Modelling Long-Term Durability Performance of Cementitious Materials under Sodium Sulphate Interaction


Abstract

1. Introduction
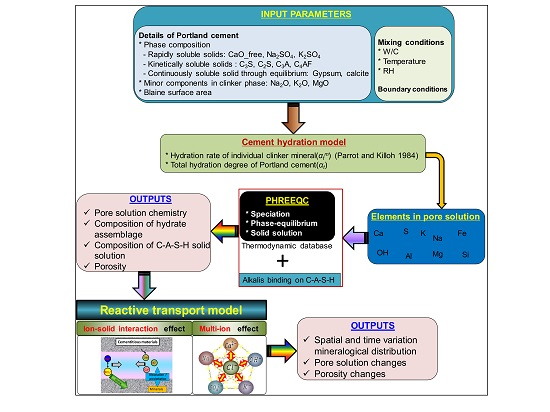
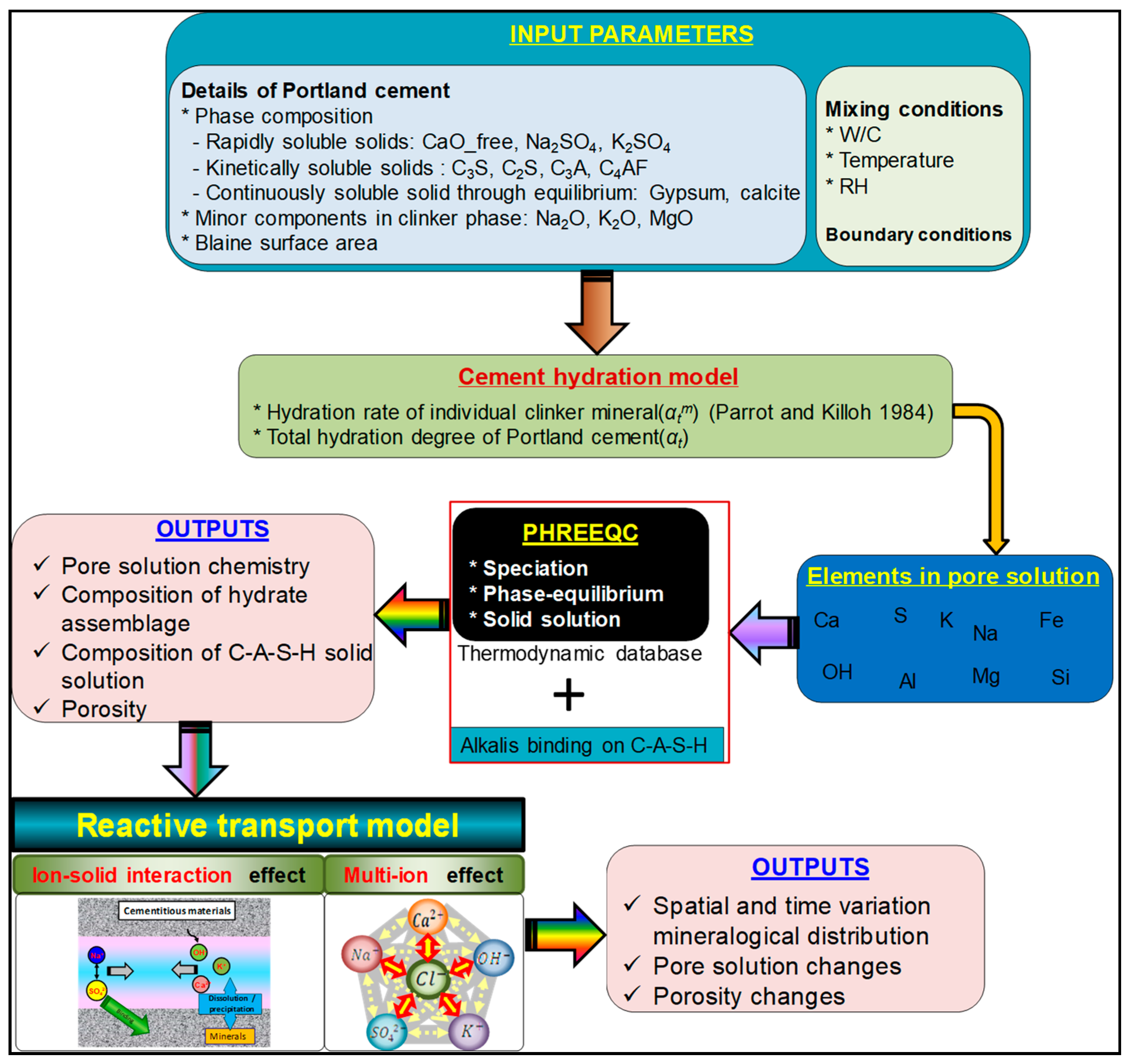
2. Calculation Procedure and Input Parameters
3. Simulation Results and Discussions
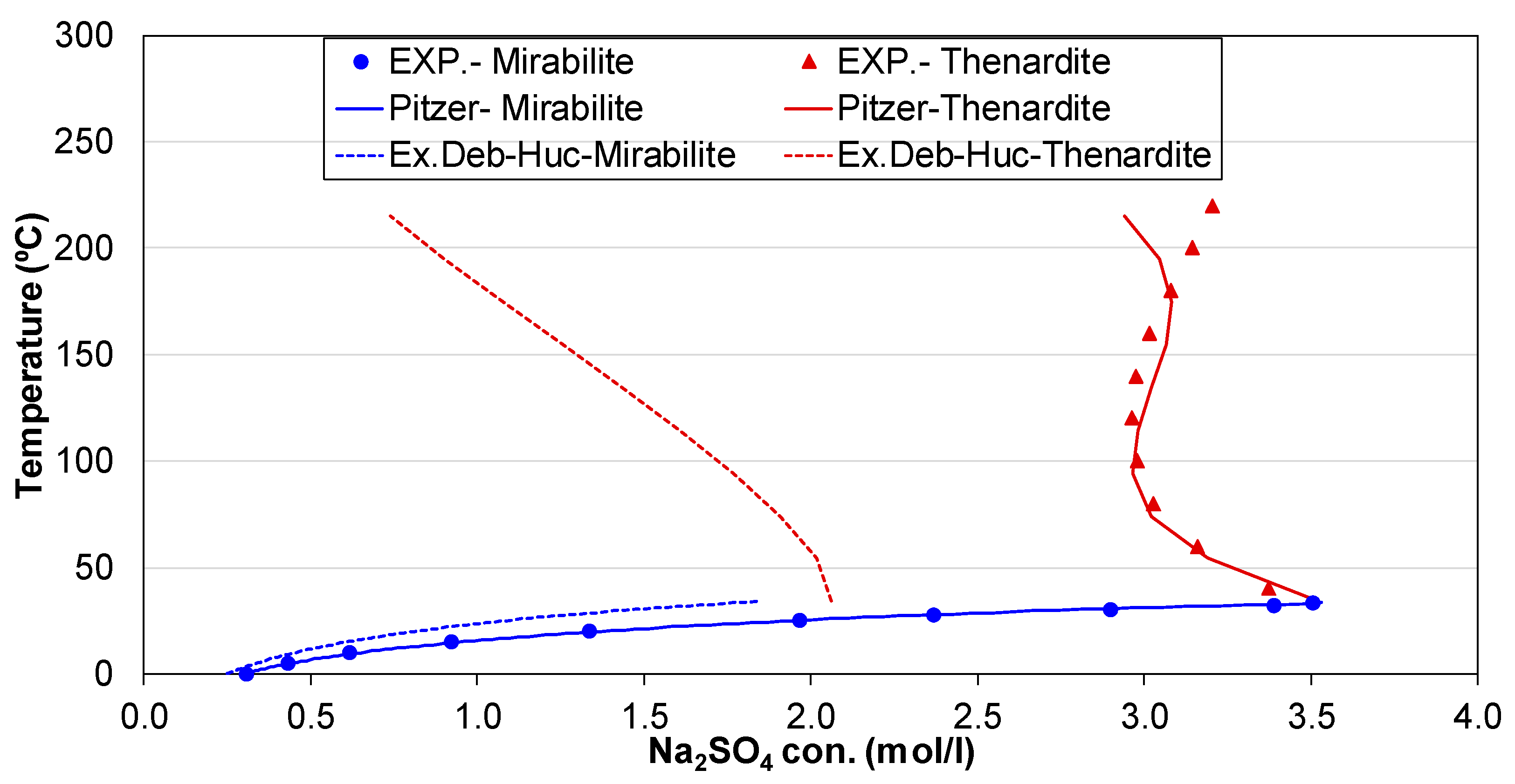
3.1. Check on Thermodynamic Database
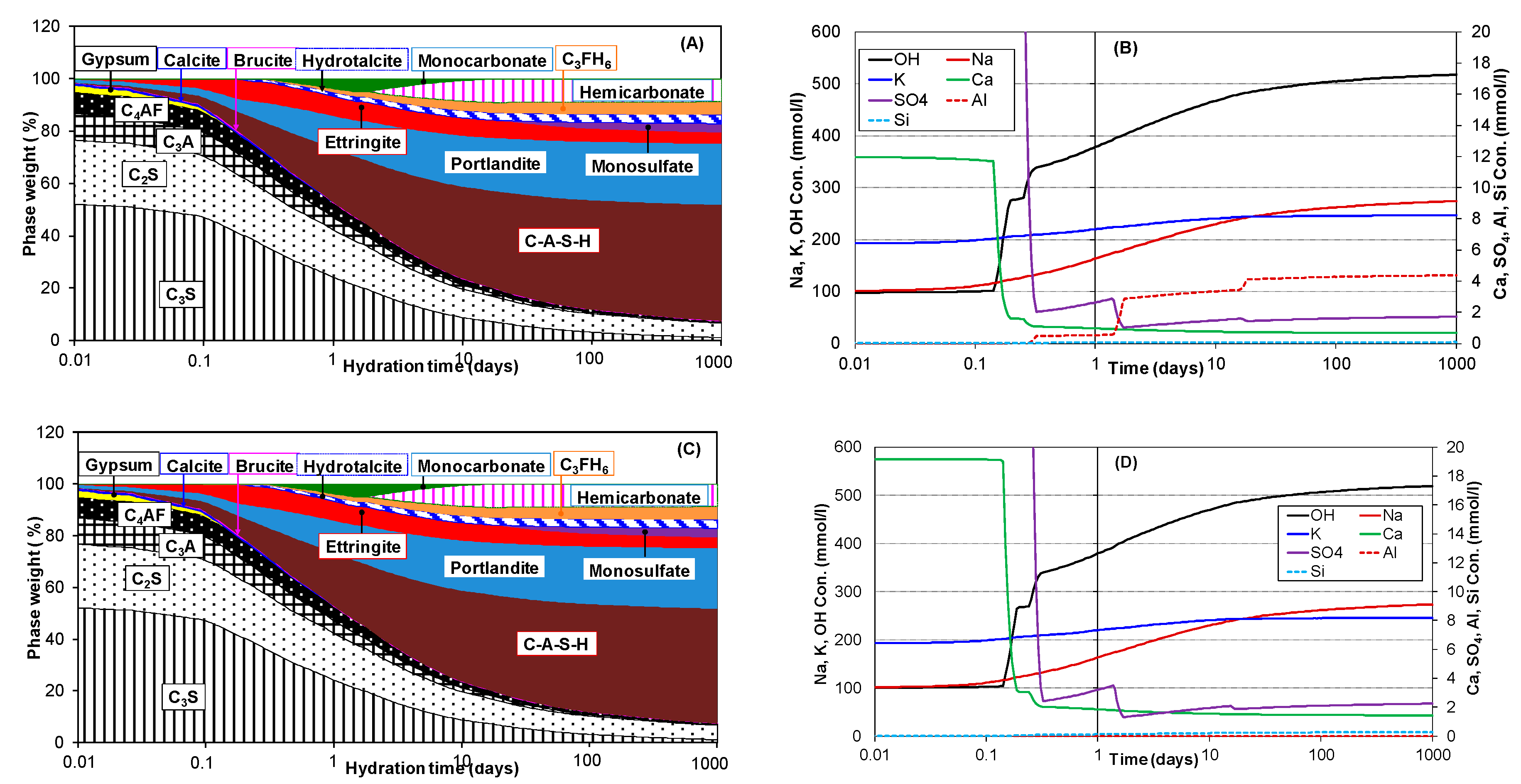
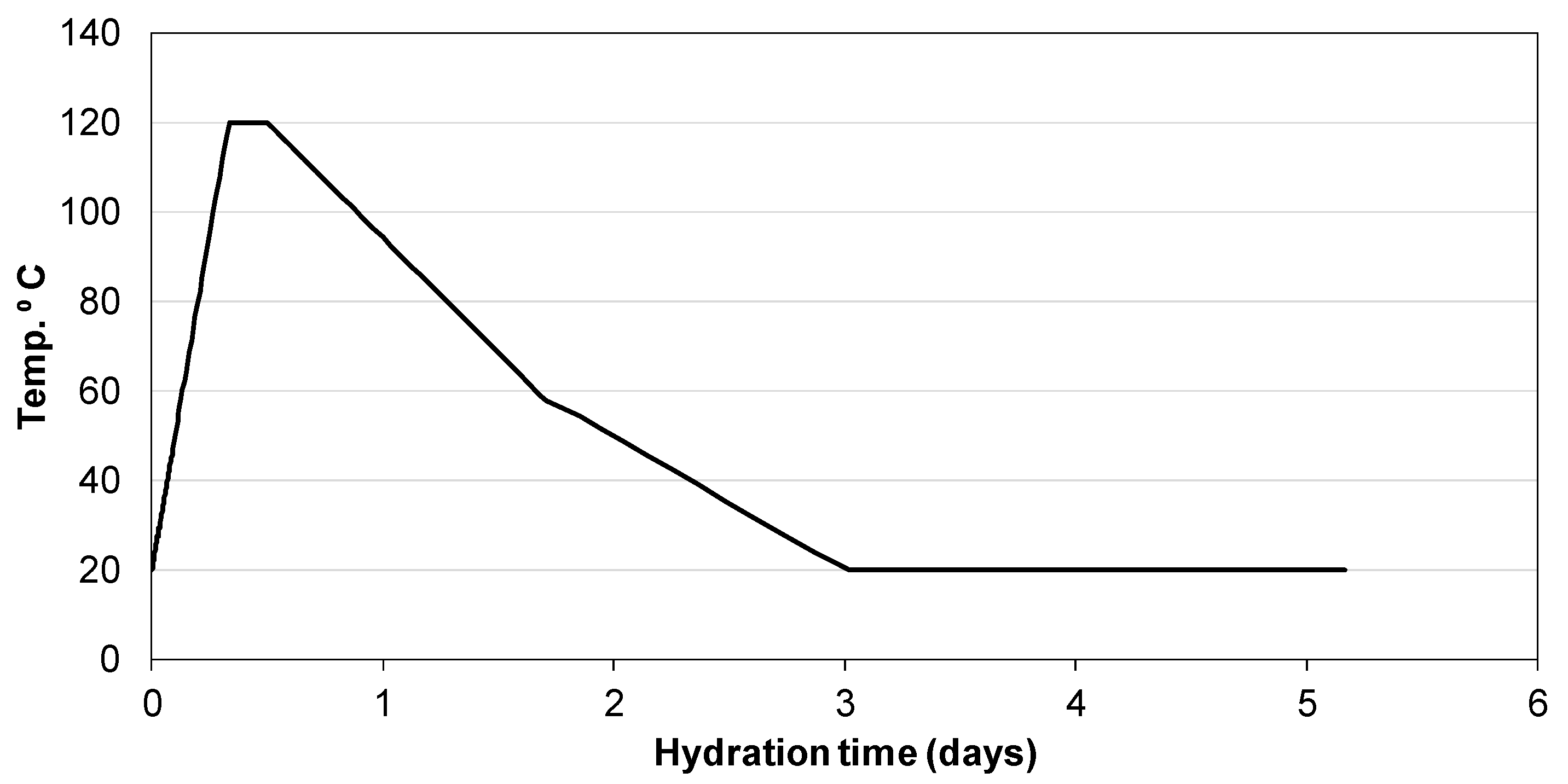
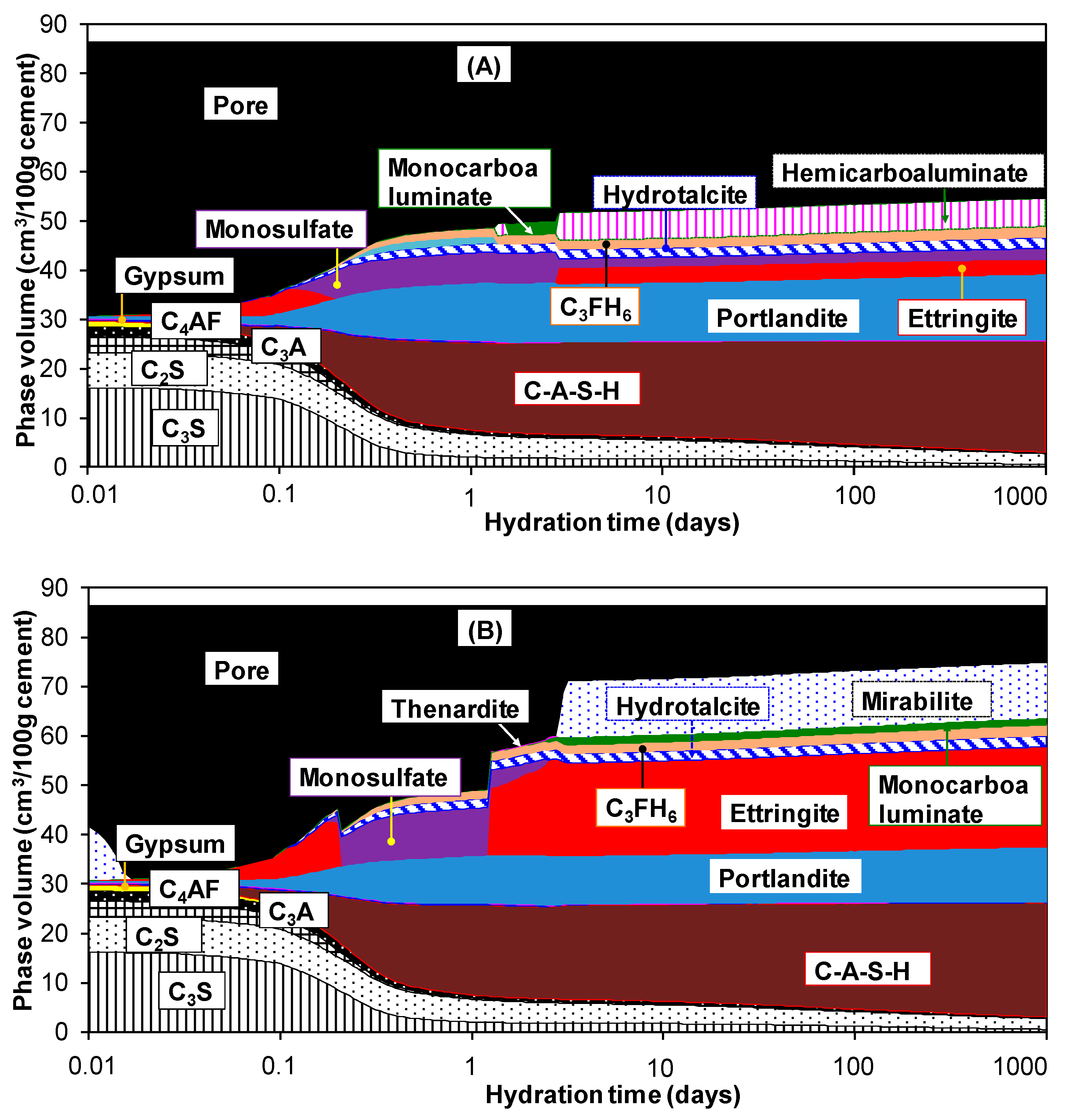
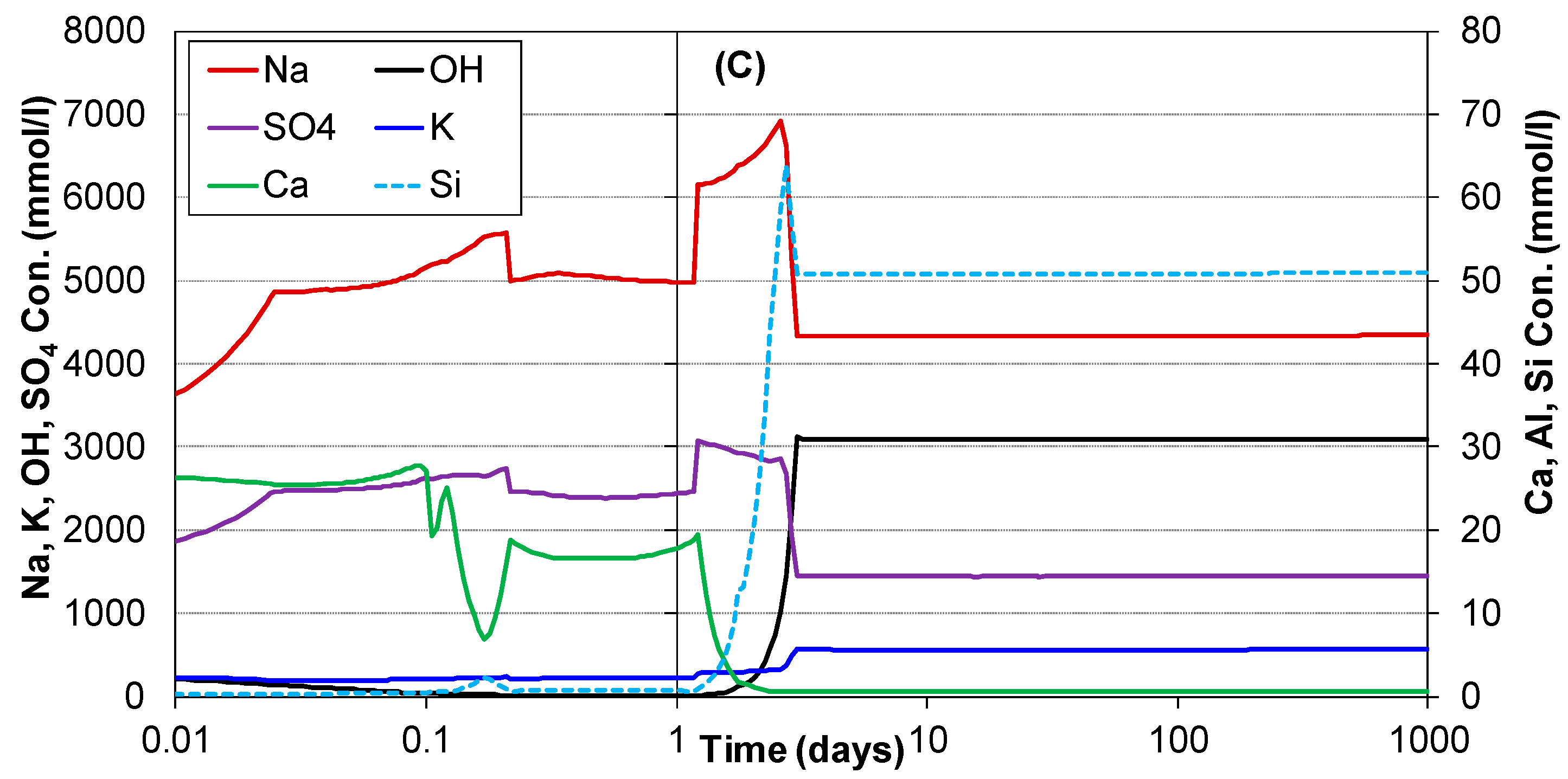
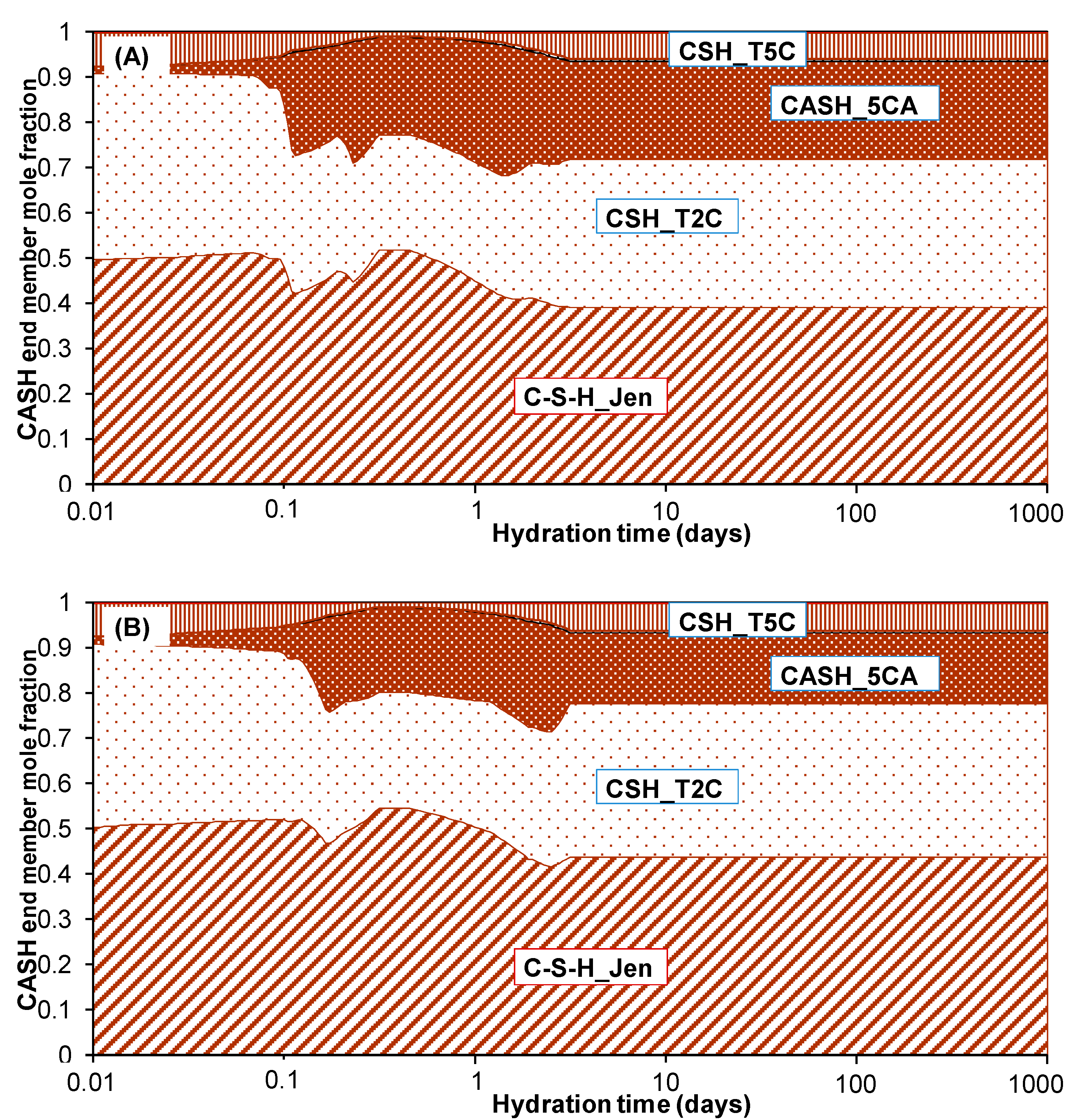
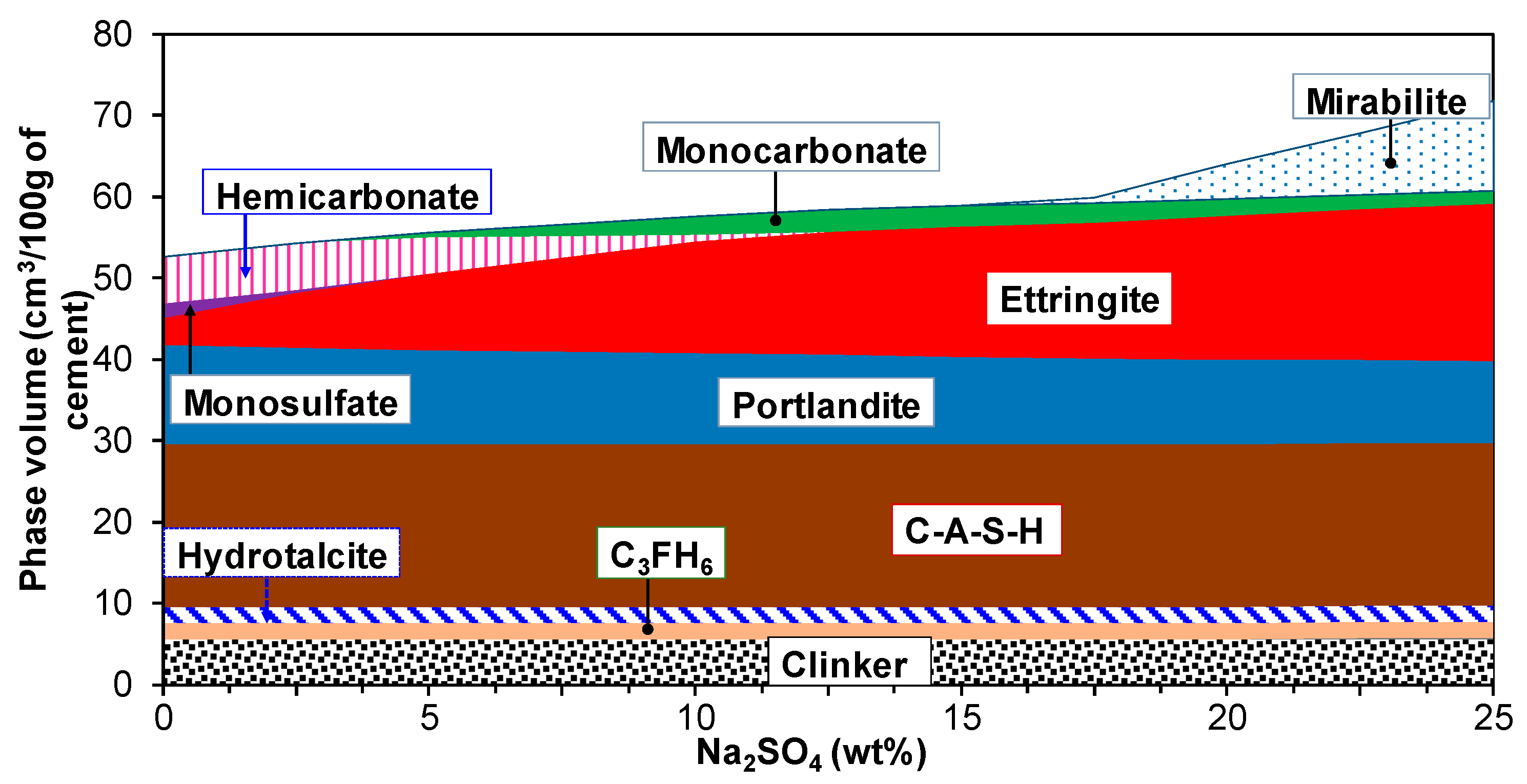
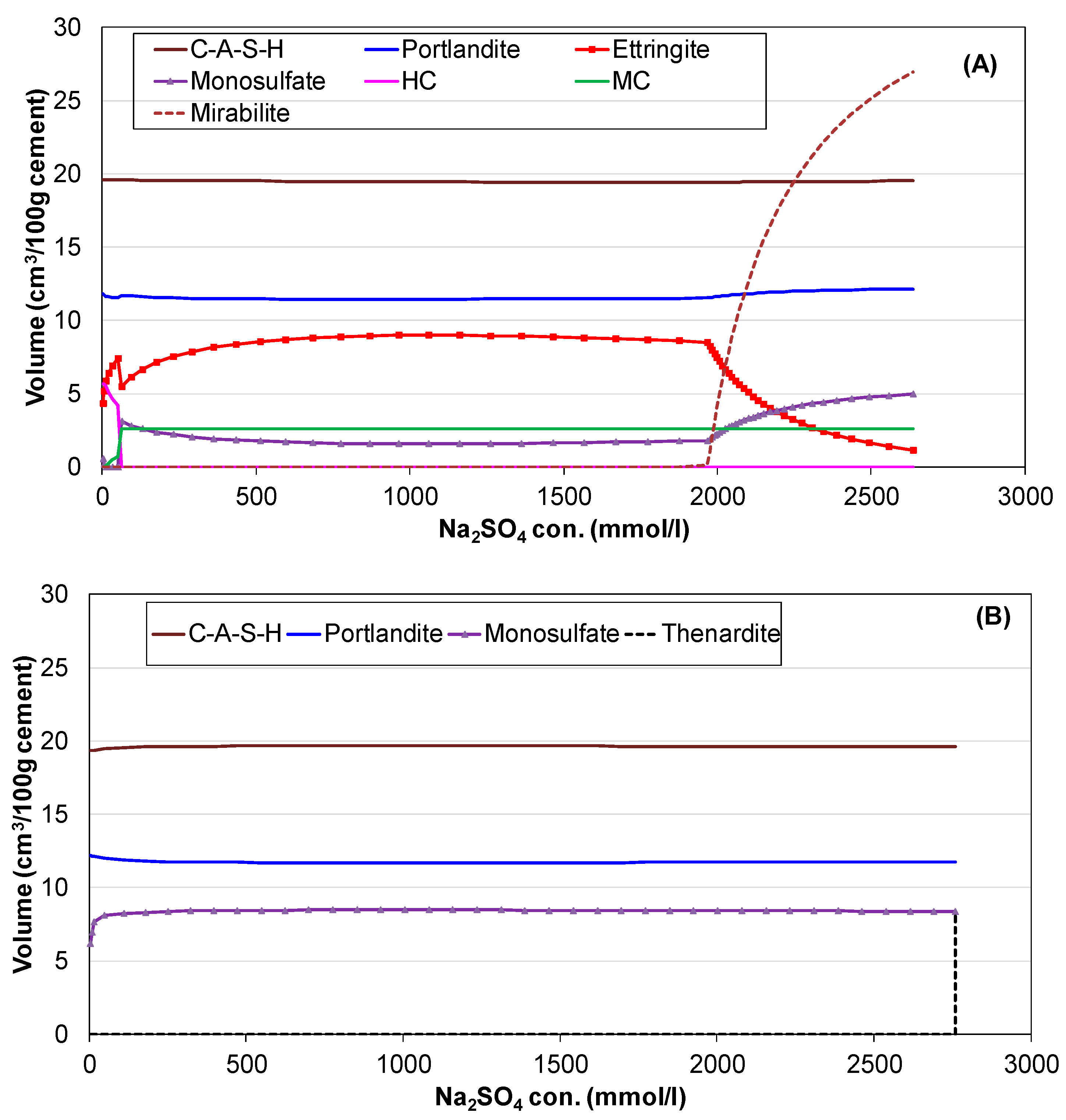
3.2. Hydration of Portland Cement with Sodium Sulphate
3.3. Influence of Na2SO4 Concentration and Temperature
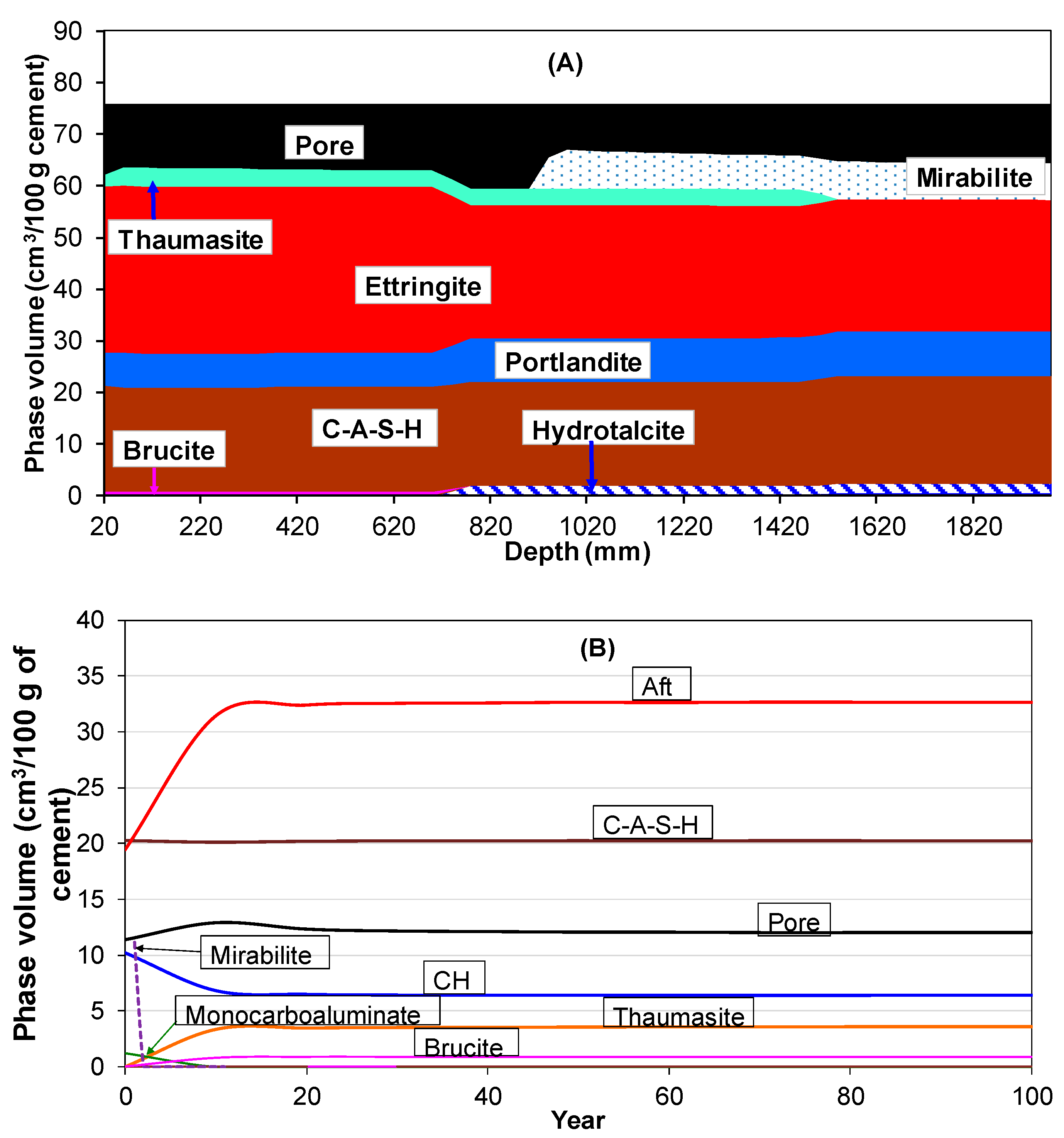
3.4. Sulphate Ingress into Hydrated Portland Cement
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A. Model Description
Appendix A.1. Phase-Equilibrium Model

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phase | Reactions | logKp | △rH0 | Ref. |
|---|---|---|---|---|
| Anhydrite | CaSO4↔ Ca2+ + SO42- | −4.36 | −1.71 | [23] |
| Brucite | Mg(OH)2 + 2H+ ↔ Mg2+ + 2H2O | 17.07 | −115.66 | [27] |
| C3AH6 | Ca3Al2(OH)12 + 12H+ ↔ 3Ca2+ + 2Al3+ + 12H2O | 82.22 | −595.76 | [27] |
| C3FH6 | Ca3Fe2(OH)12 + 12H+ ↔ 3Ca2+ + 2Fe3+ + 12H2O | 73.65 | −516.96 | [27] |
| Calcite | CaCO3 ↔ Ca2+ + CO32− | −8.48 | −2.297 | [23] |
| CASH_5CA | (CaO)1.25(Al2O3)0.125(SiO2):1.625H2O + 3.25H+ ↔ 1.25Ca2+ + 0.25Al3+ + H4SiO4 + 1.25H2O | 22.00 | −141.58 | [29] |
| CASH_INFCA | (CaO)(Al2O3)0.15625(SiO2)1.1875:1.65625H2O + 2.9375H+ ↔ Ca2+ + 0.3125Al3+ + 1.1875H4SiO4 + 0.75H2O | 16.60 | −110.67 | [29] |
| CSH_T2C | (CaO)1.5(SiO2):2.5H2O + 3H+ ↔ 1.5Ca2+ + H4SiO4 + 2H2O | 25.88 | −127.10 | [29] |
| CSH_T5C | (CaO)1.25(SiO2)1.25:2.5H2O + 2.5H+ ↔1.25Ca2+ + 1.25H4SiO4 + 1.25H2O | 18.74 | −83.46 | [29] |
| CSH_Jen | (CaO)1.667(SiO2):2.1H2O + 3.334H+ ↔ 1.667Ca2+ + H4SiO4 + 1.767H2O | 29.60 | −148.44 | [27] |
| CSH_TobH | (CaO)(SiO2)1.5:2.5H2O + 2H+ ↔ Ca2+ + 1.5H4SiO4 + 0.5H2O | 13.18 | −47.83 | [29] |
| Ettringite | Ca6Al2(SO4)3(OH)12:26H2O + 12H+ ↔ 6Ca2+ + 2Al3+ + 3SO42− + 38H2O | 57.73 | −389.36 | [27] |
| Gypsum | CaSO4:2H2O ↔ Ca2+ + SO42− + 2 H2O | −4.58 | −0.109 | [23] |
| Hemicarboaluminate | Ca4Al2(CO3)0.5(OH)13:5.5H2O + 13H+ ↔ 4Ca2+ + 2Al3+ + 0.5CO32− + 18.5H2O | 87.88 | −604.27 | [27] |
| Hydrotalcite | Mg4Al2(OH)14:3H2O + 14H+ ↔ 2Al3+ + 4Mg2+ + 17H2O | 75.97 | −607.91 | [27] |
| Mirabilite | Na2SO4:10H2O ↔ 2Na+ + SO42− + 10H2O | −1.22 | 79.471 | [28] |
| Monocarboaluminate | Ca4Al2(CO3)(OH)12:5H2O + 12H+ ↔ 4Ca2+ + 2Al3+ + CO32− + 17H2O | 71.54 | −533.14 | [27] |
| Monosulfoaluminate | Ca4Al2(SO4)(OH)12:6H2O + 12H+ ↔ 4Ca2+ + 2Al3+ + SO42− + 18H2O | 73.68 | −553.08 | [27] |
| Portlandite | Ca(OH)2 + 2H+ ↔ Ca2+ + 2H2O | 22.79 | −129.66 | [27] |
| Thaumasite | Ca6(SiO3)2(SO4)2(CO3)2: 30H2O + 4H+ ↔ 6Ca2+ + 2H4SiO4 +2CO32− + 2SO42− +28H2O | −1.655 | 20.23 | [27] |
| Thenardite | Na2SO4 ↔ 2Na+ + SO42− | −0.34 | −2.46 | [28] |
Appendix A.2. Cement Hydration Model
| C3S | C2S | C3A | C4AF | |
|---|---|---|---|---|
| K1 | 1.5 | 0.5 | 1 | 0.37 |
| N1 | 0.7 | 1 | 0.85 | 0.7 |
| K2 | 0.05 | 0.006 | 0.04 | 0.015 |
| K3 | 1.1 | 0.2 | 1 | 0.4 |
| N3 | 3.3 | 5 | 3.2 | 3.7 |
| H | 1.8 | 1.35 | 1.60 | 1.45 |
| Ea (J/mol) | 41,570 | 20,785 | 54,040 | 34,087 |
Appendix A.3. Multispecies Transport Model
References
- Metcalfe, R.; Walker, C. NUMO-TR-04-05. In Proceedings of the International Workshop on Bentonite-Cement Interaction in Repository Environments, Tokyo, Japan, 14–16 April 2004. [Google Scholar]
- IAEA. Near Surface Disposal of Radioactive Waste. In IAEA Safety Standards Series, Requirements; IAEA: Vienna, Austria, 1999. [Google Scholar]
- Atkins, M.; Glasser, F.P. Application of portland cement-based materials to radioactive waste immobilization. Waste Manag. 1992, 12, 105–131. [Google Scholar] [CrossRef]
- Glasser, F.P. Progress in the immobilization of radioactive wastes in cement. Cem. Concr. Res. 1992, 22, 201–216. [Google Scholar] [CrossRef]
- Jantzen, C.; Johnson, A. Cements in waste management. Adv. Cem. Res. 2010, 22, 225–231. [Google Scholar] [CrossRef]
- Lin, W.-S.; Liu, C.-W.; Li, M.-H. Influences of specific ions in groundwater on concrete degradation in subsurface engineered barrier system. Springerplus 2016, 5, 745. [Google Scholar] [CrossRef] [PubMed]
- Owaki, E.; Niwase, K.; Akatsuka, M.; Kudo, J. Effect of curing temperature on cement paste containing sodium sulphate. JSCE Ann. Meet. 2012, 67, 57–58. [Google Scholar]
- Divet, L.; Randriambololona, R. Delayed Ettringite Formation: The Effect of Temperature and Basicity on the Interaction of Sulphate and C-S-H Phase. Cem. Concr. Res. 1998, 28, 357–363. [Google Scholar] [CrossRef]
- Scrivener, K.; Skalny, J. International RILEM Workshop on Internal Sulfate Attack and Delayed Ettringite Formation. Mater. Struct. 2004, 37, 71–72. [Google Scholar] [CrossRef]
- Alexander, M.; Bertron, A.; De Belie, N. Performance of Cement-Based Materials in Aggressive Aqueous Environments, RILEM TC 211, ed.; Springer: New York, NY, USA, 2013. [Google Scholar]
- Sarkar, S.; Mahadevan, S.; Meeussen, J.C.L.; van der Sloot, H.; Kosson, D.S. Numerical simulation of cementitious materials degradation under external sulfate attack. Cem. Concr. Compos. 2010, 32, 241–252. [Google Scholar] [CrossRef]
- Marchand, J.; Samson, E.; Maltais, Y.; Beaudoin, J.J. Theoretical analysis of the effect of weak sodium sulfate solutions on the durability of concrete. Cem. Concr. Compos. 2002, 24, 317–329. [Google Scholar] [CrossRef]
- Rozière, E.; Loukili, A.; El Hachem, R.; Grondin, F. Durability of concrete exposed to leaching and external sulphate attacks. Cem. Concr. Res. 2009, 39, 1188–1198. [Google Scholar] [CrossRef]
- Rodriguez-Navarro, C.; Doehne, E.; Sebastian, E. How does sodium sulfate crystallize? Implications for the decay and testing of building materials. Cem. Concr. Res. 2000, 30, 1527–1534. [Google Scholar] [CrossRef]
- Soive, A.; Tran, V.Q. External sulfate attack of cementitious materials: New insights gained through numerical modeling including dissolution/precipitation kinetics and surface complexation. Cem. Concr. Compos. 2017, 83, 263–272. [Google Scholar] [CrossRef]
- Maekawa, K.; Ishida, T.; Kishi, K. Multi-Scale Modelling of Concrete; Taylor Francis: Abingdon-on-Thames, UK, 2009. [Google Scholar]
- Elakneswaran, Y.; Ishida, T. Development and verification of an integrated physicochemical and geochemical modelling framework for performance assessment of cement-based materials. J. Adv. Concr. Technol. 2014, 12, 111–126. [Google Scholar] [CrossRef]
- Geiker, M.R.; Michel, A.; Lepech, M.D.; Wu, J.; Stang, H. Multi-scale and multi-physics deterioration for design and assessment of reinforced concrete structures. RILEM Tech. Lett. 2017, 2, 119–128. [Google Scholar] [CrossRef]
- Jensen, M.M. A Coupled Transport and Chemical Model for Durability Predictions of Cement Based Materials. Ph.D. Thesis, Technical University of Denmark, Copenhagen, Denmark, 2014. [Google Scholar]
- Marty, N.C.M.; Bildstein, O.; Blanc, P.; Claret, F.; Cochepin, B.; Gaucher, E.C.; Jacques, D.; Lartigue, J.-E.; Liu, S.; Mayer, K.U.; et al. Benchmarks for multicomponent reactive transport across a cement/clay interface. Comput. Geosci. 2015, 19, 635–653. [Google Scholar] [CrossRef]
- Elakneswaran, Y.; Owaki, E.; Miyahara, S.; Ogino, M.; Maruya, T.; Nawa, T. Hydration study of slag-blended cement based on thermodynamic considerations. Constr. Build. Mater. 2016, 124, 615–625. [Google Scholar] [CrossRef]
- Appelo, C.A.J.; Postma, D. Geochemistry, Groundwater and Pollution; CRC Press Taylor & Francis Group: Boca Raton, FL, USA, 2009. [Google Scholar]
- Parkhust, D.L.; Appelo, C.A.J. A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport and Inverse Geochemical Calculations; USGS: Middleton, WI, USA, 1999.
- Pitzer, K.S. Ion Interaction Approach: Theory and Data Correlation. In Activity Coefficients in Electrolyte Solutions; Pitzer, K.S., Ed.; CRC Press: Boca Raton, FL, USA, 1991. [Google Scholar]
- Elakneswaran, Y.; Iwasa, A.; Nawa, T.; Sato, T.; Kurumisawa, K. Ion-cement hydrate interactions govern multi-ionic transport model for cementitious materials. Cem. Concr. Res. 2010, 40, 1756–1765. [Google Scholar] [CrossRef]
- Kari, O.P.; Elakneswaran, Y.; Nawa, T.; Puttonen, J.A. A model for a long-term diffusion of multispecies in concrete based on ion-cement-hydrate interaction. J. Mater. Sci. 2013, 48, 4243–4259. [Google Scholar] [CrossRef]
- Lothenbach, B.; Matschei, T.; Möschner, G.; Glasser, F.P. Thermodynamic modelling of the effect of temperature on the hydration and porosity of Portland cement. Cem. Concr. Res. 2008, 38, 1–18. [Google Scholar] [CrossRef]
- BRGM. THERMODDEM. Available online: http://thermoddem.brgm.fr/ (accessed on 2 October 2018).
- Myers, R.J.; Bernal, S.A.; Provis, J.L. A thermodynamic model for C-(N-)A-S-H gel: CNASH_ss. Derivation and validation. Cem. Concr. Res. 2014, 66, 27–47. [Google Scholar] [CrossRef]
- Appelo, C.A.J. Principles, caveats and improvements in databases for calculating hydrogeochemical reactions in saline waters from 0 to 200 °C. and 1 to 1000 atm. Appl. Geochem. 2015, 55, 62–71. [Google Scholar] [CrossRef]
- Accornero, M.; Marini, L. Empirical prediction of the Pitzer’s interaction parameters for cationic Al species with both SiO2 (aq) and CO2 (aq): Implications for the geochemical modelling of very saline solutions. Appl. Geochem. 2009, 24, 747–759. [Google Scholar] [CrossRef]
- Boily, J.F.; Qafoku, O.; Felmy, A.R. A potentiometric, spectrophotometric and pitzer ion-interaction study of reaction equilibria in the aqueous H+-Al3+, H+-oxalate and H+-Al3+-oxalate systems up to 5 mol/dm-3 NaCl. J. Solut. Chem. 2007, 36, 1727–1743. [Google Scholar] [CrossRef]
- Li, G.; Le Bescop, P.; Moranville-Regourd, M. Synthesis of the U phase (4CaO·0.9Al2O3·1.1SO3·0.5Na2O·16H2O). Cem. Concr. Res. 1997, 27, 7–13. [Google Scholar] [CrossRef]
- Li, G.; Le Bescop, P.; Moranville, M. The U phase formation in cement-based systems containing high amounts of Na2SO4. Cem. Concr. Res. 1996, 26, 27–33. [Google Scholar] [CrossRef]
- Champenois, J.B.; Dhoury, M.; Coumes, C.C.D.; Mercier, C.; Revel, B.; Le Bescop, P.; Damidot, D. Influence of sodium borate on the early age hydration of calcium sulfoaluminate cement. Cem. Concr. Res. 2015, 70, 83–93. [Google Scholar] [CrossRef]
- Morinaga, Y.; Kajio, T.; Elakneswaran, Y.; Nawa, T.; Owaki, E. U-phase[(CaO)x(Al2O3) (SO3)y(Na2O)z:nH2O] in hydrating cement with sodium sulfate. In Proceedings of the The International Conference on Advances in Construction Materials and Systems, Chennai, India, 3–8 September 2017. [Google Scholar]
- Lothenbach, B.; Bary, B.; Le Bescop, P.; Schmidt, T.; Leterrier, N. Sulfate ingress in Portland cement. Cem. Concr. Res. 2010, 40, 1211–1225. [Google Scholar] [CrossRef]
- Plummer, L.N.; Parkhurst, D.L.; Fleming, G.W. A Computer Program Incorporating Pitzer’s Equations for Calculation of Geochemical Reactions in Brines. US Geolog. Surv. Water-Resour. Investig. Rep. 1988, 88, 310. [Google Scholar]
- Parrot, L.J.; Killoh, D.C. Prediction of cement hydration. Br. Ceram. Proc. 1984, 35, 41–53. [Google Scholar]
- Lothenbach, B.; Winnefeld, F. Thermodynamic modelling of the hydration of Portland cement. Cem. Concr. Res. 2006, 36, 209–226. [Google Scholar] [CrossRef]










| Oxide Composition (wt %) | Mineralogical Composition (wt %) | ||
|---|---|---|---|
| SiO2 | 21.60 | C3S | 50.61 |
| Al2O3 | 5.40 | C2S | 23.83 |
| Fe2O3 | 2.60 | C3A | 9.92 |
| CaO | 64.00 | C4AF | 7.90 |
| SO3 | 2.00 | Gypsum | 2.09 |
| MgO | 1.80 | Calcite | 1.00 |
| Na2O | 0.38 | CaO_free | 1.00 |
| K2O | 0.55 | ||
| Physical properties | Mixing conditions | ||
| Density (g/cm3) | 3.18 | W/B | 0.55 |
| Blaine (m2/kg) | 334 | Temp. (°C) | 25 |
| RH | Fully saturated | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elakneswaran, Y.; Owaki, E.; Nawa, T. Modelling Long-Term Durability Performance of Cementitious Materials under Sodium Sulphate Interaction. Appl. Sci. 2018, 8, 2597. https://doi.org/10.3390/app8122597
Elakneswaran Y, Owaki E, Nawa T. Modelling Long-Term Durability Performance of Cementitious Materials under Sodium Sulphate Interaction. Applied Sciences. 2018; 8(12):2597. https://doi.org/10.3390/app8122597
Chicago/Turabian StyleElakneswaran, Yogarajah, Eiji Owaki, and Toyoharu Nawa. 2018. "Modelling Long-Term Durability Performance of Cementitious Materials under Sodium Sulphate Interaction" Applied Sciences 8, no. 12: 2597. https://doi.org/10.3390/app8122597
APA StyleElakneswaran, Y., Owaki, E., & Nawa, T. (2018). Modelling Long-Term Durability Performance of Cementitious Materials under Sodium Sulphate Interaction. Applied Sciences, 8(12), 2597. https://doi.org/10.3390/app8122597