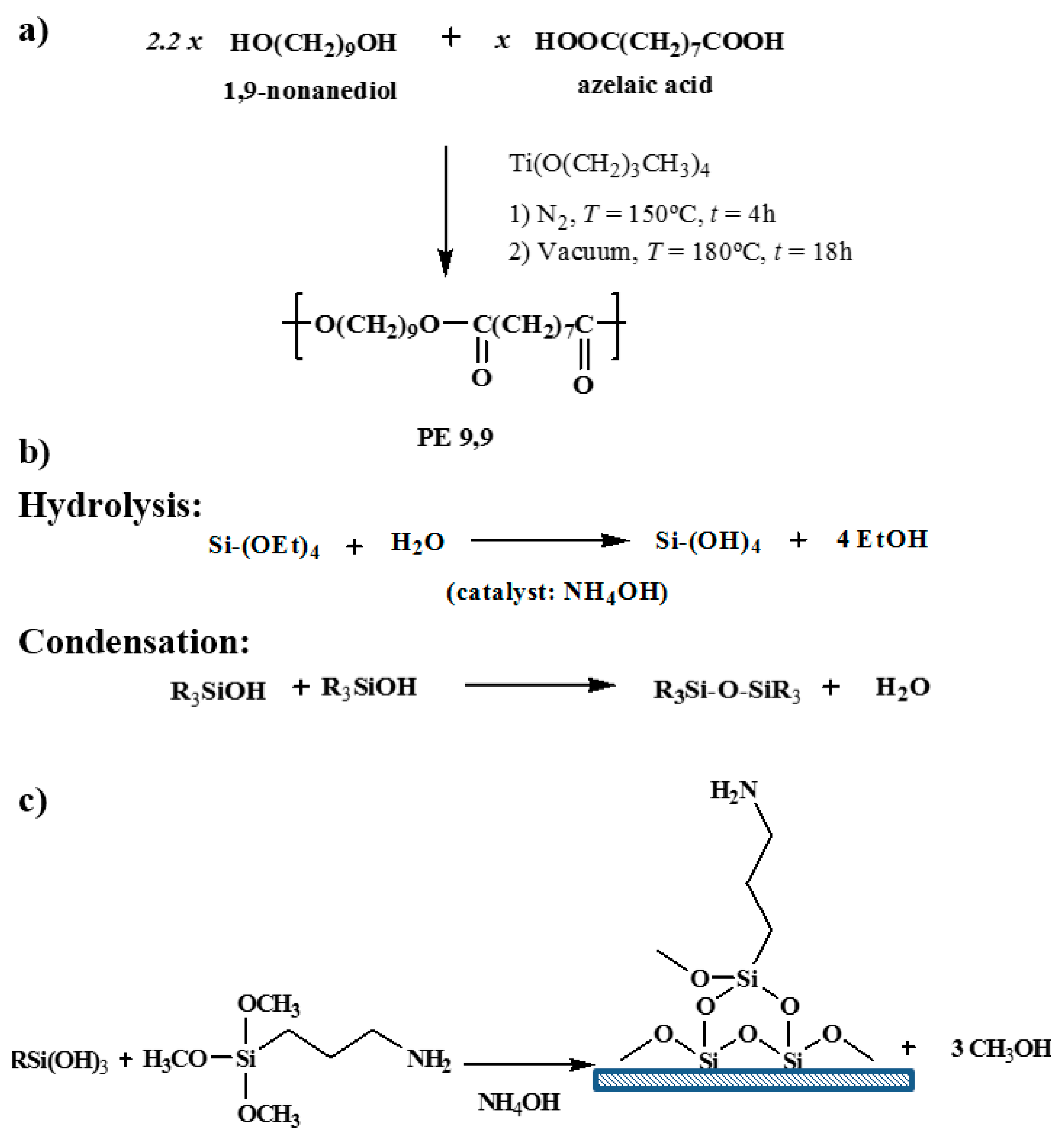
3.1. Characterization of Functionalized Silica Particles
Silica particles were effectively prepared using tetraethoxysilane as a precursor. TEM micrographs clearly revealed a highly regular spherical form (
Figure 3a) for silica particles obtained at a low stirring speed. These particles showed a highly homogeneous diameter that varied in a narrow range (
i.e., between 570 and 650 nm). Particles were slightly more irregular in form (
Figure 3b) when prepared at a higher stirring speed. Diameter size was considerably reduced but a homogeneous distribution could still be observed (
i.e., values were always within the 20–30 nm interval). The two kinds of silica preparations will be designated as micro (M) and nano (N) particles.
Figure 3.
TEM micrographs of functionalized microspheres (a); and nanospheres (b).
Figure 3.
TEM micrographs of functionalized microspheres (a); and nanospheres (b).
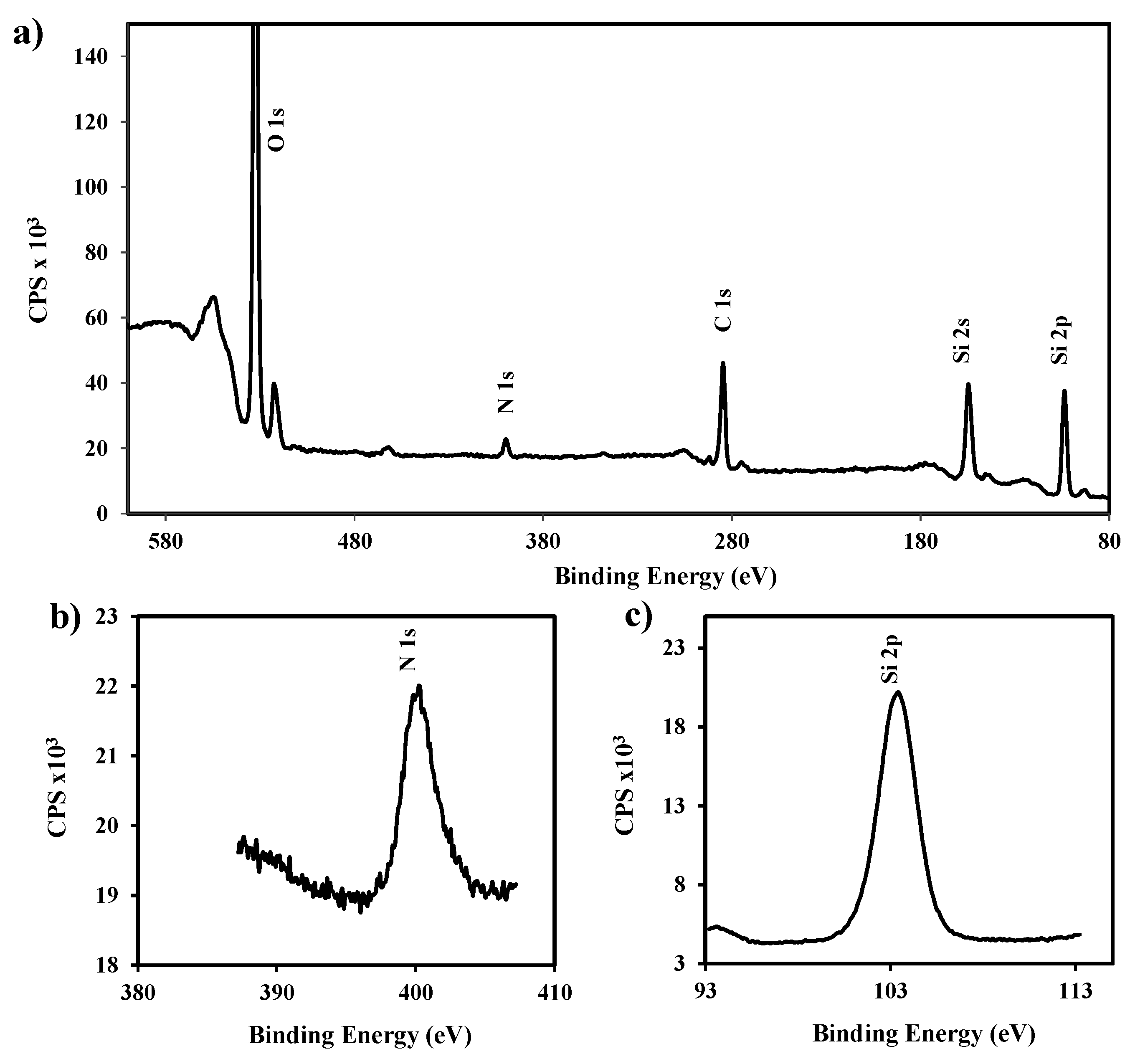
Figure 4.
Full scale XPS spectra (a) and details corresponding to silicon (b) and nitrogen (c) XPS signals detected in the functionalized silica nanoparticles.
Figure 4.
Full scale XPS spectra (a) and details corresponding to silicon (b) and nitrogen (c) XPS signals detected in the functionalized silica nanoparticles.
Surface functionalization by reaction with (3-aminopropyl) triethoxysilane was verified by analysis of XPS spectra (
Figure 4), which allowed for determining a ratio between N and Si atoms of close to 8% (
i.e., 8.8% and 7.3% for micro- and nanoparticles, respectively).
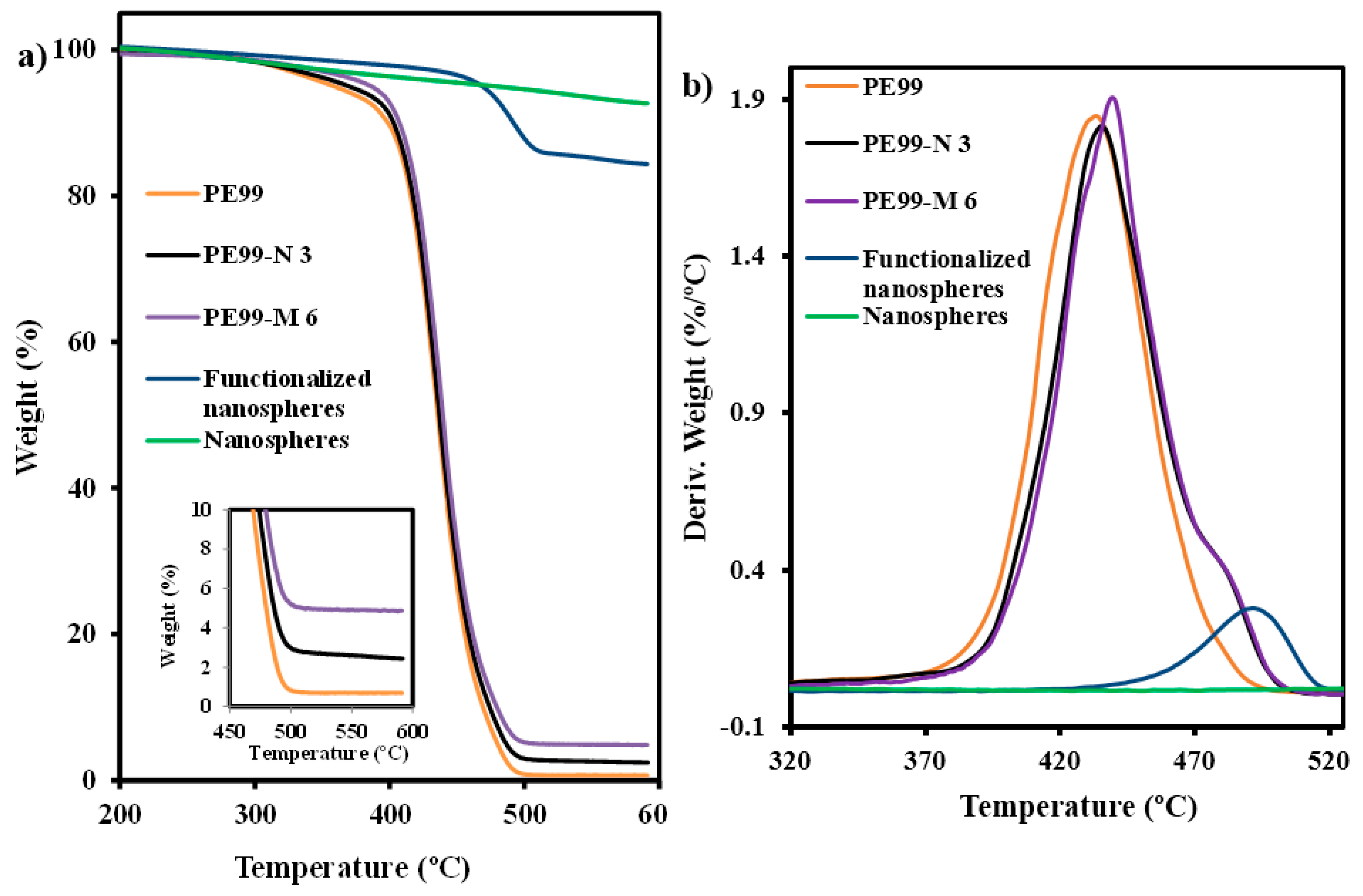
Thermogravimetric analyses (
Figure 5) also showed a significant weight loss of functionalized silica particles because of decomposition of grafted AMPS groups, which took place at around 500 °C. This decomposition step corresponded to an approximate weight loss of 9% and, logically, was not detected in non-functionalized particles. Gradual weight loss leading to a value of only 8%–9% at 590 °C was observed for all particles. Thus, at this temperature the total loss was 17% and 8% for functionalized and non-functionalized particles, respectively. Practically no differences were found in the TGA (
Figure 5a) and DTGA (
Figure 5b) curves of micro- and nanoparticles.
Figure 5.
TGA (a) and DTGA (b) curves of silica nanospheres, functionalized silica nanospheres, and micro-molded PE99, PE99-N 3, and PE99-M 6 specimens.
Figure 5.
TGA (a) and DTGA (b) curves of silica nanospheres, functionalized silica nanospheres, and micro-molded PE99, PE99-N 3, and PE99-M 6 specimens.
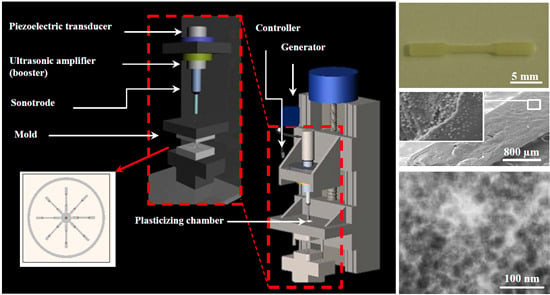
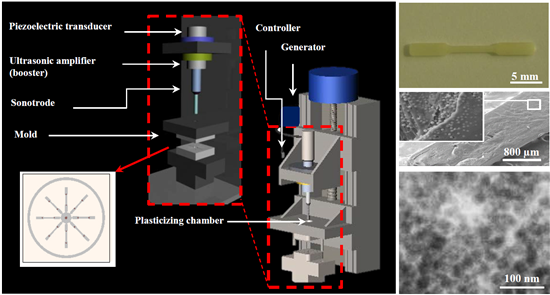
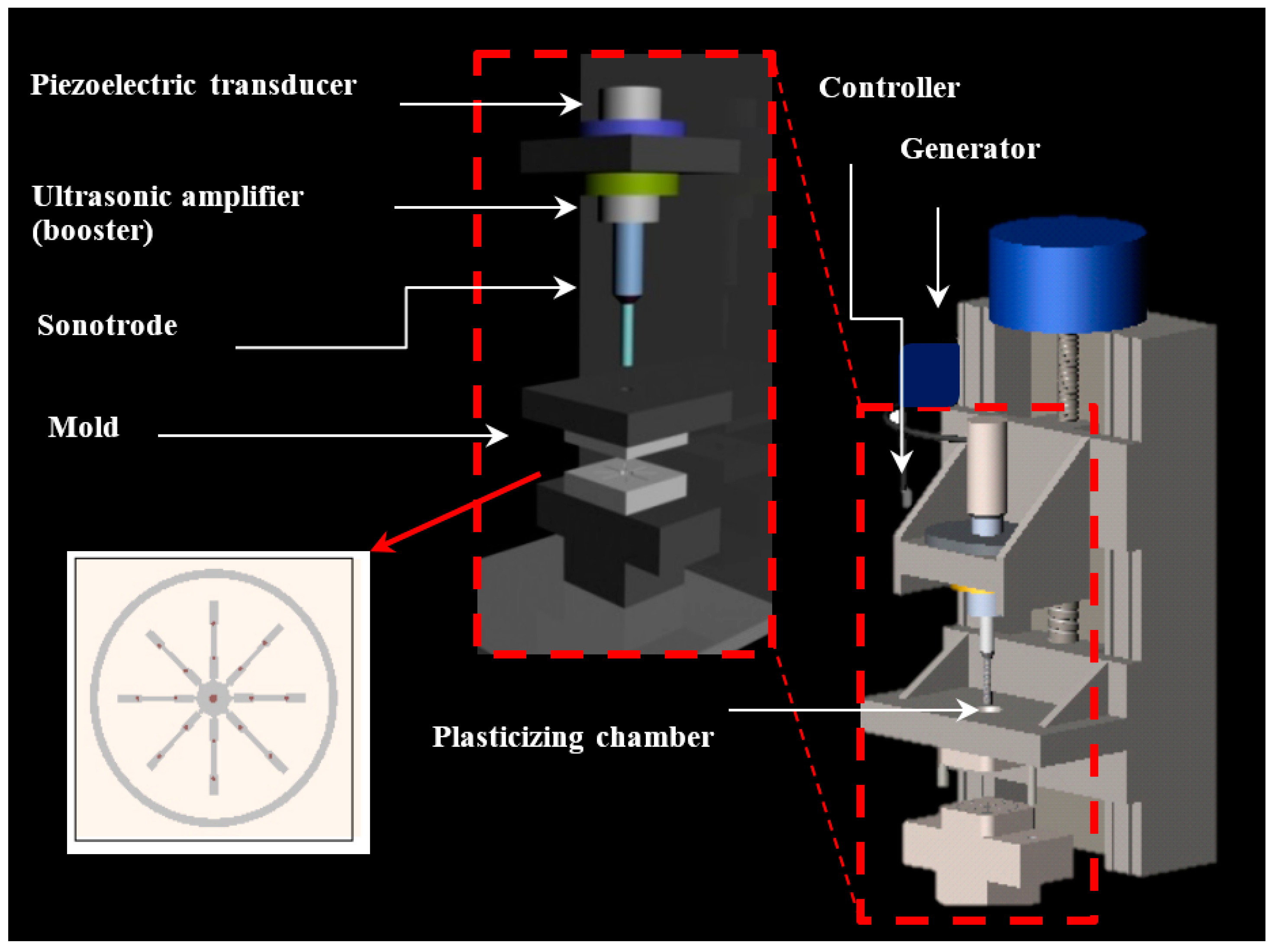
3.2. Dispersion of Functionalized Silica Micro- and Nanoparticles by Ultrasound Micro-Molding Technology
PE99 samples could be processed under relatively mild conditions, as previously established [
15]. Thus, a minimum irradiation time of 1.2 s, low amplitude of 24 μm, and a moderate molding force of 300 N were sufficient to guarantee a 100% molding efficiency. Experimental conditions could be maintained for processing mixtures with functionalized micro- and nanospheres up to the maximum test load of 6 wt. %.
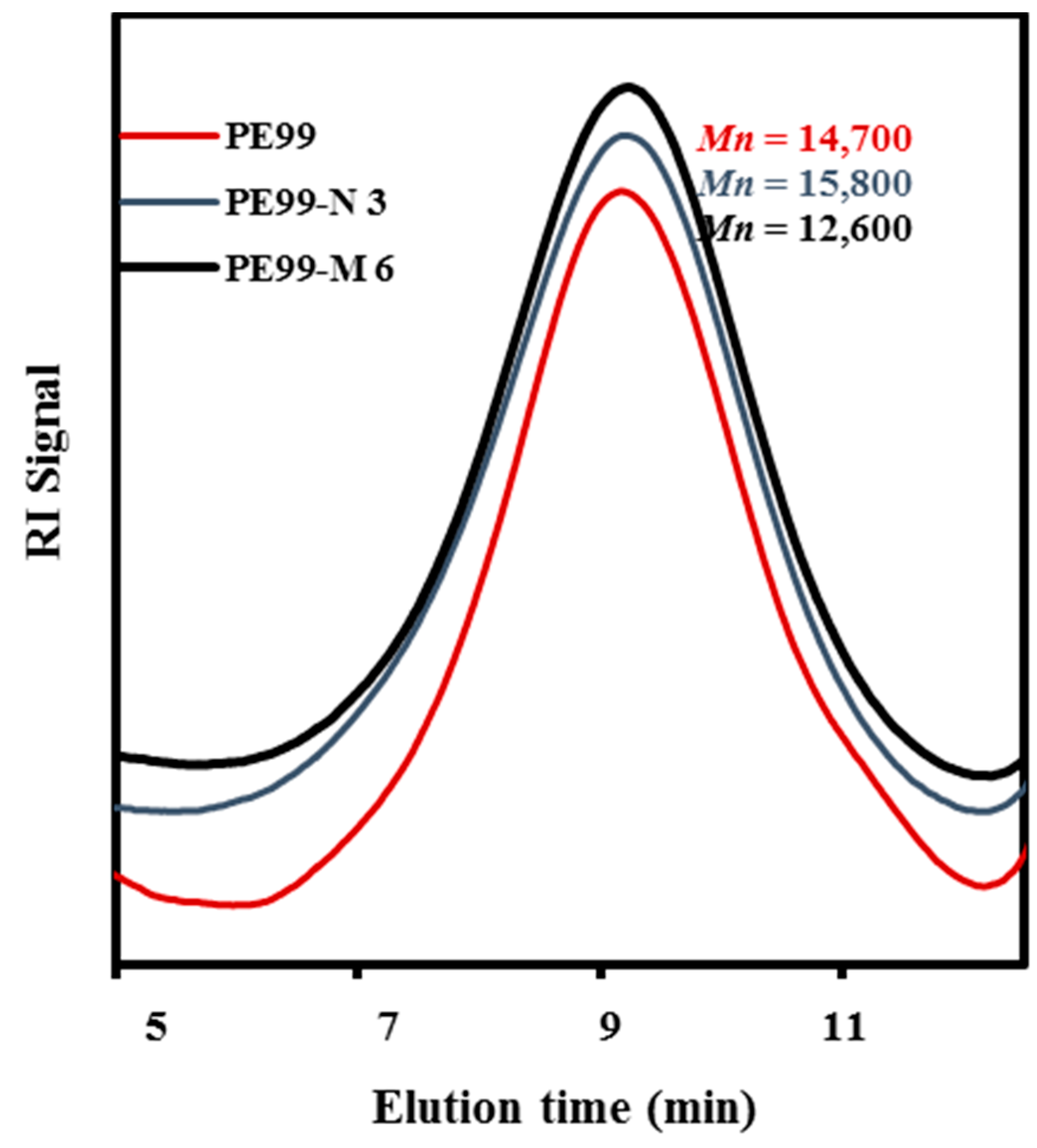
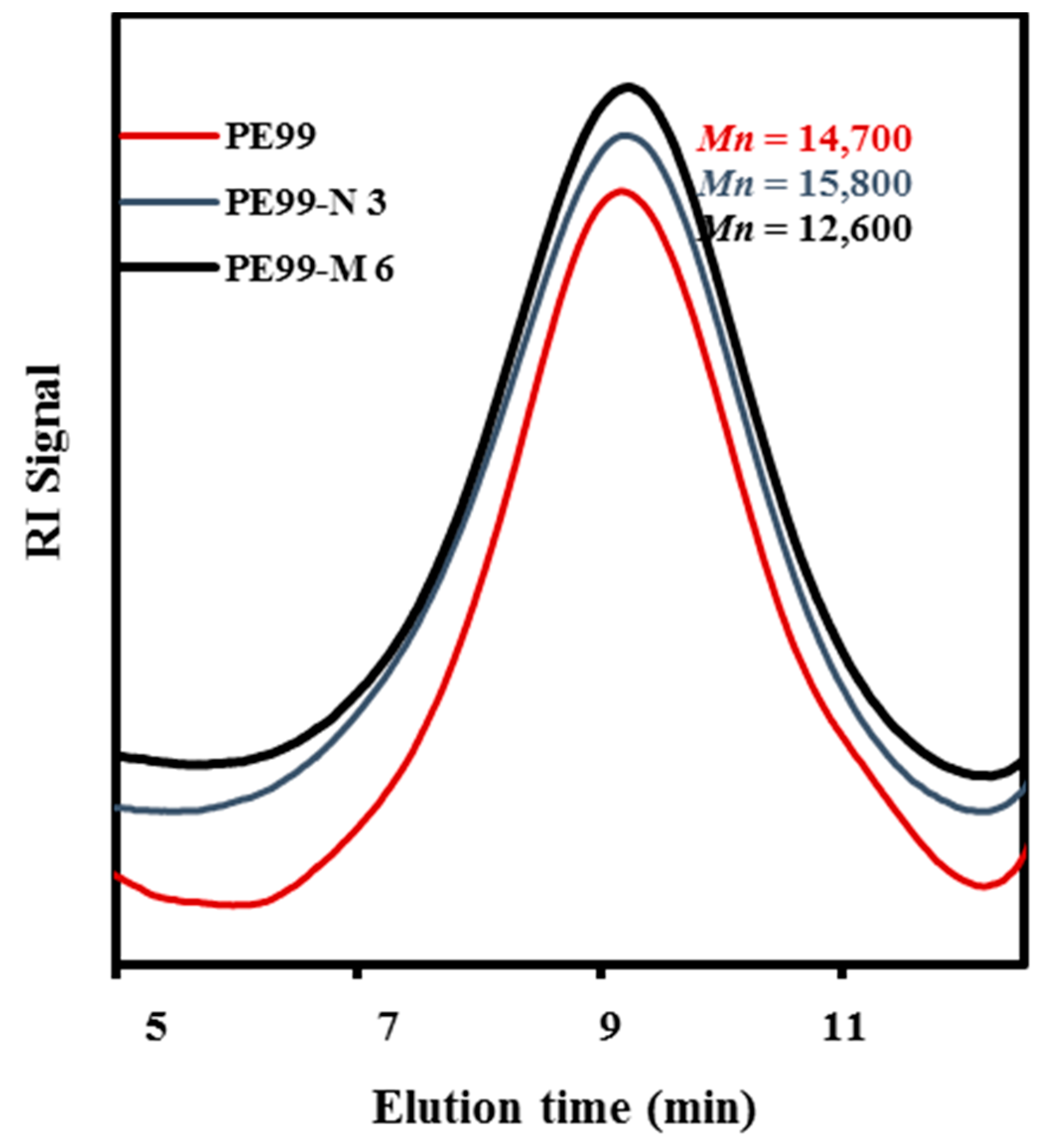
Molecular weights of raw PE99 and specimens processed under the above optimized conditions are summarized in
Table 1, whereas
Figure 6 compares the GPC traces of PE99, PE99-N 3, and PE99-M 6 specimens.
Table 1.
Molecular weights of processed PE 99 and their mixtures with functionalized micro- and nanoparticles a.
Table 1.
Molecular weights of processed PE 99 and their mixtures with functionalized micro- and nanoparticles a.
| Sample | Mn (g/mol) | Mw (g/mol) | Mw/Mn |
|---|
| PE99 (raw) | 13,300 | 35,900 | 2.7 |
| PE99 | 14,700 | 37,200 | 2.5 |
| PE99-M 3 | 14,800 | 36,800 | 2.5 |
| PE99-M 6 | 12,600 | 33,800 | 2.7 |
| PE99-N 3 | 15,800 | 39,200 | 2.5 |
| PE99-N 6 | 14,200 | 37,900 | 2.7 |
Figure 6.
GPC molecular weight distribution curves determined for micro-molded PE99, PE-N 3, and PE99-M 6 specimens.
Figure 6.
GPC molecular weight distribution curves determined for micro-molded PE99, PE-N 3, and PE99-M 6 specimens.
No statistically significant differences were found between samples before and after processing or upon addition of functionalized nanospheres. Only a not highly significant decrease was detected for samples loaded with the maximum percentage (6 wt. %) of microspheres. The new technology appears fully adequate to obtain micropieces with negligible degradation of composites constituted by PE99 and functionalized silica micro/nanoparticles.
Figure 7a,b show optical and SEM images of a representative silica loaded specimen for which a regular texture is detected in the longitudinal section micrographs. All specimens were highly homogeneous, without the presence of cavities that could affect the final properties of the material.
Figure 7.
Image of a processed PE99 specimen (a); SEM micrograph of details of a micro-molded PE99-M 3 specimen (b). Microparticles can be observed in the inset.
Figure 7.
Image of a processed PE99 specimen (a); SEM micrograph of details of a micro-molded PE99-M 3 specimen (b). Microparticles can be observed in the inset.
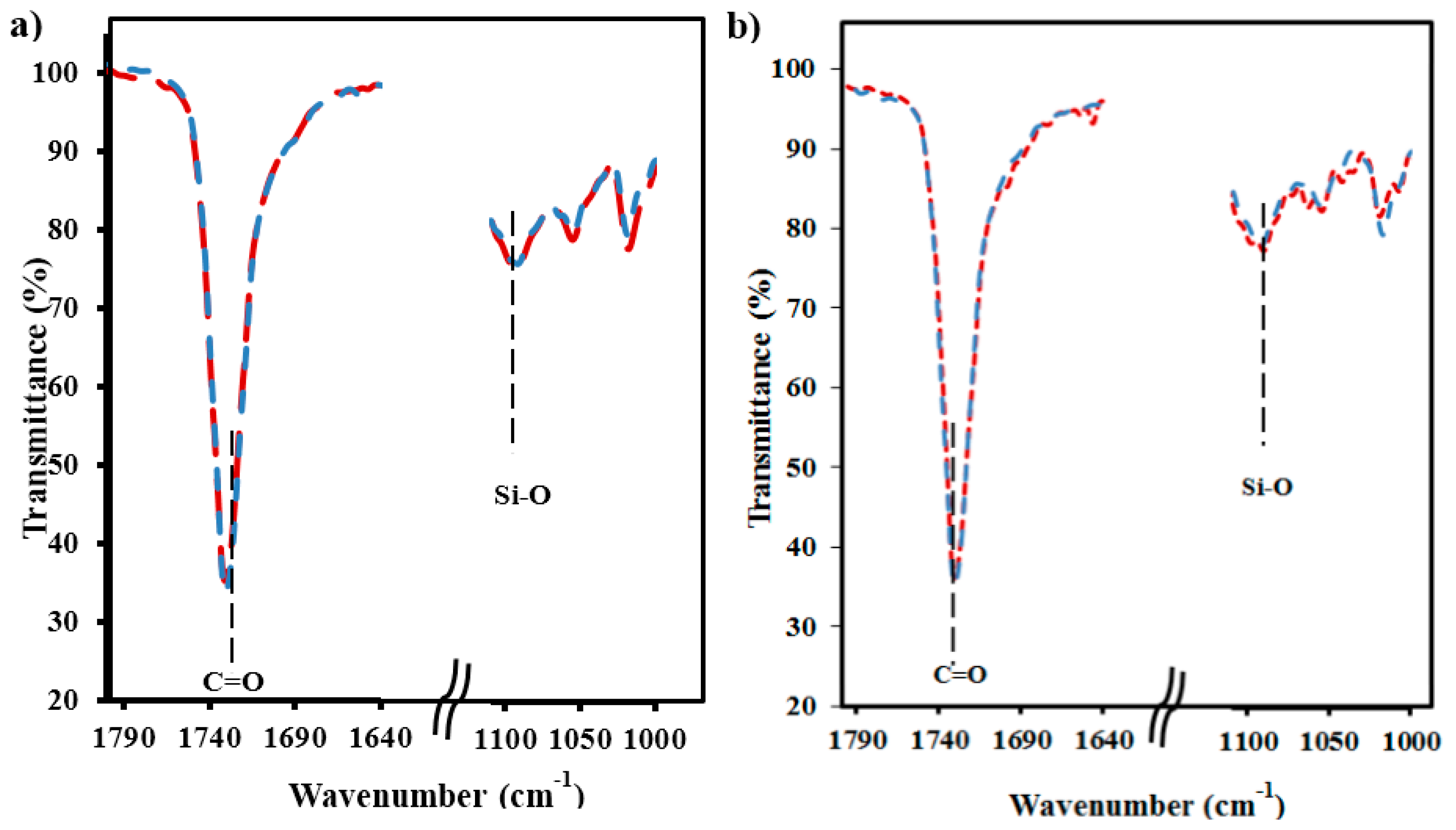
FTIR spectra were also useful to verify the uniform incorporation of silica particles into micro-molded specimens and to discard concentration in the sprue. Thus, the typical Si–O stretching band at 1075 cm
−1 was observed in every part of all loaded specimens. Hence, the spectra of specimen zones close to (proximal part) and distant from (distal part) the feeding channel showed a similar ratio between the intensity of the band associated with the C=O stretching band of the polyester and the band associated with the silica particles (see
Figure 8 for representative samples containing micro- and nanoparticles).
Figure 8.
FTIR spectra of the characteristic C=O and Si–O stretching bands of micro-molded PE-N 3 (a) and PE-M 3 (b) specimens. Blue and red lines correspond to the proximal and distal parts of the specimens, respectively.
Figure 8.
FTIR spectra of the characteristic C=O and Si–O stretching bands of micro-molded PE-N 3 (a) and PE-M 3 (b) specimens. Blue and red lines correspond to the proximal and distal parts of the specimens, respectively.
Analysis of particle dispersion in the processed composites was also carried out by transmission electron microscopy observation of ultrathin sections of micro-molded specimens. Flawless ultrasections could be obtained, as can be seen in the images for representative samples in
Figure 9. These micrographs show the distribution of micro- and nanoparticles in the specimen, which is not completely homogeneous since density appears to be locally variable when observed at this scale.
Figure 9.
TEM micrographs of the dispersion of functionalized silica microspheres (a,b) and nanospheres (c,d) in the micro-molded PE99-M 3 and PE99-N 3 specimens.
Figure 9.
TEM micrographs of the dispersion of functionalized silica microspheres (a,b) and nanospheres (c,d) in the micro-molded PE99-M 3 and PE99-N 3 specimens.
Thermogravimetric analysis also gave information about the percentage of silica particles incorporated into the polymer matrix and their effect on thermal stability. In all cases, a constant char yield was attained at high temperatures, as shown by representative PE99-N 3 and PE99-M 6 specimens in
Figure 5a. Furthermore, the remaining weight percentage was always in full agreement with the expected silica content (
i.e., close to 3 and 6 wt. %). The result is meaningful since it has demonstrated again that particles were not generally retained in the sprue. Silica was effectively led through the feeding channels by the molten polymer, giving rise to well-dispersed specimens. A close resemblance between the TGA and DTGA traces of the micro-molded PE99 sample and the silica-loaded composites was also found during the first stages of degradation (probably also as a consequence of the low content of added particles). The added particles even seem to slightly stabilize the sample (
Table 2 and
Figure 5). In any case, functionalization of silica particles did not have a negative impact on the ultrasound micro-molding process, as was also observed by GPC measurement. Differences were noticeable only at the end of the degradation process; the presence of shoulders around 483 °C in the DTGA plots (
Figure 5b) seems to be due to decomposition of grafted AMPS groups.
Table 2.
Characteristic TGA temperatures and remaining weight percentages for the decomposition of the studied micro-molded specimens.
Table 2.
Characteristic TGA temperatures and remaining weight percentages for the decomposition of the studied micro-molded specimens.
| Polymer | Tonset (°C) | T10% (°C) | T20% (°C) | T40% (°C) | Tmax (°C) | Remaining Weight (%) |
|---|
| PE 99 | 367 | 397 | 411 | 426 | 433 | 0 |
| PE 99-N 3 | 377 | 405 | 415 | 431 | 436 | 2.7 |
| PE 99-N 6 | 377 | 405 | 415 | 432 | 438 | 5.7 |
| PE 99-M 3 | 377 | 405 | 417 | 431 | 437 | 2.9 |
| PE 99-M 6 | 377 | 404 | 419 | 432 | 439 | 5.8 |
3.3. Calorimetric Studies on the Influence of Functionalized Silica Nanoparticles on the Isothermal Crystallization of Poly(nonamethylene Azelate)
Kinetic analysis was only performed for melt crystallization processes because of the impossibility of obtaining amorphous samples by cooling the melted nanocomposite at the maximum rate allowed by the equipment.
Crystallization experiments were therefore carried out in a narrow temperature interval (
i.e., between 56 and 59 °C) due to experimental limitations.
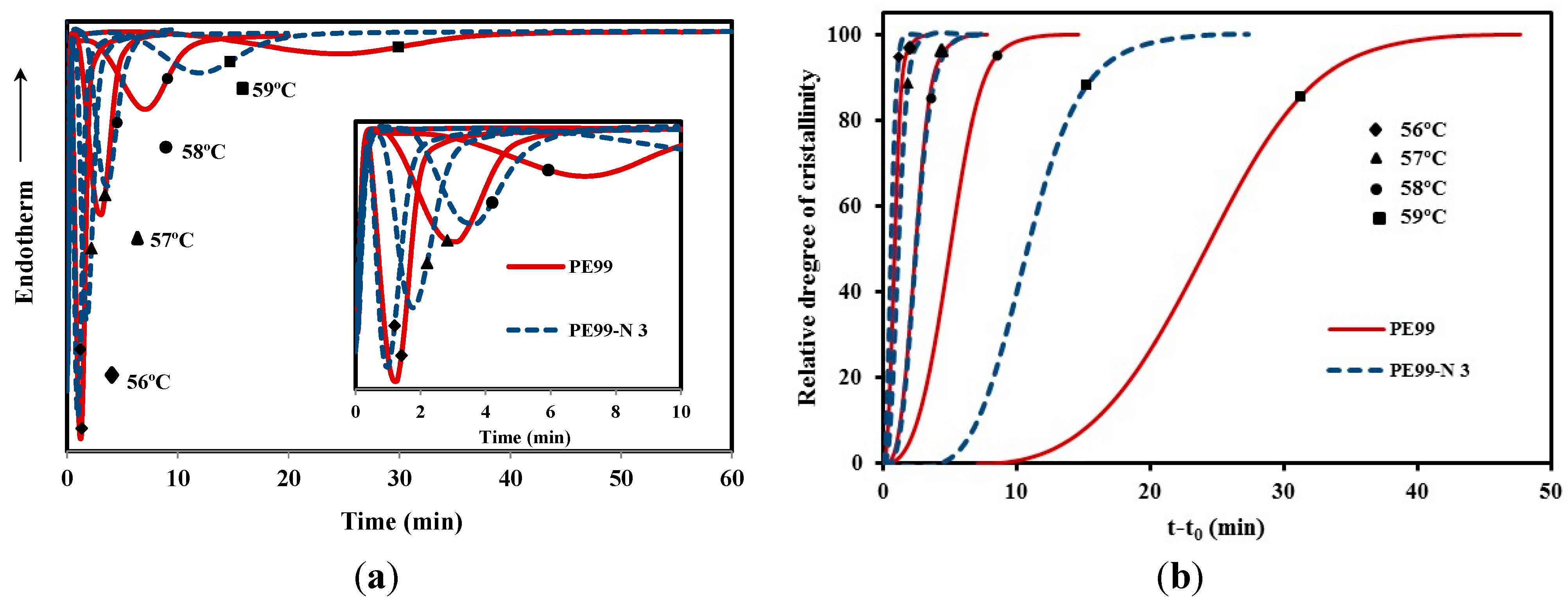
Figure 10a shows the crystallization exotherms of the neat polyester and the PE99-N 3 nanocomposite, which allowed for determining the time evolution of the relative degree of crystallinity, χ(
t). The last was calculated according to Equation (1):
where
t0 is the induction time and d
H/d
t corresponds to the heat flow rate. The evolution of crystallinity always showed a sigmoidal dependence on time for the five melt crystallization experiments performed for the different samples (
Figure 10b). Experimental data were analyzed considering the typical Avrami equation [
27,
28]:
where
Z is a temperature-dependent rate constant and
n the Avrami exponent whose value depends on the mechanism and geometry of the crystallization process. A normalized rate constant,
k =
Z1/n, can also be calculated for comparison purposes since corresponding units (time
−1) are independent of the specific value of the Avrami exponent.
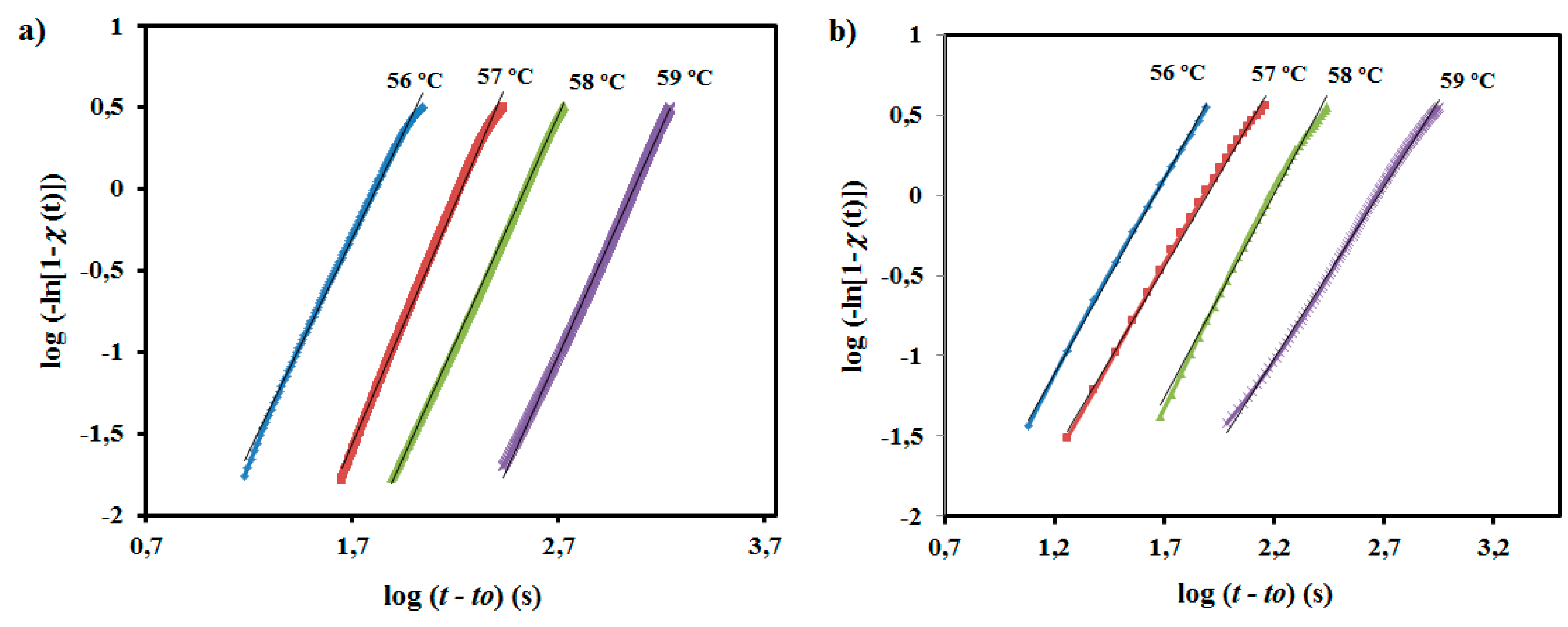
Plots of log(−ln(1 − χ(
t −
t0))) against log(
t −
t0) (
Figure 11) allowed for determining the indicated crystallization parameters, which are summarized in
Table 3. Avrami exponents for the neat polyester remain in a narrow range (
i.e., 2.60–2.94) and have an average value of 2.78. The determined Avrami exponent indicates a predetermined (heterogeneous) nucleation and a spherical growth under geometric constraints. Note that a slight deviation is observed with respect to the theoretical value of 3 and that a value close to 4 should be expected for a sporadic (heterogeneous) and homogeneous nucleations. It should also be pointed out than homogeneous nucleation usually requires high undercooling, which does not correspond with the performed experiments. The exponent slightly decreased for the nanocomposite (
i.e., 2.54–2.14, with 2.33 being the average value), suggesting an increase in geometric constraints upon incorporation of the well-dispersed nanospheres. Exponents determined for both samples were found to vary without a well-defined trend within the selected narrow temperature interval.
Reciprocal crystallization half-times (1/
τ1/2) are also summarized in
Table 3. This parameter is directly determined from DSC isotherms (
i.e., it corresponds to the inverse of the difference between thr crystallization start time and half-crystallization time) and can be useful to test the accuracy of the Avrami parameters since an estimated value can be obtained from them (
i.e., 1/
τ1/2 = (
Z/ln2)
1/n)).
Figure 10.
(a) Exothermic DSC peaks of isothermal crystallizations of PE99 (garnet) and PE99-N 3 (blue) samples at temperatures between 56 and 59 °C; (b) development of the relative degree of crystallinity of PE99 (garnet) and PE99-N 3 (blue) samples at different crystallization temperatures.
Figure 10.
(a) Exothermic DSC peaks of isothermal crystallizations of PE99 (garnet) and PE99-N 3 (blue) samples at temperatures between 56 and 59 °C; (b) development of the relative degree of crystallinity of PE99 (garnet) and PE99-N 3 (blue) samples at different crystallization temperatures.
Figure 11.
Avrami plots of isothermal crystallization of PE99 (a) and PE99-N 3 (b) at the indicated temperatures.
Figure 11.
Avrami plots of isothermal crystallization of PE99 (a) and PE99-N 3 (b) at the indicated temperatures.
Table 3.
Main crystallization kinetic parameters determined by DSC for the neat polyester and its nanocomposite with 3 wt. % of functionalized silica nanospheres.
Table 3.
Main crystallization kinetic parameters determined by DSC for the neat polyester and its nanocomposite with 3 wt. % of functionalized silica nanospheres.
| Sample | T (°C) | Z × 106 (s−n) | n | k × 103 (s−1) | 1/τ1/2 × 103 (s−1) | (Z/ln2)1/n × 103 (s−1) |
|---|
| PE99 | 56 | 18.48 | 2.60 | 15.23 | 17.93 | 17.5 |
| 57 | 0.28 | 2.94 | 5.89 | 6.82 | 6.67 |
| 58 | 0.077 | 2.81 | 2.92 | 3.31 | 3.32 |
| 59 | 0.0028 | 2.79 | 0.86 | 0.97 | 0.99 |
| PE99-N 3 | 56 | 98.56 | 2.42 | 21.98 | 25.97 | 25.58 |
| 57 | 42.03 | 2.31 | 12.84 | 15.12 | 15.05 |
| 58 | 2.69 | 2.54 | 6.37 | 7.62 | 7.36 |
| 59 | 1.89 | 2.14 | 2.12 | 2.54 | 2.52 |
Variation of the overall rate constant with temperature for the neat polymer and its nanocomposites was also evaluated (
Table 3). The rate for the nanocomposite increased (
i.e., from 2.12 × 10
−3 s
−1 to 21.98 × 10
−3 s
−1) when crystallization temperature decreased (
i.e., from 59 °C to 56 °C), a trend that was also logically observed for PE99. More interestingly, the nanocomposite showed a remarkably higher crystallization rate than the neat polyester at all test crystallization temperatures (e.g., 21.98 × 10
−3 s
−1 and 15.23 × 10
−3 s
−1 were determined at 56 °C for PE99-N 3 and PE99, respectively). Therefore, incorporation of functionalized silica nanoparticles had a significant influence on the crystallization process and, logically, on the final material properties. It should be pointed out that the ratio between the two kinetic constants decreased (
i.e., between 2.5 and 1.4) as crystallization temperature decreased, which justifies further studies on nucleation and crystal growth processes.
3.4. Optical Microscopy Studies on the Influence of Functionalized Silica Nanoparticles on the Isothermal Crystallization of Poly(nonamethylene Azelate)
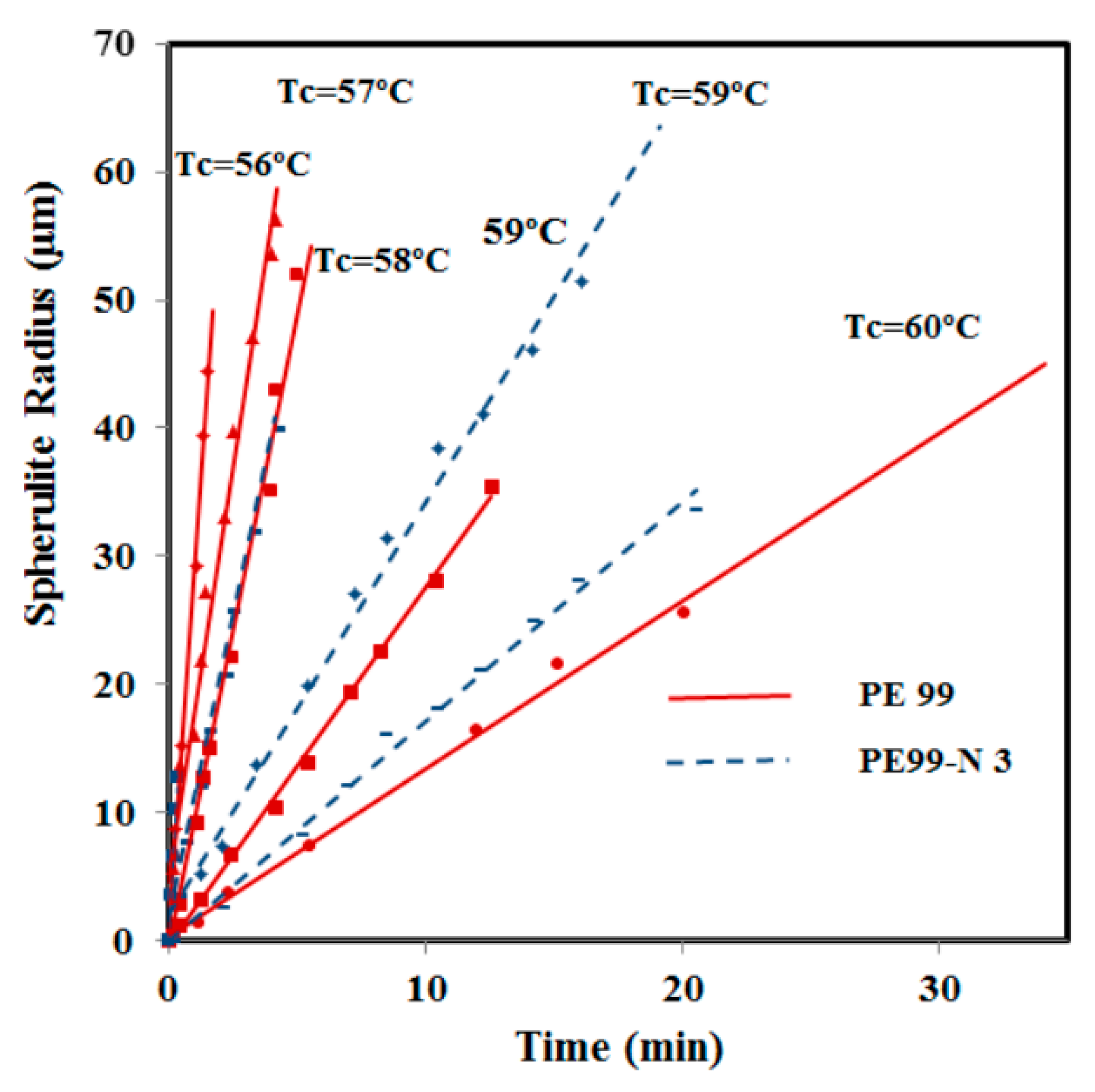
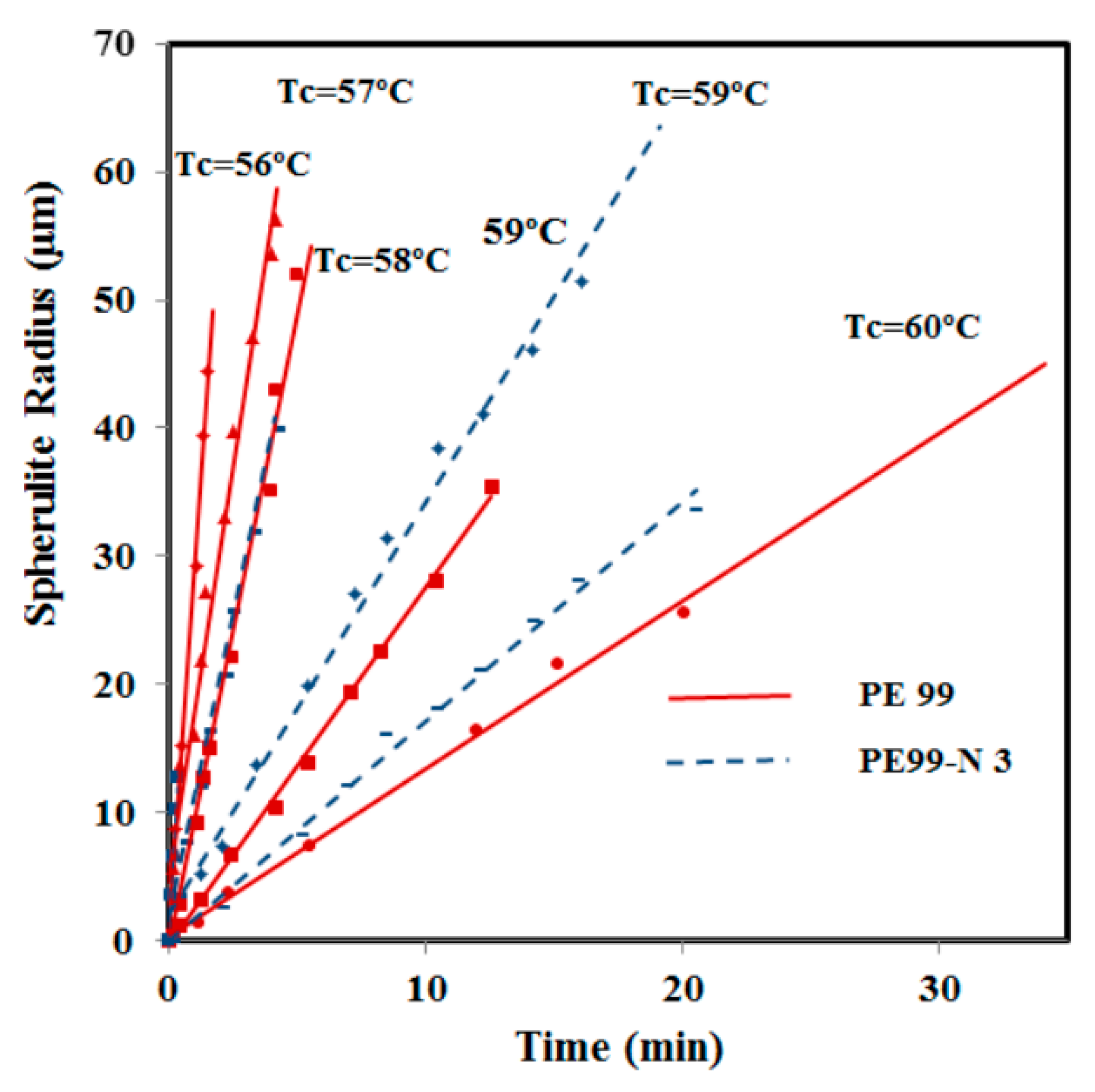
Crystallization kinetics from the melt state was also studied for micro-molded samples with and without functionalized silica nanospheres by optical microscopy. Spherulite radii grew linearly with time until impingement in both cases, as shown in
Figure 12. Crystal growth rates were clearly higher for the nanocomposite at high crystallization temperatures, whereas differences were minimal when this temperature decreased. The relatively high growth rate allowed for collecting experimental data only over a narrow temperature range where crystallization was mainly governed by secondary nucleation (
i.e., the typical bell curve of crystal growth rate
versus temperature could not be obtained). The increase in the crystal growth rate for the nanocomposite is peculiar and suggests favored deposition of molecules onto existing crystal surfaces, as will be discussed. At lower temperatures, this effect seems to be counterbalanced by reduced chain mobility in the presence of silica nanoparticles and spatial constraints imposed by confinement [
29,
30].
Figure 12.
Variation of PE99 (garnet) and PE99-N 3 (blue) spherulite radii with time for isothermal crystallizations performed at the indicated temperatures.
Figure 12.
Variation of PE99 (garnet) and PE99-N 3 (blue) spherulite radii with time for isothermal crystallizations performed at the indicated temperatures.
Pristine polyester and nanocomposite spherulites with similar morphological features were formed (
Figure 13). Thus, both samples crystallized from the melt into ringed spherulites with negative birefringence. The spacing between rings decreased significantly with decreasing crystallization temperatures (
i.e., 13 μm at 60 °C and 5 μm at 56 °C) and decreased slightly upon incorporation of nanospheres (
i.e., 5 μm as opposed to 4 μm for crystallizations at 56 °C). A more confusing texture of less defined rings was detected at an intermediate temperature (
i.e., 59 °C) for both samples. Note that pristine polyester and nanocomposite spherulites fill the field of view and have a relatively uniform size, suggesting athermal nucleation (
i.e., the number of nuclei remains constant during crystallization). More interestingly, this result indicates good dispersion of silica nanoparticles in the polyester matrix. In fact, adsorption of PE99 molecules onto the functionalized surface of silica nanoparticles may hinder particle-particle agglomeration and enhance colloidal stability.
Figure 13.
Optical micrographs of PE99 (a,c,e) and PE99-N 3 (b,d,f) spherulites isothermally grown at 60 °C (a,b); first step at 59 °C and second step at 54 °C (c,d); and 56 °C (e,f). Well-defined rings with interspacing between 3 and 2 μm were detected (see arrows) in the outer part of spherulites grown at 54 °C.
Figure 13.
Optical micrographs of PE99 (a,c,e) and PE99-N 3 (b,d,f) spherulites isothermally grown at 60 °C (a,b); first step at 59 °C and second step at 54 °C (c,d); and 56 °C (e,f). Well-defined rings with interspacing between 3 and 2 μm were detected (see arrows) in the outer part of spherulites grown at 54 °C.
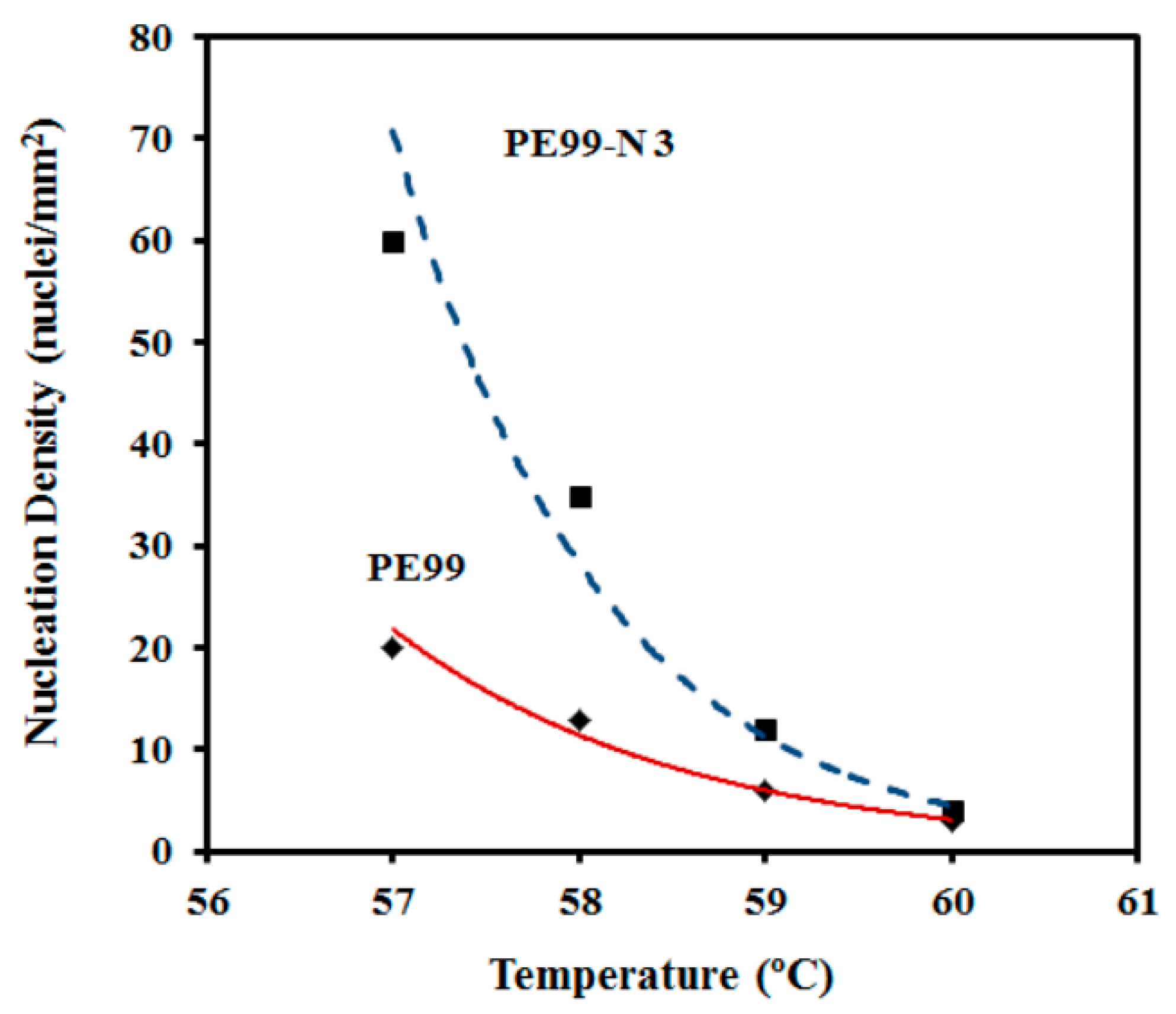
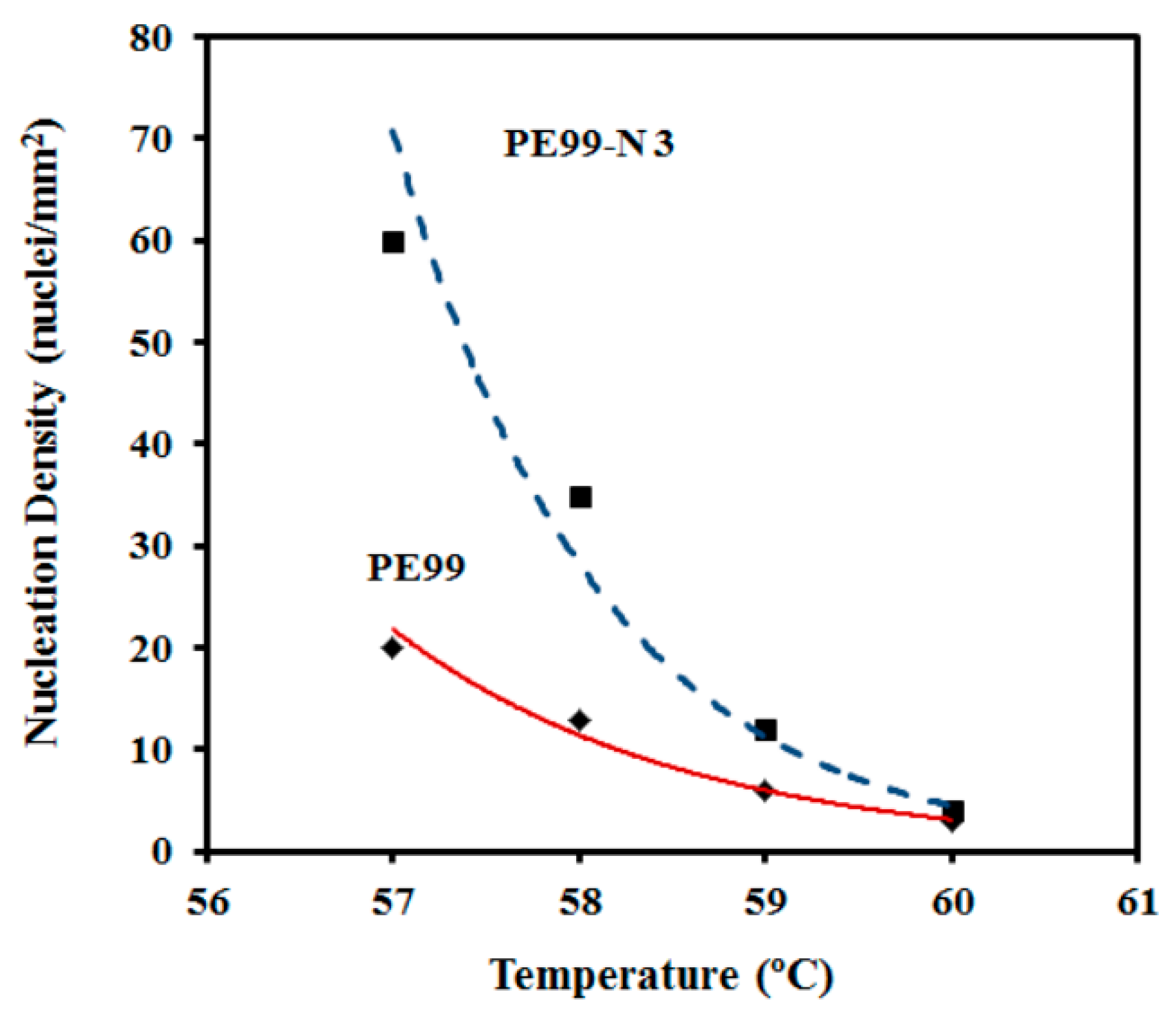
Despite the morphological similarities, great differences were detected in the primary nucleation (
Figure 14). They were more remarkable at low crystallization temperatures; for example, nucleation densities of 60 and 20 nucleus/mm
2 were determined from the optical micrographs taken at 57 °C. Note that crystal growth rates were similar at this temperature; consequently, the differences determined for the overall crystallization rate (e.g., 0.01284 and 0.00589 s
−1 for PE99 and PE99-N 3 at 57 °C, respectively) were mainly attributed to a nucleation effect. On the contrary, nucleation densities were similar at high temperatures, while crystal growth rates were clearly different. Therefore, the incorporation of nanoparticles had a strong impact on the overall crystallization rate due to differences in crystal growth rate and primary nucleation densities, which became more significant at high and low crystallization temperatures, respectively.
Figure 14 also shows that the nucleation density increased exponentially for the two samples at lower crystallization temperatures. These changes in nucleation logically affected the final spherulite size. Thus, a diameter decrease from 120 μm to 35 μm and from 80 μm to 15 μm was observed for PE99 and PE99-N 3 samples, respectively, when the temperature decreased from 60 °C to 56 °C.
Figure 14.
Change in the nucleation density with isothermal crystallization temperature for PE99 (garnet) and PE99-N 3 (blue) samples.
Figure 14.
Change in the nucleation density with isothermal crystallization temperature for PE99 (garnet) and PE99-N 3 (blue) samples.
The logarithmic form of the Lauritzen and Hoffman equation [
31] was employed to estimate the secondary nucleation constant (
Kg):
where
G is the radial growth rate,
Tc is the crystallization temperature,
T∞ is the temperature below molecular motion ceases, Δ
T is the degree of supercooling,
f is a correction factor calculated as 2
Tc/(
Tm +
Tc),
U* is the activation energy,
G0 is a constant preexponential factor,
R is the gas constant, and
Kg is the secondary nucleation constant.
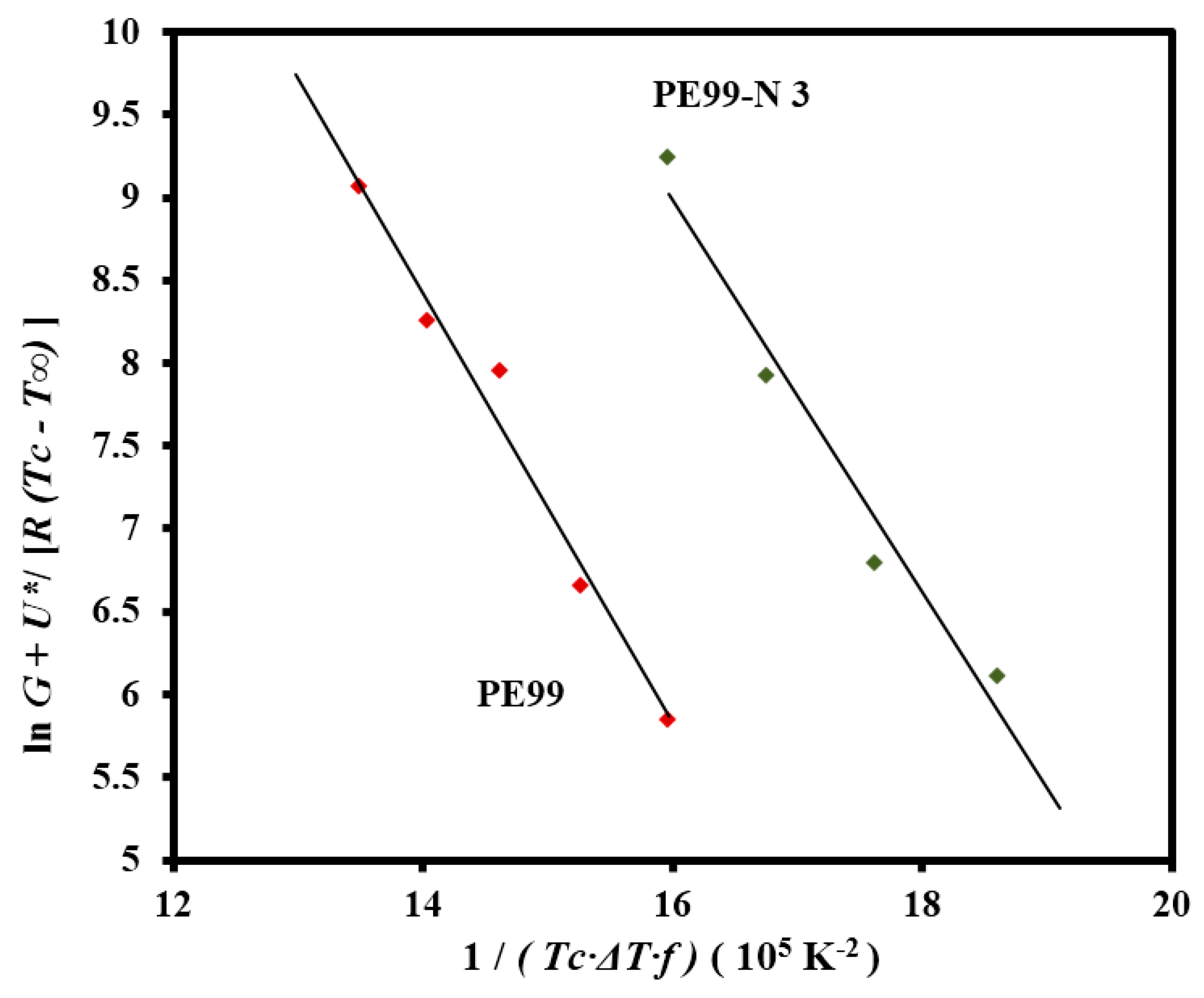
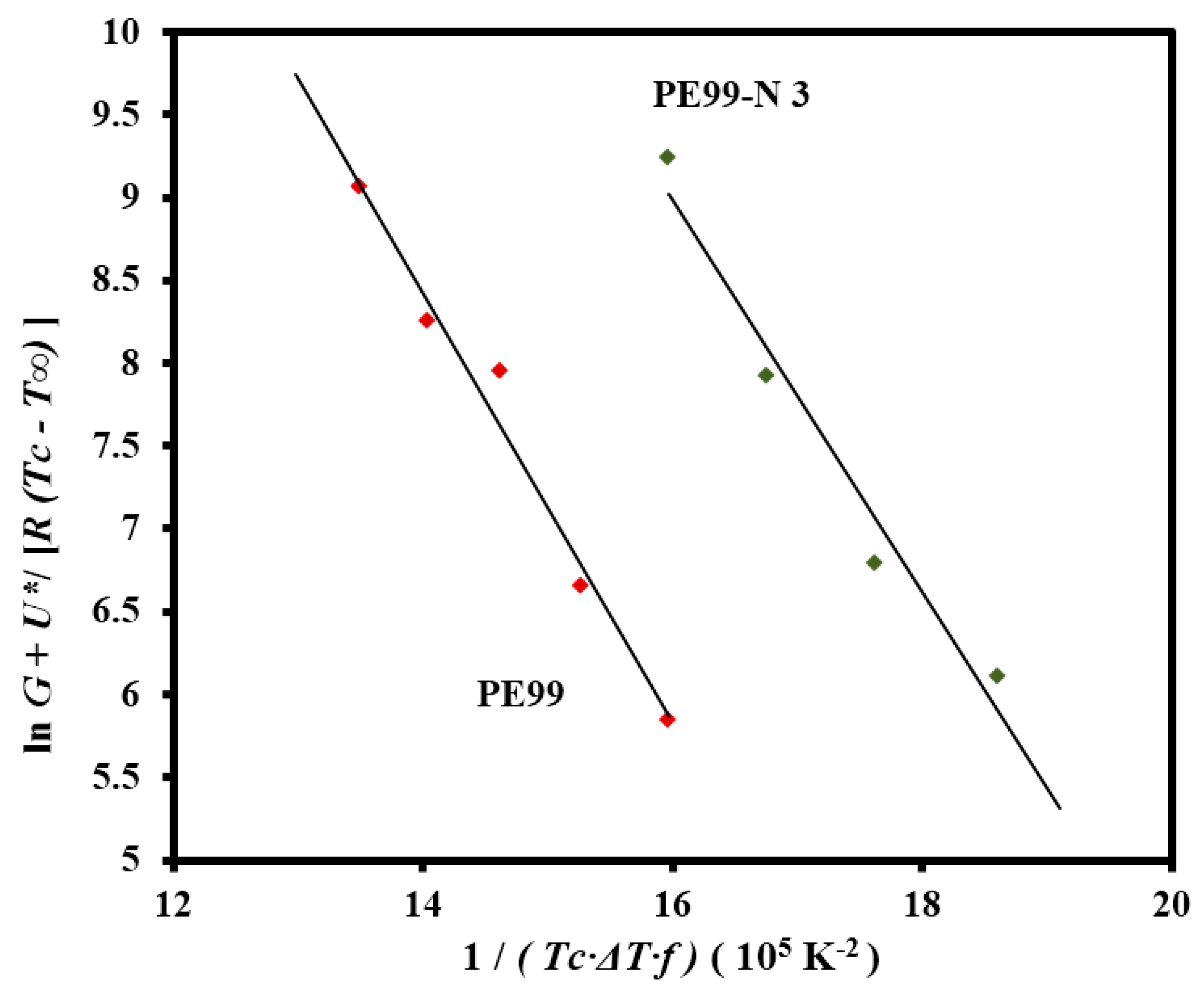
The Lauritzen-Hoffman plot was fitted with straight lines (
r2 > 0.97) for micro-molded PE99 and PE99-N 3 samples when the “universal” values reported by Suzuki and Kovacs [
32] (
i.e.,
U* = 1500 cal/mol and
T∞ =
Tg − 30 K) and the experimental
Tg of PE99 (
i.e., −45 °C) were used in the calculation (
Figure 15).
The nucleation term (deduced from the slope of the plot) mainly influenced the kinetic features at low supercoolings in such a way that crystallization rates could become relatively insensitive to the
U* and
T∞ parameters. Therefore, the equilibrium melting temperature was determined since it influenced the degree of supercooling, and consequently the nucleation term. Typical Hoffman-Weeks plots [
33] were made with samples crystallized at different temperatures (not shown), leading to equilibrium temperatures of 79.2 °C and 76.4 °C for PE99 and PE99-N 3 samples, respectively. The slight change suggests less perfect lamellae upon addition of functionalized silica nanoparticles. The Lauritzen-Hoffman plot allowed for estimating secondary nucleation constants of 1.30 × 10
5 and 1.18 × 10
5 K
2 for PE99 and PE99-N 3 samples, respectively. These values indicate that the presence of functionalized nanoparticles favored the crystallization process. Thus, enhanced PE99/SiO
2 interfacial interaction may decrease the energy involved in the folding of polyester chains and promote their deposition on existing crystal surfaces during the secondary nucleation process [
26,
34].
Figure 15.
Plot of lnG + U*/R(Tc − T∞) versus 1/Tc(∆T)f to determine the Kg secondary nucleation parameter of PE99 (garnet) and PE99-N 3 (blue) samples.
Figure 15.
Plot of lnG + U*/R(Tc − T∞) versus 1/Tc(∆T)f to determine the Kg secondary nucleation parameter of PE99 (garnet) and PE99-N 3 (blue) samples.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}