Influence of Calcium Sulfate Type on Evolution of Reaction Products and Strength in NaOH- and CaO-Activated Ground Granulated Blast-Furnace Slag




Abstract
1. Introduction
2. Materials and Experimental Program
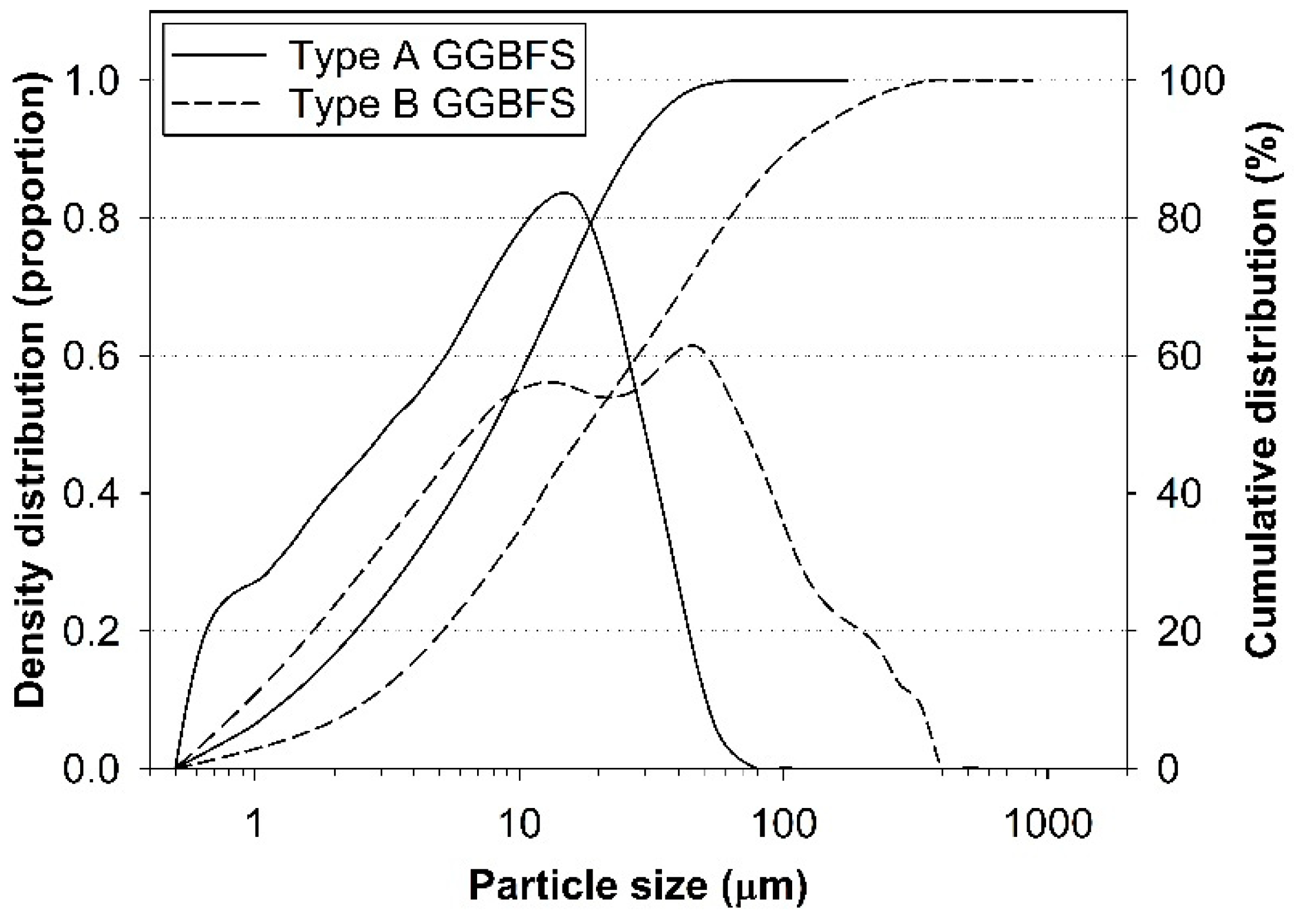
2.1. Materials
2.2. Sample Preparation
2.3. Tests
3. Results and Discussion
3.1. Strength Development
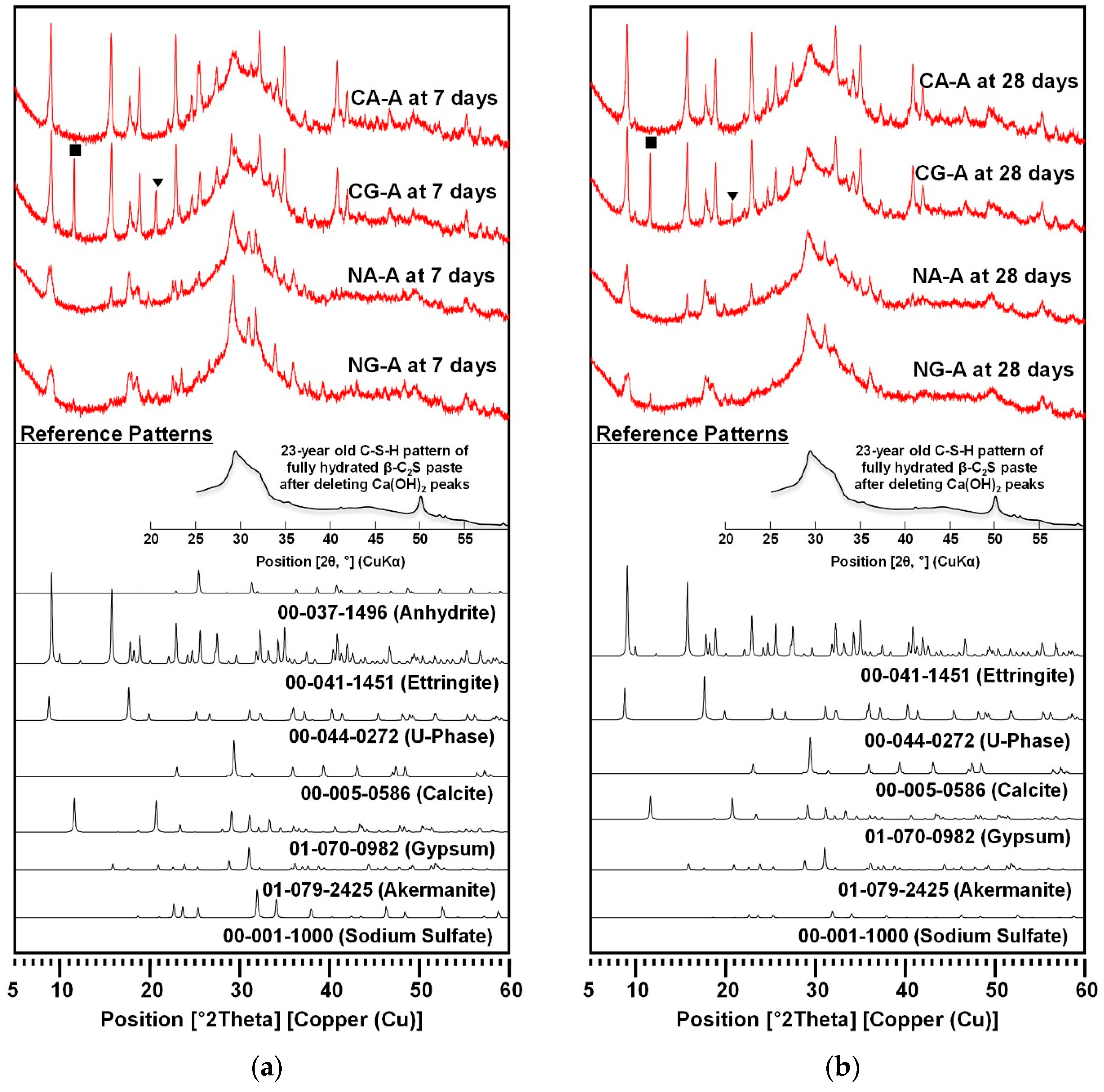
3.2. XRD
3.3. TGA
3.4. MIP
4. Conclusions
- (1)
- The CaO-activated GGBFS achieved a great strength enhancement from the addition of CaSO4 sources, and the type of calcium sulfates and source of GGBFS did not significantly affect the strength development in this study, while the NaOH-activated GGFBS showed its complicated dependency on the choice of CaSO4 type and GGBFS source for strength development; thus, the use of CaO was found to be better than that of NaOH for quality control (meaning less change in strength by input parameters) when calcium sulfates were present in raw GGBFS.
- (2)
- In the ANOVA study, the influential degree of each variable for strength was found to be greater in the following order: activator type > CaSO4 type > GGBFS source; in particular, the type of calcium sulfate was highly influential in NaOH-activation, while it was relatively insignificant in CaO-activation.
- (3)
- When calcium sulfate sources were added, the NaOH-activated samples showed significantly complex phase composition of reaction products, while the CaO-activated ones exhibited a much simpler composition.
- (4)
- In the NaOH-activation, due to the substantial formation of U-phase, the ettringite formation seemed to be suppressed. As the formation of ettringite generally contributes to strength evolution to some extent, more formation of ettringite in CaO-activation may be the cause for the higher strength of CaO-activated samples than those of NaOH-activation.
- (5)
- The TGA results indicated that the use of calcium sulfate sources in the CaO-activation clearly increased the quantity of reaction products; however, in the NaOH-activation, this barely increased, or even reduced, the quantity of reaction products.
- (6)
- In the MIP results, in the CaO-activation, the use of calcium sulfates significantly reduced the pore size and the total porosity from 7 to 28 days, while, in the NaOH-activation, it showed almost no reducing effect on total porosity and pore size; even the use of gypsum increased the porosity.
Author Contributions
Funding
Conflicts of Interest
Appendix A
- -
- Compressive strength: the measured values of all 144 samples are listed in Table A1.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Activator | Source of CaSO4 | Curing (days) | Type A GGBFS | Type B GGBFS | ||||
|---|---|---|---|---|---|---|---|---|
| Sample 1 (MPa) | Sample 2 (MPa) | Sample 3 (MPa) | Sample 1 (MPa) | Sample 2 (MPa) | Sample 3 (MPa) | |||
| None | 1 | 1.77 | 1.79 | 1.80 | 1.16 | 1.14 | 1.05 | |
| 3 | 6.33 | 6.26 | 6.52 | 7.76 | 7.99 | 8.19 | ||
| 7 | 19.37 | 17.33 | 19.42 | 17.19 | 17.51 | 18.31 | ||
| 28 | 31.66 | 29.57 | 31.30 | 30.15 | 27.85 | 29.09 | ||
| CaO | Gypsum | 1 | 4.82 | 4.71 | 4.82 | 5.45 | 5.48 | 5.07 |
| 3 | 10.24 | 9.78 | 10.17 | 12.34 | 11.71 | 12.05 | ||
| 7 | 17.03 | 17.45 | 17.25 | 19.16 | 19.40 | 19.86 | ||
| 28 | 41.72 | 44.60 | 41.39 | 45.24 | 44.22 | 46.62 | ||
| Anhydrite | 1 | 4.96 | 4.84 | 4.50 | 3.47 | 3.40 | 3.51 | |
| 3 | 11.88 | 11.42 | 12.34 | 12.69 | 13.89 | 13.30 | ||
| 7 | 19.25 | 18.11 | 18.56 | 17.18 | 16.80 | 17.55 | ||
| 28 | 44.07 | 44.38 | 48.34 | 45.91 | 51.61 | 51.10 | ||
| None | 1 | 9.63 | 9.77 | 9.77 | 11.08 | 11.15 | 11.05 | |
| 3 | 13.31 | 14.02 | 13.85 | 17.04 | 16.44 | 17.30 | ||
| 7 | 20.18 | 19.03 | 18.84 | 22.87 | 22.73 | 22.87 | ||
| 28 | 29.84 | 30.01 | 29.56 | 33.52 | 32.93 | 34.02 | ||
| NaOH | Gypsum | 1 | 6.09 | 6.27 | 5.60 | 4.57 | 4.35 | 4.45 |
| 3 | 12.33 | 12.39 | 12.80 | 10.63 | 11.14 | 10.94 | ||
| 7 | 17.26 | 17.57 | 16.88 | 14.26 | 13.91 | 13.48 | ||
| 28 | 26.29 | 25.76 | 26.35 | 20.95 | 19.94 | 21.80 | ||
| Anhydrite | 1 | 3.56 | 3.50 | 3.60 | 2.48 | 2.45 | 2.39 | |
| 3 | 19.24 | 19.20 | 19.20 | 12.68 | 11.71 | 13.46 | ||
| 7 | 27.24 | 27.08 | 26.60 | 19.16 | 19.40 | 19.86 | ||
| 28 | 35.16 | 29.73 | 36.07 | 26.70 | 24.71 | 22.25 | ||
- -
- ANOVA test: all calculated values of the factorial designs from ANOVA are listed in Table A2.
| Data | Explanatory Factor | Sum F Squares | Mean Square | F Statistic | p-Value | |
|---|---|---|---|---|---|---|
| Entire data calculation | Time | 2.01 | 1.00 | 229.02 | 0.00 | |
| Activator type | 0.74 | 0.74 | 169.51 | 0.00 | ||
| GGBFS type | 0.0023 | 0.0023 | 0.53 | 0.47 | ||
| CaSO4 type | 0.0042 | 0.0042 | 0.95 | 0.33 | ||
| Residual | 0.29 | 0.0044 | ||||
| Separate data | Group of CaO-activation | Time | 1.93 | 0.96 | 287.02 | 0.00 |
| GGBFS type | 0.0015 | 0.0015 | 0.44 | 0.52 | ||
| CaSO4 type | 0.0016 | 0.0016 | 0.48 | 0.49 | ||
| Residual | 0.10 | 0.0034 | ||||
| Group of NaOH-activation | Time | 0.53 | 0.26 | 523.87 | 0.00 | |
| GGBFS type | 0.00023 | 0.00023 | 0.47 | 0.50 | ||
| CaSO4 type | 0.0029 | 0.0029 | 5.68 | 0.02 | ||
| Residual | 0.016 | 0.00050 | ||||
References
- Flower, D.J.M.; Sanjayan, J.G. Greenhouse gas emissions due to concrete manufacture. Int. J. Life Cycle Assess. 2007, 12, 282–288. [Google Scholar] [CrossRef]
- Ali, M.; Saidur, R.; Hossain, M. A review on emission analysis in cement industries. Renew. Sustain. Energy Rev. 2011, 15, 2252–2261. [Google Scholar] [CrossRef]
- Pacheco-Torgal, F.; Castro-Gomes, J.; Jalali, S. Alkali-activated binders: A review: Part 1. Historical background, terminology, reaction mechanisms and hydration products. Constr. Build. Mater. 2008, 22, 1305–1314. [Google Scholar] [CrossRef]
- Pacheco-Torgal, F.; Castro-Gomes, J.; Jalali, S. Alkali-activated binders: A review. Part 2. About materials and binders manufacture. Constr. Build. Mater. 2008, 22, 1315–1322. [Google Scholar] [CrossRef]
- Oh, J.E.; Monteiro, P.J.; Jun, S.S.; Choi, S.; Clark, S.M. The evolution of strength and crystalline phases for alkali-activated ground blast furnace slag and fly ash-based geopolymers. Cem. Concr. Res. 2010, 40, 189–196. [Google Scholar] [CrossRef]
- Richardson, I.; Brough, A.; Groves, G.; Dobson, C. The characterization of hardened alkali-activated blast-furnace slag pastes and the nature of the calcium silicate hydrate (CSH) phase. Cem. Concr. Res. 1994, 24, 813–829. [Google Scholar] [CrossRef]
- Van Deventer, J.S.; Provis, J.L.; Duxson, P. Technical and commercial progress in the adoption of geopolymer cement. Miner. Eng. 2012, 29, 89–104. [Google Scholar] [CrossRef]
- Yang, K.-H.; Cho, A.-R.; Song, J.-K.; Nam, S.-H. Hydration products and strength development of calcium hydroxide-based alkali-activated slag mortars. Constr. Build. Mater. 2012, 29, 410–419. [Google Scholar] [CrossRef]
- Yang, K.-H.; Sim, J.-I.; Nam, S.-H. Enhancement of reactivity of calcium hydroxide-activated slag mortars by the addition of barium hydroxide. Constr. Build. Mater. 2010, 24, 241–251. [Google Scholar] [CrossRef]
- Kim, M.S.; Jun, Y.; Lee, C.; Oh, J.E. Use of CaO as an activator for producing a price-competitive non-cement structural binder using ground granulated blast furnace slag. Cem. Concr. Res. 2013, 54, 208–214. [Google Scholar] [CrossRef]
- Jeon, D.; Jun, Y.; Jeong, Y.; Oh, J.E. Microstructural and strength improvements through the use of Na2CO3 in a cementless Ca(OH)2-activated Class F fly ash system. Cem. Concr. Res. 2015, 67, 215–225. [Google Scholar] [CrossRef]
- Jeong, Y.; Oh, J.E.; Jun, Y.; Park, J.; Ha, J.-h.; Sohn, S.G. Influence of four additional activators on hydrated-lime [Ca(OH)2] activated ground granulated blast-furnace slag. Cem. Concr. Compos. 2016, 65, 1–10. [Google Scholar] [CrossRef]
- Poon, C.; Kou, S.; Lam, L.; Lin, Z. Activation of fly ash/cement systems using calcium sulfate anhydrite (CaSO4). Cem. Concr. Res. 2001, 31, 873–881. [Google Scholar] [CrossRef]
- Ma, W.; Liu, C.; Brown, P.W.; Komarneni, S. Pore structures of fly ashes activated by Ca(OH)2 and CaSO4· 2H2O. Cem. Concr. Res. 1995, 25, 417–425. [Google Scholar] [CrossRef]
- Park, H.; Jeong, Y.; Jeong, J.-H.; Oh, J.E. Strength Development and Hydration Behavior of Self-Activation of Commercial Ground Granulated Blast-Furnace Slag Mixed with Purified Water. Materials 2016, 9, 185. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Jeong, Y.; Jun, Y.; Jeong, J.-H.; Oh, J.E. Strength enhancement and pore-size refinement in clinker-free CaO-activated GGBFS systems through substitution with gypsum. Cem. Concr. Compos. 2016, 68, 57–65. [Google Scholar] [CrossRef]
- Chang, J.J.; Yeih, W.; Hung, C.C. Effects of gypsum and phosphoric acid on the properties of sodium silicate-based alkali-activated slag pastes. Cem. Concr. Compos. 2005, 27, 85–91. [Google Scholar] [CrossRef]
- Charola, A.E.; Pühringer, J.; Steiger, M. Gypsum: A review of its role in the deterioration of building materials. Environ. Geol. 2007, 52, 339–352. [Google Scholar] [CrossRef]
- Standards, B. BS EN 15167-1, Ground Granulated Blast Furnace Slag for Use in Concrete, Mortar and Grout—Part 1: Definitions, Specifications and Conformity Criteria; BSI Milton Keynes: Milton Keynes, UK, 2006. [Google Scholar]
- Starndards, K. KS F 2563, Ground Granulated Blast-Furnace Slag for Use in Concrete; British Standards Institution: London, UK, 2009. [Google Scholar]
- Zhang, J.; Scherer, G.W. Comparison of methods for arresting hydration of cement. Cem. Concr. Res. 2011, 41, 1024–1036. [Google Scholar] [CrossRef]
- PANalytical. X’Pert HighScore Plus Software; PANalytical: Almelo, The Netherlands, 2012. [Google Scholar]
- ICDD. International Centre for Diffraction Data; International Centre for Diffraction Data: Newtown Square, PA, USA, 2000. [Google Scholar]
- Taylor, H.F.W. Cement Chemistry; Thomas Telford: London, UK, 1997. [Google Scholar]
- Li, G.; Le Bescop, P.; Moranville, M. The U phase formation in cement-based systems containing high amounts of Na2SO4. Cem. Concr. Res. 1996, 26, 27–33. [Google Scholar] [CrossRef]
- Sánchez-Herrero, M.J.; Fernández-Jiménez, A.; Palomo, A.; Viehland, D. Alkaline Hydration of Tricalcium Aluminate. J. Am. Ceram. Soc. 2012, 95, 3317–3324. [Google Scholar] [CrossRef]
- Jeong, Y.; Park, H.; Jun, Y.; Jeong, J.-H.; Oh, J.E. Microstructural verification of the strength performance of ternary blended cement systems with high volumes of fly ash and GGBFS. Constr. Build. Mater. 2015, 95, 96–107. [Google Scholar] [CrossRef]
- Chaunsali, P.; Peethamparan, S. Evolution of strength, microstructure and mineralogical composition of a CKD–GGBFS binder. Cem. Concr. Res. 2011, 41, 197–208. [Google Scholar] [CrossRef]
- Dweck, J.; Melchert, M.; Viana, M.; Cartledge, F.; Büchler, P. Importance of quantitative thermogravimetry on initial cement mass basis to evaluate the hydration of cement pastes and mortars. J. Therm. Anal. Calorim. 2013, 113, 1481–1490. [Google Scholar] [CrossRef]
- Vassileva, C.G.; Vassilev, S.V. Behaviour of inorganic matter during heating of Bulgarian coals: 1. Lignites. Fuel Process. Technol. 2005, 86, 1297–1333. [Google Scholar] [CrossRef]
- Gruskovnjak, A.; Lothenbach, B.; Holzer, L.; Figi, R.; Winnefeld, F. Hydration of alkali-activated slag: comparison with ordinary Portland cement. Adv. Cem. Res. 2006, 18, 119–128. [Google Scholar] [CrossRef]
- Kanezaki, E. Thermal behavior of the hydrotalcite-like layered structure of Mg and Al-layered double hydroxides with interlayer carbonate by means of in situ powder HTXRD and DTA/TG. Solid State Ionics 1998, 106, 279–284. [Google Scholar] [CrossRef]
- Haha, M.B.; Lothenbach, B.; Le Saout, G.; Winnefeld, F. Influence of slag chemistry on the hydration of alkali-activated blast-furnace slag—Part I: Effect of MgO. Cem. Concr. Res. 2011, 41, 955–963. [Google Scholar] [CrossRef]
- Diamond, S. Mercury porosimetry: an inappropriate method for the measurement of pore size distributions in cement-based materials. Cem. Concr. Res. 2000, 30, 1517–1525. [Google Scholar] [CrossRef]






| GGBFS | Oxide (wt.%) | Atomic Ratio | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CaO | SiO2 | Al2O3 | MgO | SO3 | TiO2 | K2O | Fe2O3 | Ca/Si | Al/Si | |
| Type A | 44.6% | 34.4% | 13.8% | 3.7% | 1.4% | 0.6% | 0.5% | 0.4% | 1.3 | 0.4 |
| Type B | 44.2% | 34.3% | 14.3% | 3.3% | 1.4% | 0.7% | 0.6% | 0.5% | 1.3 | 0.4 |
| Data Group | Sample Label | Activator Type | GGBFS Source | CaSO4 Type | Water | Tests | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CaO | NaOH | Type A | Type B | Anhydrite | Gypsum | S | X | T | M | |||
| CaO-activation | C-A | 5 | - | 95 | - | - | - | 40 | ● | ● | ● | |
| C-B | 5 | - | - | 95 | - | - | 40 | ● | ||||
| CA-A | 5 | - | 90 | - | 5 | - | 40 | ● | ● | ● | ● | |
| CA-B | 5 | - | - | 90 | 5 | - | 40 | ● | ||||
| CG-A | 5 | - | 90 | - | - | 5.7 | 39.3 | ● | ● | ● | ● | |
| CG-B | 5 | - | - | 90 | - | 5.7 | 39.3 | ● | ||||
| NaOH-activation | N-A | - | 5 | 95 | - | - | - | 40 | ● | ● | ● | |
| N-B | - | 5 | - | 95 | - | - | 40 | ● | ||||
| NA-A | - | 5 | 90 | - | 5 | - | 40 | ● | ● | ● | ● | |
| NA-B | - | 5 | - | 90 | 5 | - | 40 | ● | ||||
| NG-A | - | 5 | 90 | - | - | 5.7 | 39.3 | ● | ● | ● | ● | |
| NG-B | - | 5 | - | 90 | - | 5.7 | 39.3 | ● | ||||
| Explanatory Factor | Entire Data Calculation | Separate Data Calculation | |
|---|---|---|---|
| Group of CaO-Activation | Group of NaOH-Activation | ||
| Activator type (CaO vs. NaOH) | 0.00 | - | - |
| GGBFS source (type A vs. B) | 0.47 | 0.52 | 0.50 |
| CaSO4 type (anhydrite vs. gypsum) | 0.33 | 0.49 | 0.02 |
| Phase | CaO-Activation | NaOH-Activation | ||||||
|---|---|---|---|---|---|---|---|---|
| CA-A | CG-A | NA-A | NG-A | |||||
| 7-Day | 28-Day | 7-Day | 28-Day | 7-Day | 28-Day | 7-Day | 28-Day | |
| C-S-H | C-S-H formed in all the samples, but the relative comparison of the peak intensities was not possible among samples because C-S-H is semi-amorphous. | |||||||
| Ettringite | ●●●● | ●●●● | ●●●● | ●●●● | ●● | ●● | ● | ● |
| Gypsum | - | - | ●●● | ●● | - | - | ● | ● |
| Anhydrite | ● | - | - | - | - | - | - | - |
| U-phase | - | - | - | - | ●● | ●● | ●● | ●● |
| Calcite | ● or - | ● or - | ● or - | ● or - | ●● | ●● | ●● | ● |
| Sodium sulfate | - | - | - | - | ●● | ● or - | ●● | ● or - |
| Akermanite | - | - | - | - | ● or - | ● or - | ● or - | ● or - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoon, S.; Park, H.; Yum, W.S.; Suh, J.-I.; Oh, J.E. Influence of Calcium Sulfate Type on Evolution of Reaction Products and Strength in NaOH- and CaO-Activated Ground Granulated Blast-Furnace Slag. Appl. Sci. 2018, 8, 2500. https://doi.org/10.3390/app8122500
Yoon S, Park H, Yum WS, Suh J-I, Oh JE. Influence of Calcium Sulfate Type on Evolution of Reaction Products and Strength in NaOH- and CaO-Activated Ground Granulated Blast-Furnace Slag. Applied Sciences. 2018; 8(12):2500. https://doi.org/10.3390/app8122500
Chicago/Turabian StyleYoon, Seyoon, Hyeoneun Park, Woo Sung Yum, Jung-Il Suh, and Jae Eun Oh. 2018. "Influence of Calcium Sulfate Type on Evolution of Reaction Products and Strength in NaOH- and CaO-Activated Ground Granulated Blast-Furnace Slag" Applied Sciences 8, no. 12: 2500. https://doi.org/10.3390/app8122500
APA StyleYoon, S., Park, H., Yum, W. S., Suh, J.-I., & Oh, J. E. (2018). Influence of Calcium Sulfate Type on Evolution of Reaction Products and Strength in NaOH- and CaO-Activated Ground Granulated Blast-Furnace Slag. Applied Sciences, 8(12), 2500. https://doi.org/10.3390/app8122500