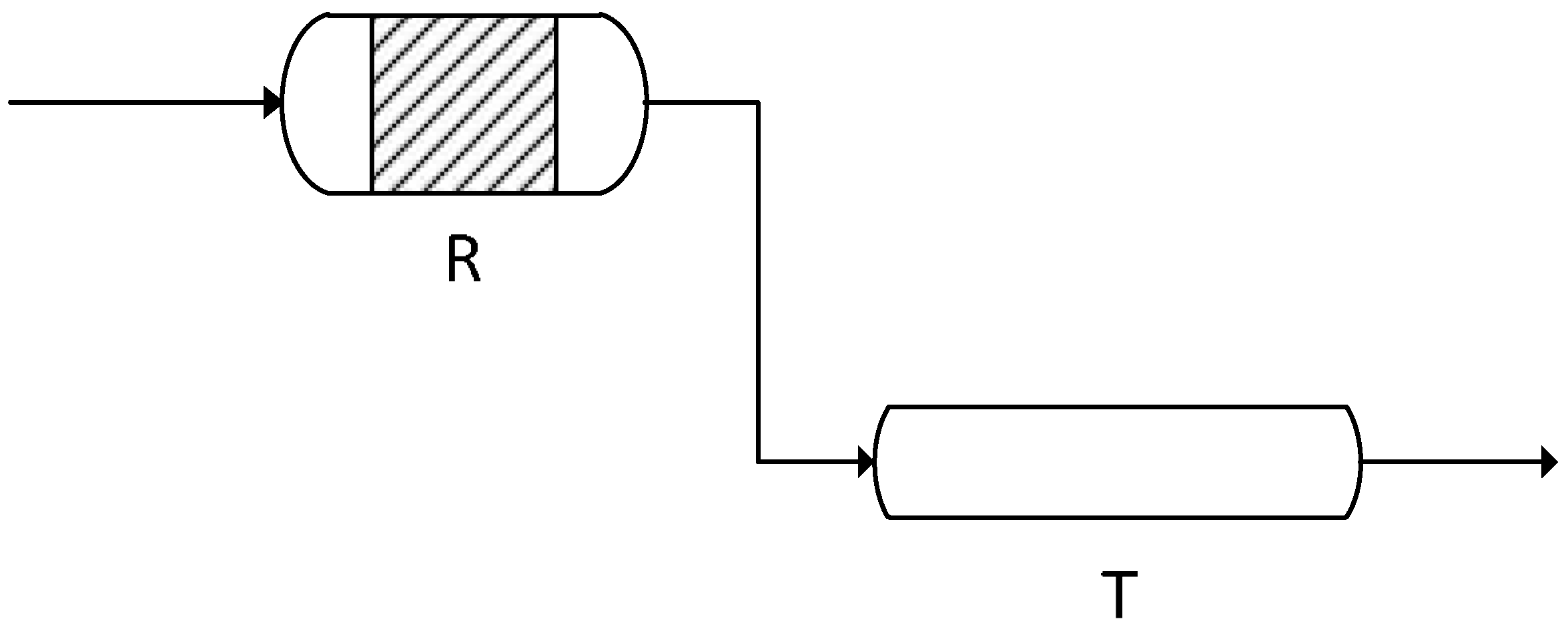
2.2. Lab Test Rig
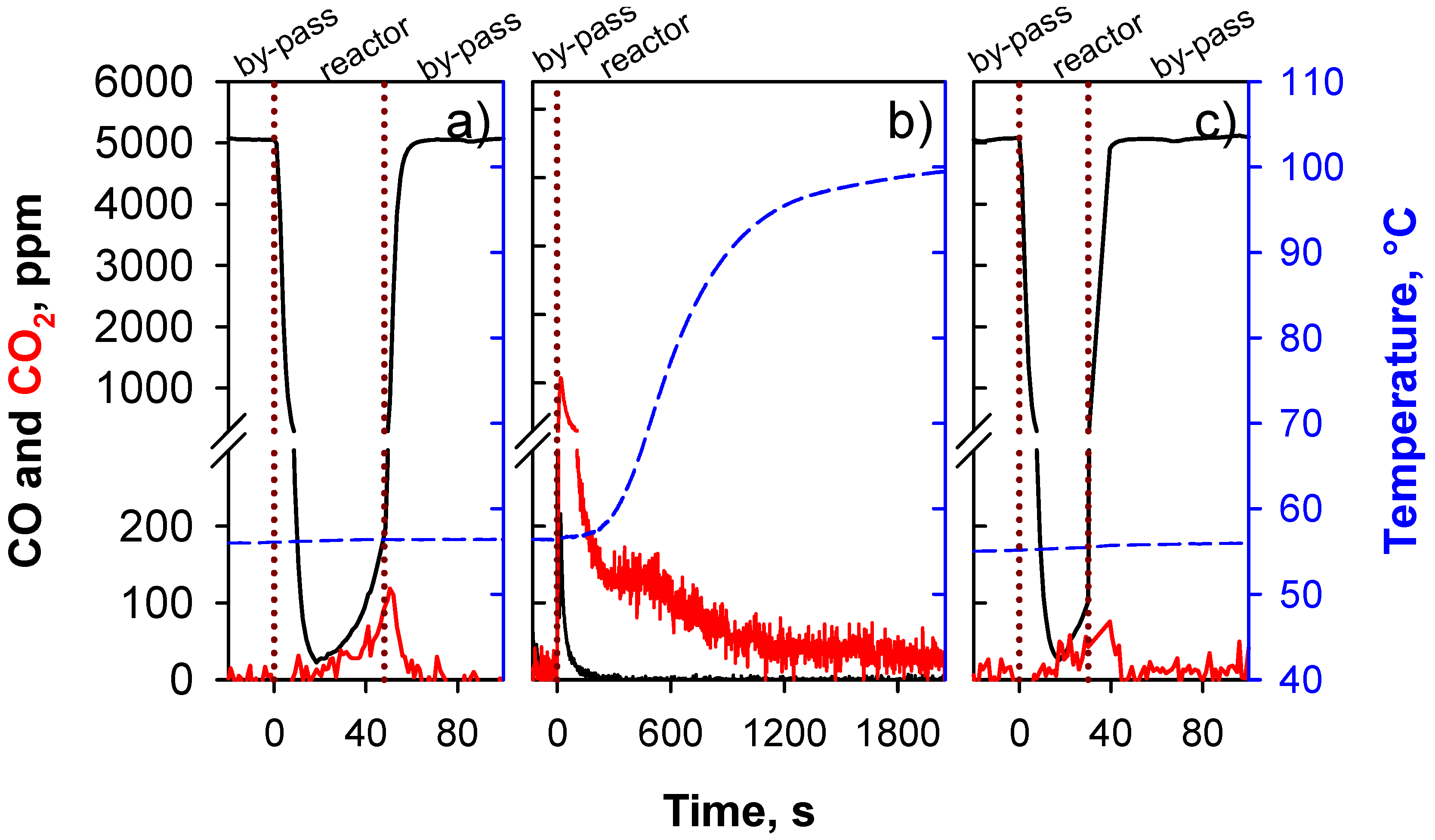
- Segment 1—Isothermal adsorption. T = 55 °C, CO = 5000 ppm in N2 flow. When the CO concentration reaches 100 ppm the reactor is by-passed.
- Segment 2—Oxidation temperature is increased up to 100 °C, heating rate = 10 °C/min, 5 vol % O2 in N2 flow to the reactor.
- Segment 3—Isothermal adsorption (repeatability test).
3. Results
4. Model
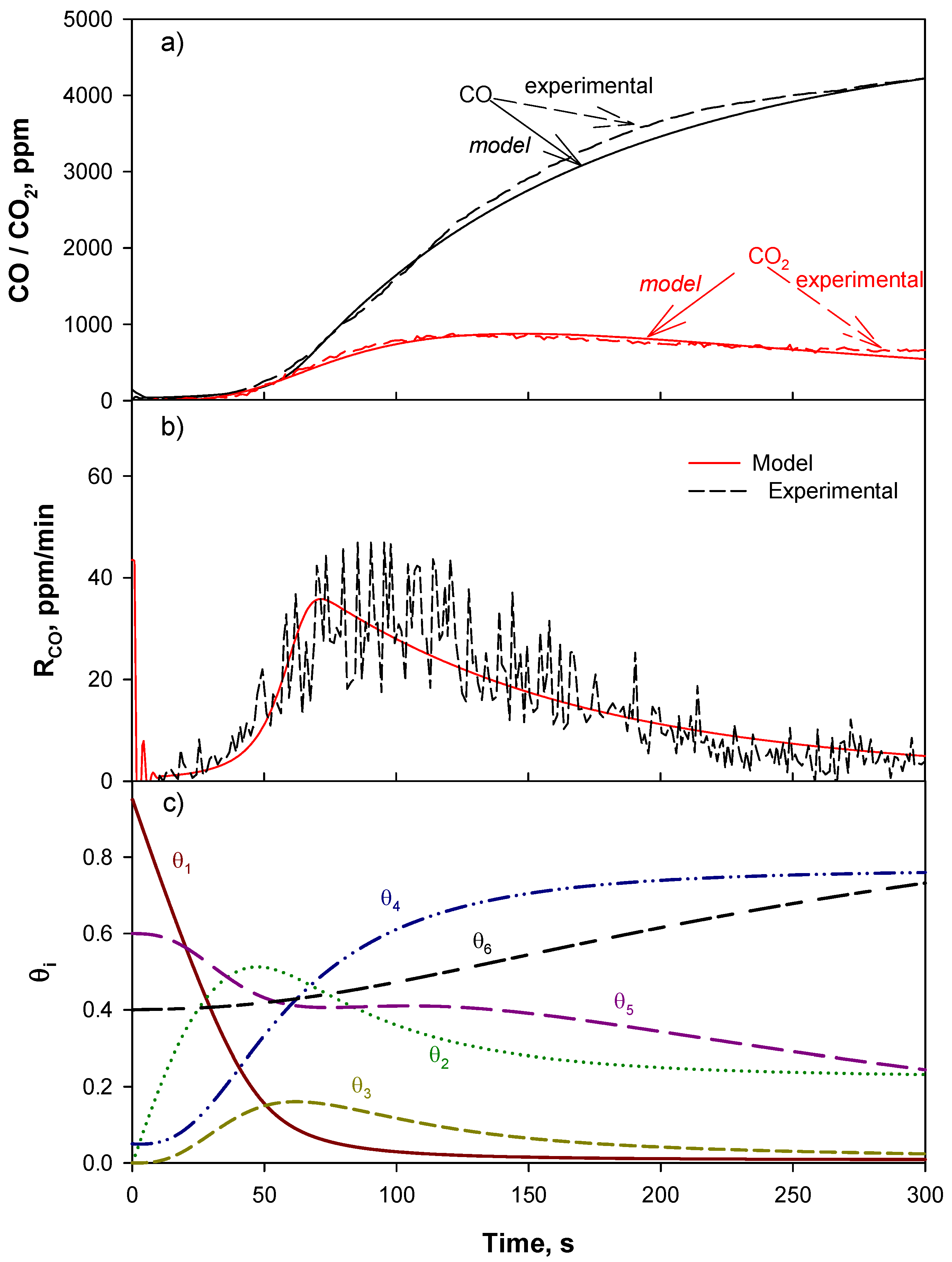
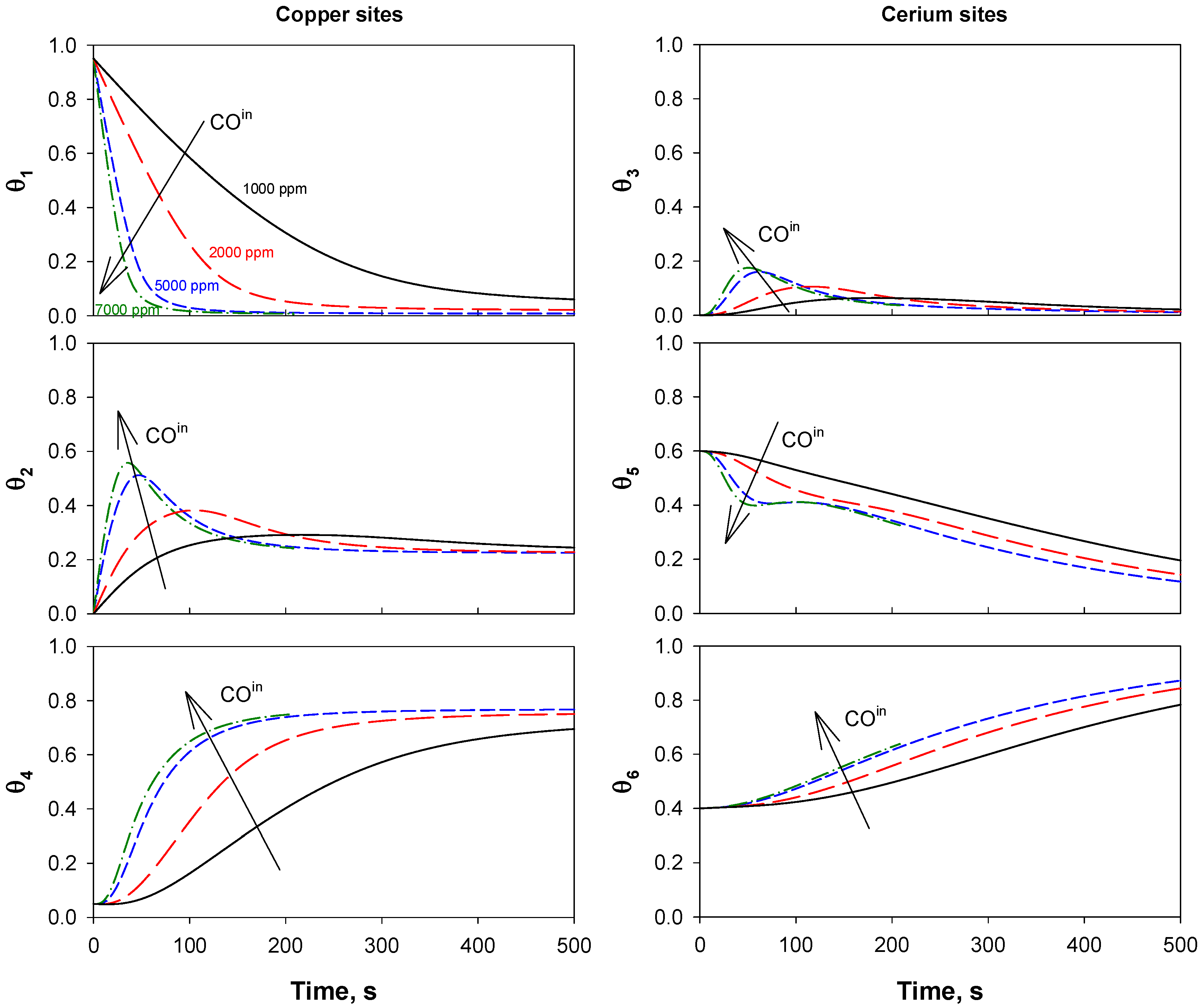
4.1. Reaction Mechanism and Kinetic Model
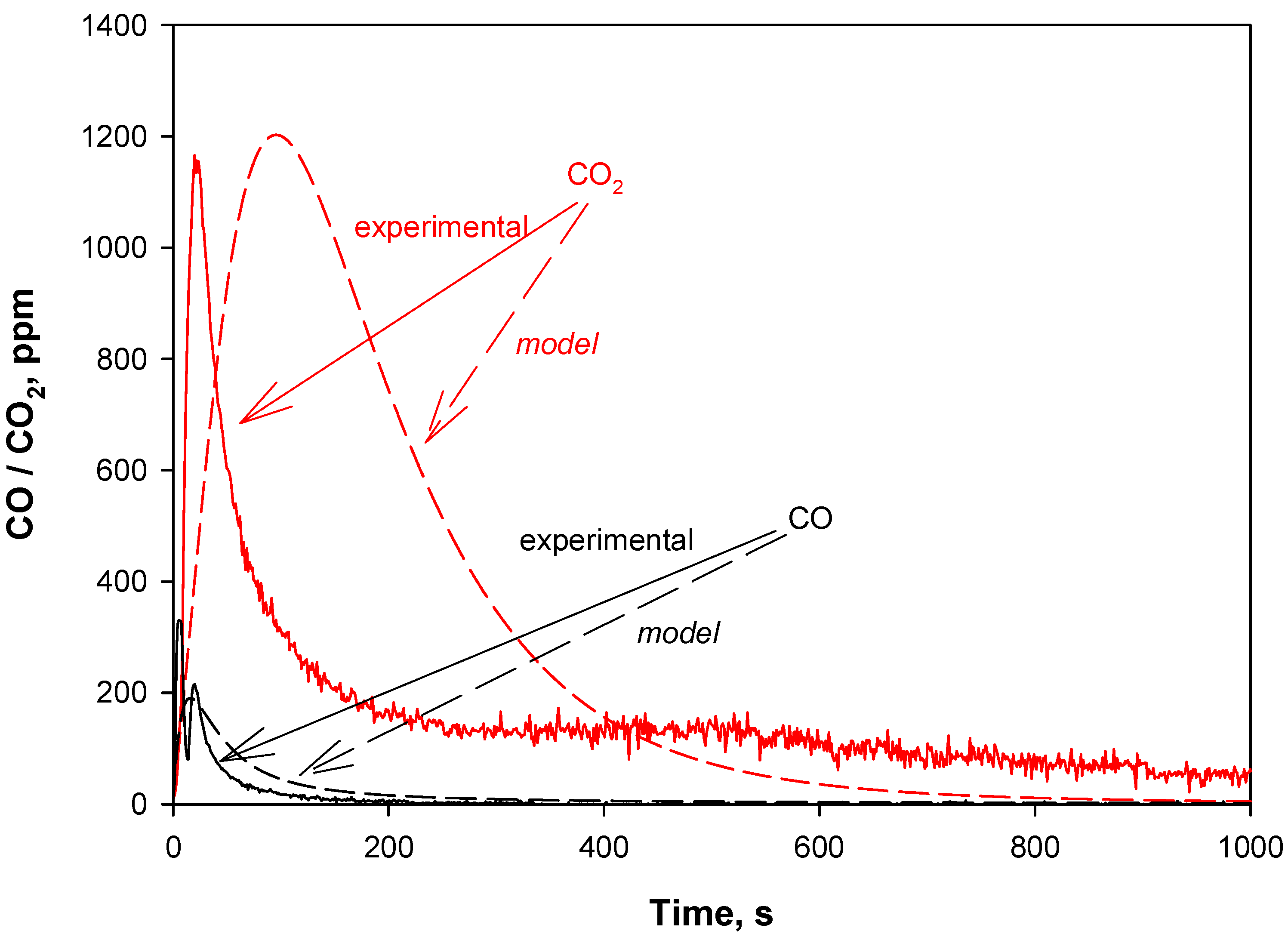
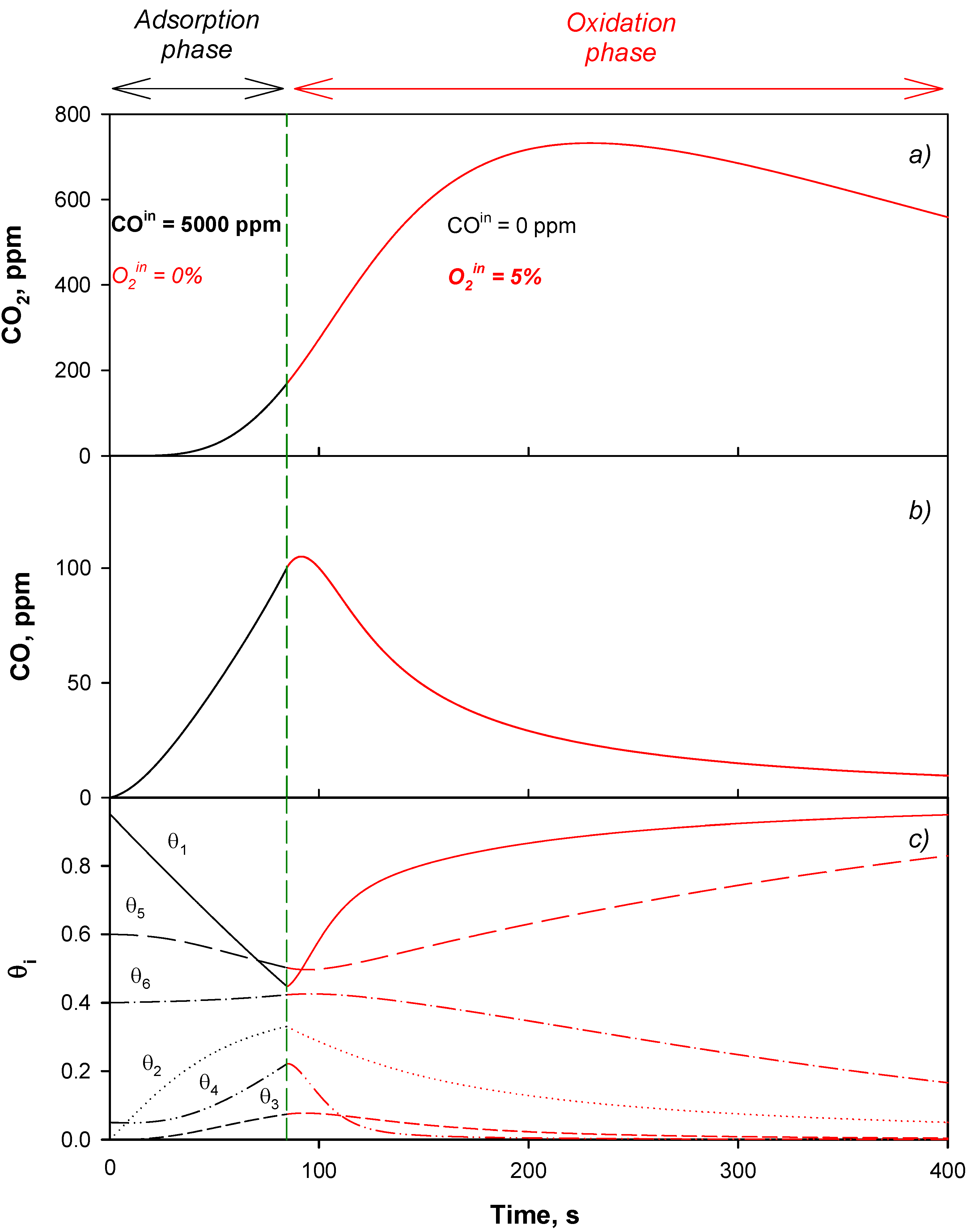
4.2. Model Validation
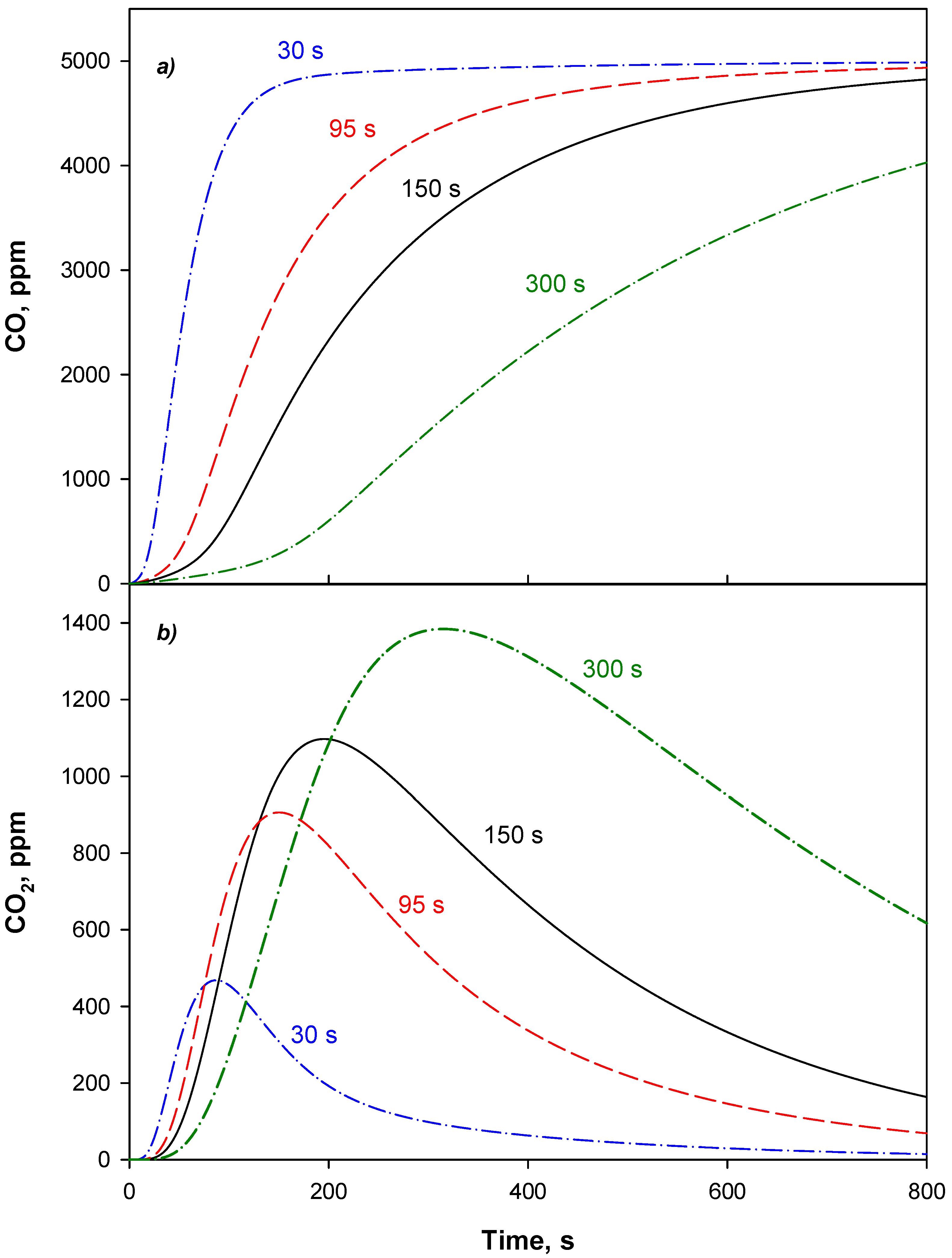
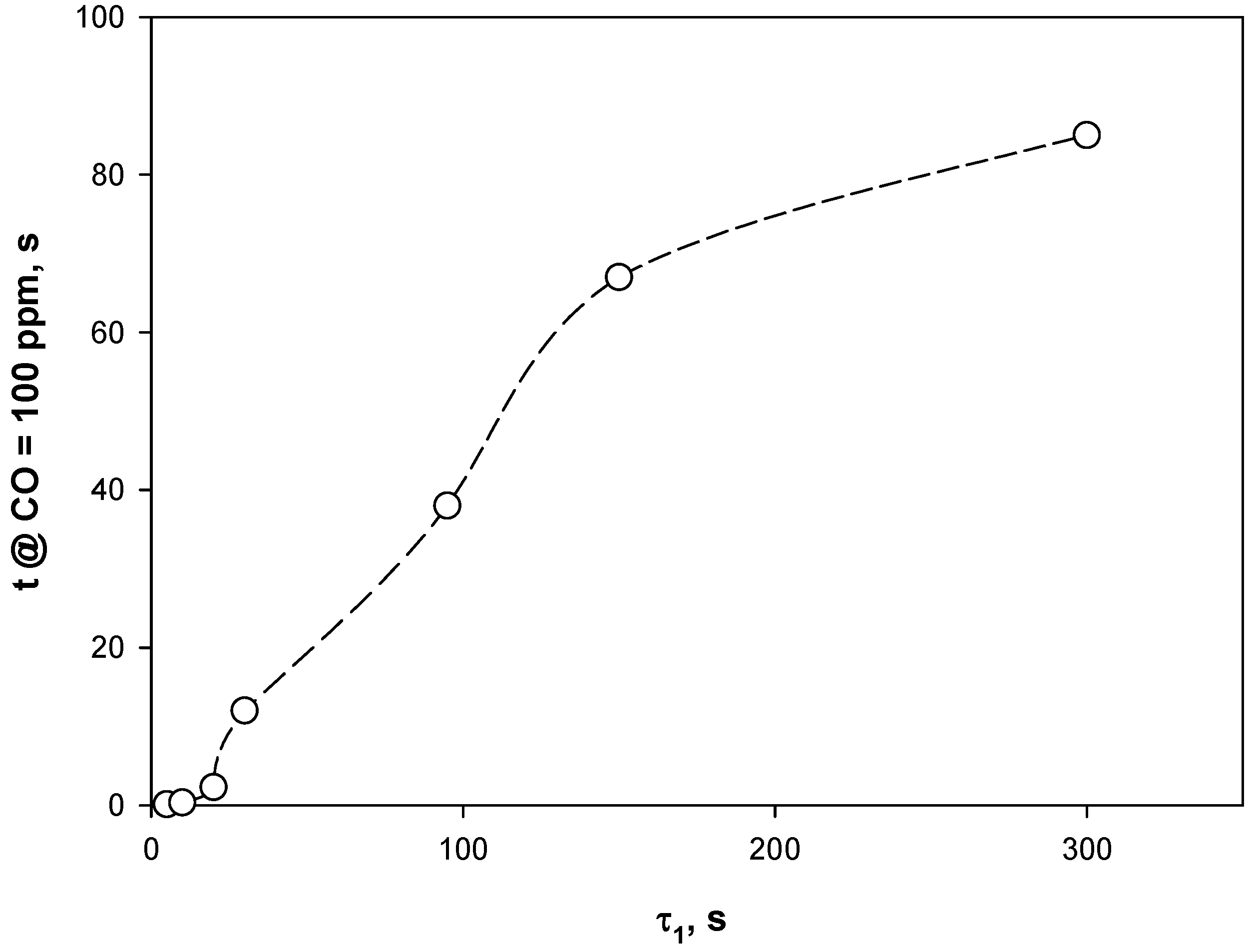
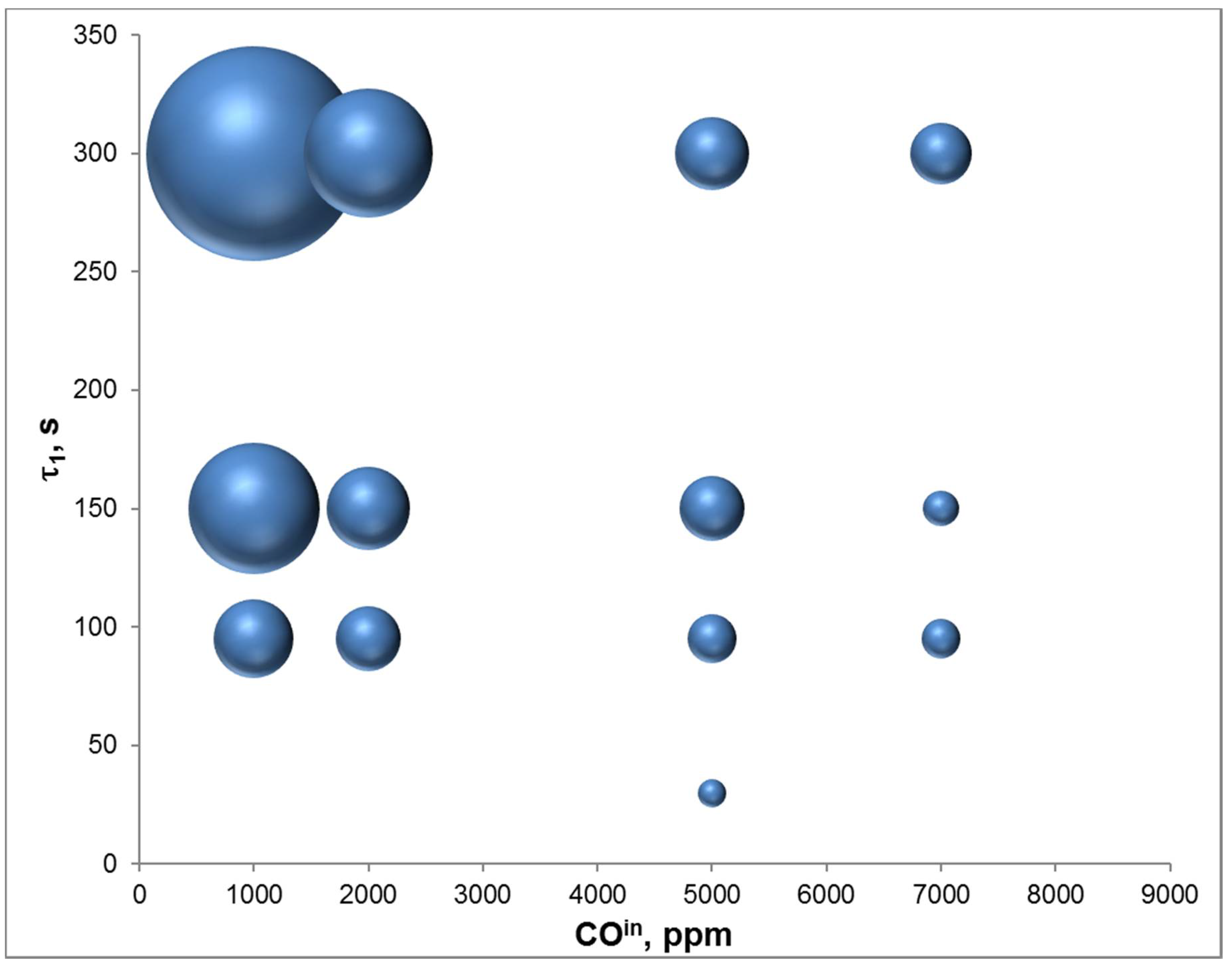
4.3. Adsorption Phase: Effect of the Operating Parameters
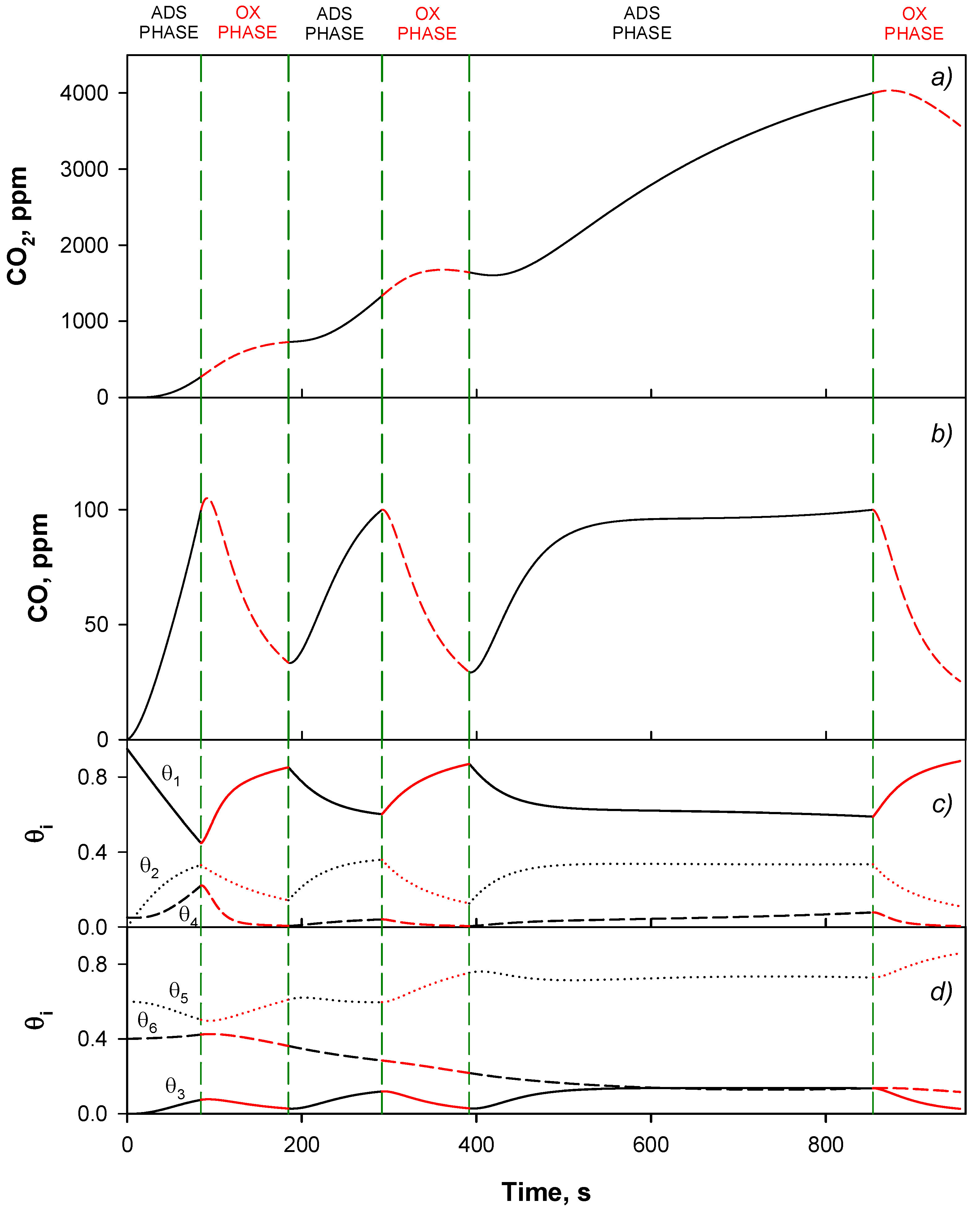
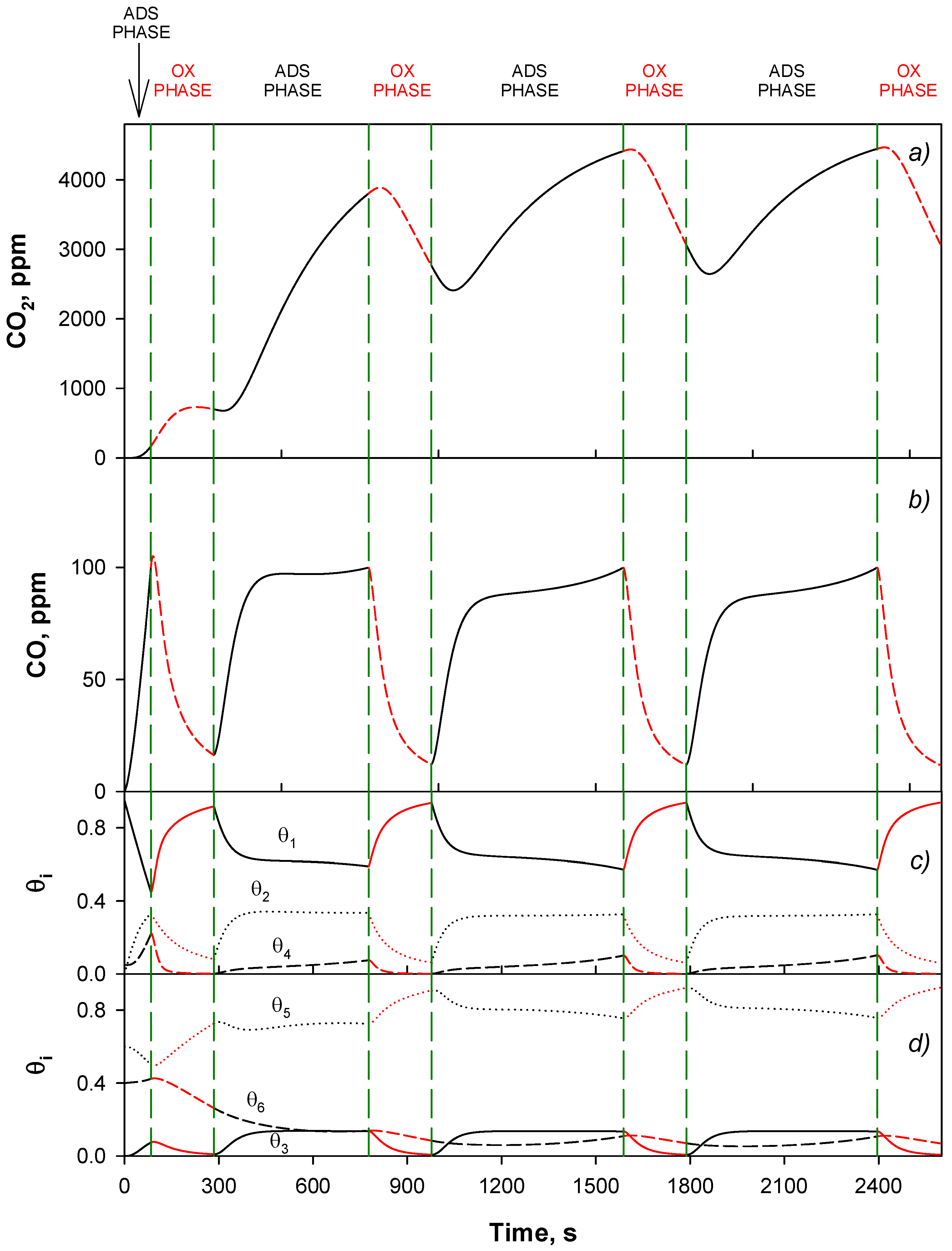
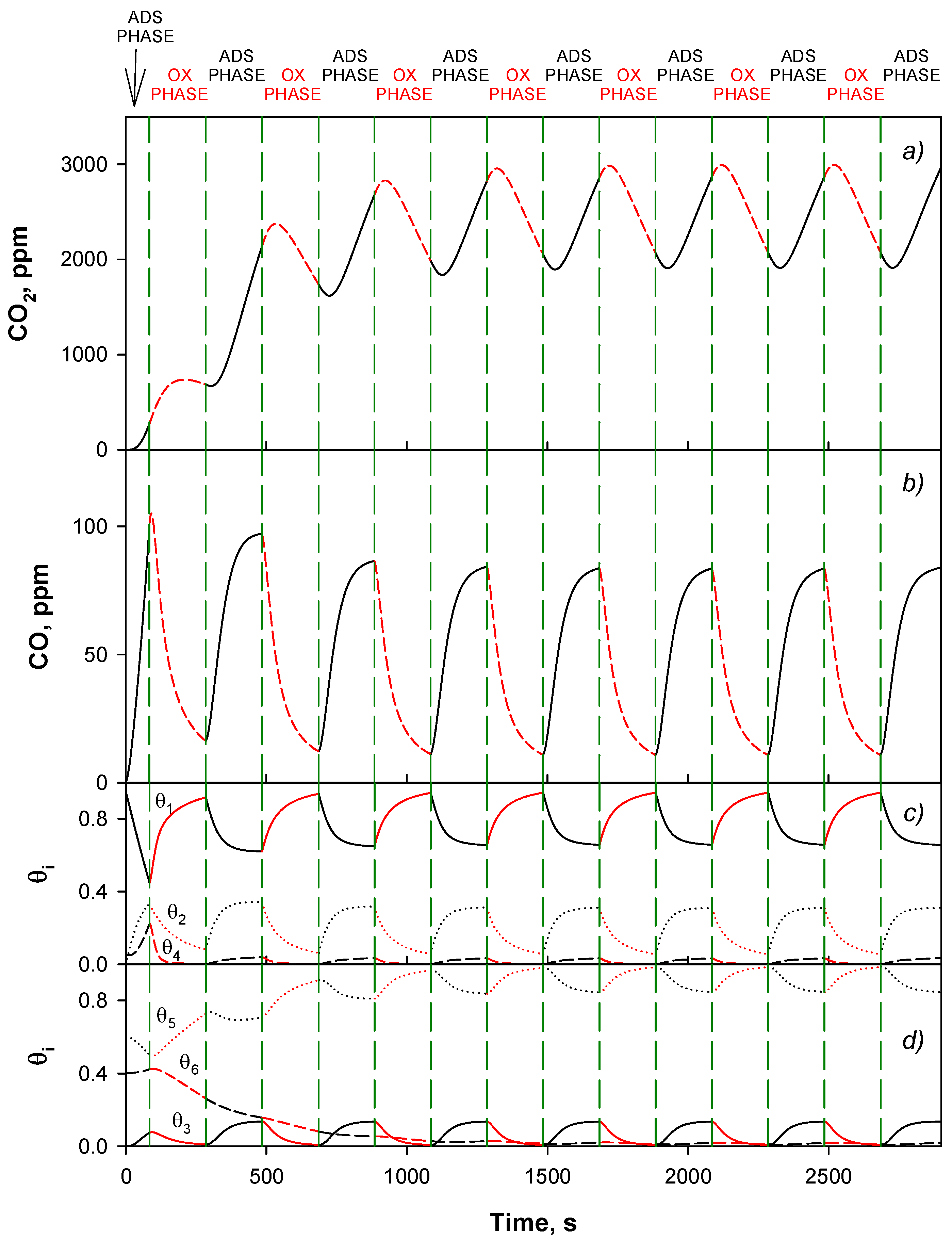
4.4. Trap Simulation
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Recupero, V.; Pino, L.; Vita, A.; Cipitì, F.; Cordaro, M.; Laganà, M. Development of a LPG fuel processor for PEFC systems: Laboratory scale evaluation of autothermal reforming and preferential oxidation subunits. Int. J. Hydrogen Energy 2005, 30, 963–971. [Google Scholar] [CrossRef]
- Pan, L.; Wang, S. A compact integrated fuel-processing system for proton exchange membrane fuel cells. Int. J. Hydrog. Energy 2006, 31, 447–454. [Google Scholar] [CrossRef]
- Men, Y.; Kolb, G.; Zapf, R.; Tiemann, D.; Wichert, M.; Hessel, V.; Lowe, H. A complete miniaturized microstructured methanol fuel processor/fuel cell system for low power applications. Int. J. Hydrogen Energy 2008, 33, 1374–1382. [Google Scholar] [CrossRef]
- Oliva, D.G.; Francesconi, J.A.; Mussati, M.C.; Aguirre, P.A. Energy efficiency analysis of an integrated glycerin processor for PEM fuel cells: Comparison with an ethanol-based system. Int. J. Hydrogen Energy 2010, 35, 709–724. [Google Scholar] [CrossRef]
- Di Bona, D.; Jannelli, E.; Minutillo, M.; Perna, A. Investigations on the behaviour of 2kW natural gas fuel processor. Int. J. Hydrogen Energy 2011, 36, 7763–7770. [Google Scholar] [CrossRef]
- Ji, H.; Bae, J.; Cho, S.; Kang, I. Start-up strategy and operational tests of gasoline fuel processor for auxiliary power unit. Int. J. Hydrogen Energy 2015, 40, 4101–4110. [Google Scholar] [CrossRef]
- Yan, C.; Hai, H.; Guo, C.; Li, W.; Huang, S.; Chen, H. Hydrogen production by steam reforming of dimethyl ether and CO-PrOx in a metal foam micro-reactor. Int. J. Hydrogen Energy 2014, 39, 10409–10416. [Google Scholar] [CrossRef]
- Engelhardt, P.; Maximini, M.; Beckmann, F.; Brenner, M.; Moritz, O. Coupled operation of a diesel steam reformer and an LT- and HT-PEFC. Int. J. Hydrogen Energy 2014, 39, 18146–18153. [Google Scholar] [CrossRef]
- Trimm, D.L.; Önsan, Z.I. Onboard Fuel Conversion for Hydrogen-Fuel-Cell-Driven Vehicles. Catal. Rev. Sci. Eng. 2001, 43, 31–84. [Google Scholar] [CrossRef]
- Zhao, Z.; Lin, X.; Jin, R.; Dai, Y.; Wang, G. High catalytic activity in CO PROX reaction of low cobalt-oxide loading catalysts supported on nano-particulate CeO2–ZrO2 oxides. Catal. Commun. 2011, 12, 1448–1451. [Google Scholar] [CrossRef]
- López, I.; Valdés-Solís, T.; Marbán, G. An attempt to rank copper-based catalysts used in the CO-PROX reaction. Int. J. Hydrogen Energy 2008, 33, 197–205. [Google Scholar] [CrossRef]
- Lukashuk, L.; Föttinger, K.; Kolar, E.; Rameshan, C.; Teschner, D.; Hävecker, M.; Knop-Gericke, A.; Yigit, N.; Li, H.; McDermotta, E. Operando XAS and NAP-XPS studies of preferential CO oxidation on Co3O4 and CeO2-Co3O4 catalysts. J. Catal. 2016, 344, 1–15. [Google Scholar] [CrossRef]
- Martinez-Arias, A.; Fernandez-Garcia, M.; Galvez, O.; Coronado, J.M.; Anderson, J.A.; Conesa, J.C.; Munuera, S.G. Comparative study on redox properties and catalytic behavior for CO oxidation of CuO/CeO2 and CuO/ZrCeO4 catalysts. J. Catal. 2000, 195, 207–216. [Google Scholar] [CrossRef]
- Sedmak, G.; Hočevar, S.; Levec, J. Kinetics of selective CO oxidation in excess of H2 over the nanostructured Cu0.1Ce0.9O2-y catalyst. J. Catal. 2003, 213, 135–150. [Google Scholar] [CrossRef]
- Caputo, T.; Lisi, L.; Pirone, R.; Russo, G.; Ii, F. Kinetics of the Preferential Oxidation of CO over CuO/CeO2 Catalysts in H2-Rich. Ind. Eng. Chem. Res. 2007, 46, 6793–6800. [Google Scholar] [CrossRef]
- Martínez-Arias, A.; Hungría, A.B.; Fernández-García, M.; Conesa, J.C.; Munuera, G. Preferential oxidation of CO in a H2-rich stream over CuO/CeO2 and CuO/(Ce,M)Ox (M = Zr, Tb) catalysts. J. Power Sources 2005, 151, 32–42. [Google Scholar] [CrossRef]
- Martínez-Arias, A.; Hungría, A.B.; Munuera, G.; Gamarra, D. Preferential oxidation of CO in rich H2 over CuO/CeO2: Details of selectivity and deactivation under the reactant stream. Appl. Catal. B Environ. 2006, 65, 207–216. [Google Scholar] [CrossRef]
- Moretti, E.; Lenarda, M.; Riello, P.; Storaro, L.; Talon, A.; Frattini, R.; Reyes-Carmon, A.; Jiménez-López, A.; Rodríguez-Castellón, E. Influence of synthesis parameters on the performance of CeO2-CuO and CeO2-ZrO2-CuO systems in the catalytic oxidation of CO in excess of hydrogen. Appl. Catal. B Environ. 2013, 129, 556–565. [Google Scholar] [CrossRef]
- Avgouropoulos, G.; Ioannides, T.; Matralis, H. Influence of the preparation method on the performance of CuO-CeO2 catalysts for the selective oxidation of CO. Appl. Catal. B Environ. 2005, 56, 87–93. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, R.; Zheng, X. Influence of rare-earth metal doping on the catalytic performance of CuO-CeO2 for the preferential oxidation of CO in excess hydrogen. J. Nat. Gas. Chem. 2008, 17, 283–287. [Google Scholar] [CrossRef]
- Reddy, L.H.; Reddy, G.K.; Devaiah, D.; Reddy, B.M. A rapid microwave-assisted solution combustion synthesis of CuO promoted CeO2-MxOy (M = Zr, La, Pr and Sm) catalysts for CO oxidation. Appl. Catal. A Gen. 2012, 445-446, 297–305. [Google Scholar] [CrossRef]
- Avgouropoulos, G.; Ioannides, T.; Matralis, H.K.; Batista, J.; Hocevar, S. CuO–CeO2 mixed oxide catalysts for the selective oxidation of carbon monoxide in excess hydrogen. Catal. Lett. 2001, 73, 33–40. [Google Scholar] [CrossRef]
- Zou, H.; Dong, X.; Lin, W. Selective CO oxidation in hydrogen-rich gas over CuO/CeO2 catalysts. Appl. Surf. Sci. 2006, 253, 2893–2898. [Google Scholar] [CrossRef]
- Avgouropoulos, G.; Ioannides, T. Selective CO oxidation over CuO-CeO2 catalysts prepared via the urea–nitrate combustion method. Appl. Catal. A Gen. 2003, 244, 155–167. [Google Scholar] [CrossRef]
- Gurbani, A.; Ayastuy, J.L.; González-Marcos, M.P.; Gutiérrez-Ortiz, M.A. CuO-CeO2 catalysts synthesized by various methods: Comparative study of redox properties. Int. J. Hydrogen Energy 2010, 35, 11582–11590. [Google Scholar] [CrossRef]
- Marban, G.; Fuertes, A.B. Highly active and selective CuOx/CeO2 catalyst prepared by a single-step citrate method for preferential oxidation of carbon monoxide. Appl. Catal. B Environ. 2005, 57, 43–53. [Google Scholar] [CrossRef]
- Barbato, P.S.; Colussi, S.; Di Benedetto, A.; Landi, G.; Lisi, L.; Llorca, J.; Trovarelli, A. Origin of High Activity and Selectivity of CuO/CeO2 Catalysts Prepared by Solution Combustion Synthesis in CO-PROX Reaction. J. Phys. Chem. C 2016, 120, 13039–13048. [Google Scholar] [CrossRef]
- Landi, G.; Di Benedetto, A.; Colussi, S.; Barbato, P.S.; Lisi, L. Effect of carbon dioxide and water on the performances of an iron-promoted copper/ceria catalyst for CO preferential oxidation in H2-rich streams. Int. J. Hydrogen Energy 2016, 41, 7332–7341. [Google Scholar] [CrossRef]
- Barbato, P.S.; Colussi, S.; Di Benedetto, A.; Landi, G.; Lisi, L.; Llorca, J.; Trovarelli, A. CO preferential oxidation under H2-rich streams on copper oxide supported on Fe promoted CeO2. Appl. Catal. A Gen. 2015, 506, 268–277. [Google Scholar] [CrossRef]
- Wang, W.-W.; Du, P.-P.; Zou, S.-H.; He, H.-Y.; Wang, R.-X.; Jin, Z.; Shi, S.; Huang, Y.-Y.; Si, S.; Song, Q.-S. Highly Dispersed Copper Oxide Clusters as Active Species in Copper-Ceria Catalyst for Preferential Oxidation of Carbon Monoxide. ACS Catal. 2015, 5, 2088–2099. [Google Scholar] [CrossRef]
- Elias, J.S.; Artrith, N.; Bugnet, M.; Giordano, L.; Botton, G.A.; Kolpak, A.M.; Yang, S.H. Elucidating the Nature of the Active Phase in Copper/Ceria Catalysts for CO Oxidation. ACS Catal. 2016, 6, 1675–1679. [Google Scholar] [CrossRef]
- Davó-Quiñonero, A.; Navlani-García, M.; Lozano-Castelló, D.; Bueno-López, A.; Anderson, J.A. Role of Hydroxyl Groups in the Preferential Oxidation of CO over Copper Oxide-Cerium Oxide Catalysts. ACS Catal. 2016, 6, 1723–1731. [Google Scholar] [CrossRef]
- Lee, H.C.; Kim, D.H. Kinetics of CO and H2 oxidation over CuO-CeO2 catalyst in H2 mixtures with CO2 and H2O. Catal. Today 2008, 132, 109–116. [Google Scholar] [CrossRef]
- Di Benedetto, A.; Landi, G.; Lisi, L.; Russo, G. Role of CO2 on CO preferential oxidation over CuO/CeO2 catalyst. Appl. Catal. B Environ. 2013, 142-143, 169–177. [Google Scholar] [CrossRef]
- Wang, J.B.; Lin, S.C.; Huang, T.J. Selective CO oxidation in rich hydrogen over CuO/samaria-doped ceria. Appl. Catal. A Gen. 2002, 232, 107–120. [Google Scholar] [CrossRef]
- Chuang, C.; Chen, Y.; Ward, J.; Yu, C.; Liu, Y.; Lee, C. Optimal design of an experimental methanol fuel reformer. Int. J. Hydrogen Energy 2008, 33, 7062–7073. [Google Scholar] [CrossRef]
- Dudÿeld, C.D.; Chen, R.; Adcock, P.L. A carbon monoxide PROX reactor for PEM fuel cell automotive application. Int. J. Hydrogen Energy 2001, 26, 763–775. [Google Scholar] [CrossRef]
- Northrop, W.F.; Choi, S.O.; Thompson, L.T. Thermally integrated fuel processor design for fuel cell applications. Int. J. Hydrogen Energy 2012, 37, 3447–3458. [Google Scholar] [CrossRef]
- Cipitì, F.; Pino, L.; Vita, A.; Laganà, M.; Recupero, V. Experimental investigation on a methane fuel processor for polymer electrolyte fuel cells. Int. J. Hydrogen Energy 2013, 38, 2387–2397. [Google Scholar] [CrossRef]
- Ayastuy, J.L.; Gurbani, A.; González-Marcos, M.P.; Gutiérrez-Ortiz, M.A. Selective CO oxidation in H2 streams on CuO/CexZr1−xO2 catalysts: Correlation between activity and low temperature reducibility. Int. J. Hydrogen Energy 2012, 37, 1993–2006. [Google Scholar] [CrossRef]
- Sirichaiprasert, K.; Luengnaruemitchai, A.; Pongstabodee, S. Selective oxidation of CO to CO2 over Cu-Ce-Fe-O composite-oxide catalyst in hydrogen feed stream. Int. J. Hydrogen Energy 2007, 32, 915–926. [Google Scholar] [CrossRef]
- Lendzion-Bielun, Z.; Bettahar, M.M.; Monteverdi, S. Fe-promoted CuO/CeO2 catalyst: Structural characterization and CO oxidation activity. Catal. Commun. 2010, 11, 1137–1142. [Google Scholar] [CrossRef]
- Zou, H.; Chen, S.; Liu, Z.; Lin, W. Selective CO oxidation over CuO-CeO2 catalysts doped with transition metal oxides. Powder Technol. 2011, 207, 238–244. [Google Scholar] [CrossRef]
- Kydd, R.; Ferri, D.; Hug, P.; Scott, J.; Teoh, W.Y.; Amal, R. Temperature-induced evolution of reaction sites and mechanisms during preferential oxidation of CO. J. Catal. 2011, 277, 64–71. [Google Scholar] [CrossRef]
- Zhang, Q.; Shore, L.; Farrauto, R.J. Selective CO oxidation over a commercial PROX monolith catalyst for hydrogen fuel cell applications. Int. J. Hydrogen Energy 2012, 37, 10874–10880. [Google Scholar] [CrossRef]
- Caputo, T.; Lisi, L.; Pirone, R.; Russo, G. On the role of redox properties of CuO/CeO2 catalysts in the preferential oxidation of CO in H2-rich gases. Appl. Catal. A Gen. 2008, 348, 42–53. [Google Scholar] [CrossRef]
- Gamarra, D.; Munuera, G.; Hungría, A.B.; Fernández-García, M.; Conesa, J.C.; Midgley, P.A.; Wang, X.Q.; Hanson, J.C.; Rodríguez, J.A.; Martínez-Arias, A. Structure-activity relationship in nanostructured copper-ceria-based preferential CO oxidation catalysts. J. Phys. Chem C 2007, 111, 11026–11038. [Google Scholar] [CrossRef]
- Di Benedetto, A.; Landi, G.; Lisi, L. CO reactive adsorption at low temperature over CuO/CeO2 structured catalytic monolith. Int. J. Hydrogen Energy 2017. [Google Scholar] [CrossRef]
- Landi, G.; Barbato, P.S.; Di Benedetto, A.; Lisi, L. Optimization of the preparation method of CuO/CeO2 structured catalytic monolith for CO preferential oxidation in H2-rich streams. Appl. Catal. B Environ. 2016, 181, 727–737. [Google Scholar] [CrossRef]
- Barbato, P.S.; Di Benedetto, A.; Landi, G.; Lisi, L. CuO/CeO2 based monoliths for CO preferential oxidation in H2-rich streams. Chem. Eng. J. 2015, 279, 983–993. [Google Scholar] [CrossRef]
- Arango-Díaz, A.; Cecilia, J.A.; Dos Santos-Gómez, L.; Marrero-López, D.; Losilla, E.R.; Jiménez-Jiménez, J.; Rodríguez-Castellóna, E. Characterization and performance in preferential oxidation of CO of CuO-CeO2 catalysts synthesized using polymethyl metacrylate (PMMA) as template. Int. J. Hydrogen Energy 2015, 40, 11254–11260. [Google Scholar] [CrossRef]
- Moreno, M.; Bergamini, L.; Baronetti, G.T.; Laborde, M.A.; Mariño, F.J. Mechanism of CO oxidation over CuO/CeO2 catalysts. Int. J. Hydrogen Energy 2010, 35, 5918–5924. [Google Scholar] [CrossRef]
- Kanervo, J.M.; Keskitalo, T.J.; Slioor, R.I.; Krause, A.O.I. Temperature-programmed desorption as a tool to extract quantitative kinetic or energetic information for porous catalysts. J. Catal. 2006, 238, 382–393. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Segment 1 | Segment 2 (Isothermal) | Segment 2 (Total) | Segment 3 |
|---|---|---|---|---|
| (mmol) | ||||
| CO | 4.50 × 10−2 | 0.2 × 10−2 | 0.2 × 10−2 | 2.72 × 10−2 |
| CO2 | 0.04 × 10−2 | 1.65 × 10−2 | 3.02 × 10−2 | - |
| Symbol of Site | Nature of Site | Site Fraction Symbol |
|---|---|---|
| σ-O | θ1 | |
| σ-O-CO | θ2 | |
| σ’-O-CO2 | θ3 | |
| σ | θ4 | |
| σ’-O | θ5 | |
| σ’ | θ6 |
| Step | Kinetic Constant (ki, 1/s) | Activation Energy (Ei, J/K mol) |
|---|---|---|
| 1 | 120 | 30,000 |
| −1 | 60,000 | 33,000 |
| 2 | 10,000 | 32,000 |
| 3 | 6000 | 32,000 |
| 4 | 50 | 25,000 |
| Root mean square error (SRMSE) = 0.021 | ||
| Step | Kinetic Constant (ki, (m3/mol)0.5 s) | Activation Energy (Ei, J/K mol) |
|---|---|---|
| 5 | 50 | 21,000 |
| 6 | 5 | 22,000 |
| SRMSE = 0.35 | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Landi, G.; Di Benedetto, A.; Lisi, L. Two-Stage Strategy for CO Removal from H2-Rich Streams over (Nano-) CuO/CeO2 Structured Catalyst at Low Temperature. Appl. Sci. 2018, 8, 789. https://doi.org/10.3390/app8050789
Landi G, Di Benedetto A, Lisi L. Two-Stage Strategy for CO Removal from H2-Rich Streams over (Nano-) CuO/CeO2 Structured Catalyst at Low Temperature. Applied Sciences. 2018; 8(5):789. https://doi.org/10.3390/app8050789
Chicago/Turabian StyleLandi, Gianluca, Almerinda Di Benedetto, and Luciana Lisi. 2018. "Two-Stage Strategy for CO Removal from H2-Rich Streams over (Nano-) CuO/CeO2 Structured Catalyst at Low Temperature" Applied Sciences 8, no. 5: 789. https://doi.org/10.3390/app8050789
APA StyleLandi, G., Di Benedetto, A., & Lisi, L. (2018). Two-Stage Strategy for CO Removal from H2-Rich Streams over (Nano-) CuO/CeO2 Structured Catalyst at Low Temperature. Applied Sciences, 8(5), 789. https://doi.org/10.3390/app8050789