Featured Application
This proof-of-concept study proposes a new model of TGFβ induction. The demonstrated link between metabolic stress, inflammatory TGFβ and cancer progression supports a novel approach in preventing and fighting pancreatic cancer.
Abstract
Visceral fat mass is associated with a condition of chronic inflammation and can predispose the overweight to an increased cancer risk. Although it is known that adipocytes are active producers of the pro-inflammatory transforming growth factor β (TGFβ), the causes of their excessive synthesis are not clear. In this study, we reproduced two metabolic stress conditions frequently occurring in vivo, namely hypoxia and the fatty acid-driven metabolic uncoupling, and we characterized the response of an in vitro model of 3T3-L1 mouse adipocytes. For the first time, we demonstrated that the mitochondrial dysmetabolism of differentiated adipocytes induced the secretion of TGFβ. The paracrine activity of the secreted cytokine was then tested on two human pancreatic cancer cell lines. Cancer cells responded to the stimulation by increasing mitochondrial respiration, switching on the epithelial–mesenchymal transition (EMT) program and enhancing their motility. The data obtained in this proof-of-concept research show that TGFβ can be produced by dysmetabolic adipocytes, linking the altered metabolism with pro-tumorigenic inflammation. The novel observations of this study identify in metabolic stress a still unexplored cause of inflammation and cancer progression and pave the way to more detailed in vitro and clinical studies on pancreatic cancer.
1. Introduction
Adipose tissue is specialized in fat storage and its function requires an extraordinary capacity of adapting to endogenous and systemic stimuli by metabolic reprogramming [1]. The metabolic failure leads to adipocyte imbalance; in particular, the disturbed mitochondrial activity causes metabolic stress, inflammation and increased production of ROS [2]. Obesity, which is the most evident manifestation of adipose tissue strain, is associated with a decreased mitochondrial content and oxidative capacity, suggesting that mitochondrial impairment may contribute to adipocyte dysfunction [3,4].
Accumulating evidence indicates that obesity causes chronic low-grade inflammation, therefore contributing to systemic metabolic dysfunction associated with obesity-linked disorders [5]. Adipose tissue acts as a key endocrine organ by releasing adipokines with pro- or anti-inflammatory activities [6]. Among these molecules, TGFβ is produced predominantly in pathological visceral adipose tissue. Indeed, locally within adipose tissues, different types of cells, including adipocytes, adipose progenitors, preadipocytes, resident endothelial cells and macrophages, produce proteins of the TGFβ superfamily and their antagonists, creating a local microenvironment particularly sensitive to TGFβ signaling [7]. Several studies have investigated the potential contribution of altered TGFβ expression to adipose tissue dysfunction in mouse and human models of obesity, since the circulating levels of TGFβ are increased in the adipose tissue of overweight and obese subjects compared to those of normal weight, highlighting a close association between TGFβ levels and the increased body mass index during morbid obesity [8,9,10,11].
Excess visceral adipose tissue induces a state of chronic systemic inflammation and altered metabolic activity that promotes a pro-oncogenic environment [12,13]. Indeed, altered systemic release of adipokines, growth factors, and cytokines by dysfunctional adipose tissue in the overweight and obese is considered among the main players in malignant initiation and progression; the negative impact of dysfunctional adipose tissue has been described in pancreatic cancer [14,15]. Epidemiological studies have suggested that overweight and obesity were associated with a greater risk of pancreatic cancer at a younger age of disease onset [16], whereas obesity at an older age was associated with a lower overall survival in patients with pancreatic cancer [17]. However, it remains to be elucidated how interactions between adipose tissue and tumor cells may influence the local invasion and metastasis of pancreatic ductal adenocarcinoma (PDAC), the most common subtype of this cancer in which patients present locally advanced or metastatic disease at the time of diagnosis [18].
In this scenario, TGFβ could be considered one of the systemic mediators of the effects of dysfunctional and inflamed adipose tissue on pancreatic cancer cells. TGFβ acts through the activation of the resident fibroblasts, and promotes the epithelial–mesenchymal transition (EMT) and the production of extracellular matrix (ECM) structural proteins (e.g., fibrous collagen and elastin) and adhesive proteins (e.g., laminin and fibronectin) [19]. In cancer, this pleiotropic cytokine contributes to the formation of the fibrotic tumor microenvironment promoting angiogenesis, immunosuppression, invasiveness and thereby metastasis [20]. Although the involvement of TGFβ in pancreatic cancer makes the cytokine an interesting therapeutic target, the attempted preclinical and clinical investigations have shown that the pharmacological approach remains a clinical challenge [21].
The mechanisms underlying the obesity–cancer link are likely multifactorial, including metabolic and inflammation perturbations [22]. Fibrosis and EMT are two important hallmarks of tumor-related chronic inflammation progression [23,24], and in pancreatic cancer both appear to be driven by autocrine and paracrine TGFβ signaling. In addition, TGFβ modulates the lipid metabolism and the metabolic reprogramming in cancer and inflammation [25,26,27,28,29,30]. Notably, few studies have supported the strong effects of TGFβ on mitochondrial activity through several mechanisms, leading to context-specific results [31,32,33]. Despite the evidence of the metabolic effects of TGFβ, little is known about the causes that trigger its production, both in adipose tissue and cancer.
Based on the described evidence that link adiposity, inflammation and cancer, and because in obesity the dysfunctional adipose tissue displays a mitochondrial impairment [5], in this study we sought to verify whether energy dysmetabolism could be the triggering cause of TGFβ production. For this purpose, we conceived an in vitro experimental model to verify whether the adipose tissue under conditions of metabolic stress can produce the inflammatory cytokine, and to confirm the paracrine pro-tumoral activity of the secreted cytokine. Indeed, in this proof-of-concept study we demonstrate both the production of TGFβ and its stimulatory activity on the progression of two human pancreatic cancer lines.
2. Materials and Methods
2.1. Cell Cultures and Treatments
All cells were purchased from the American Type Culture Collection (ATCC; Rockville, MD, USA) and were grown in standard conditions of temperature, humidity and CO2. The mouse 3T3-L1 cell line was used as a simplified in vitro model of adipose tissue to test the effects of metabolic stress on TGFβ secretion. Several reasons led to the choice of 3T3-L1 cells; in fact, the cell line is a well-established model of differentiation to adipocytes by a standard protocol [34,35], and can be stimulated to produce TGFβ [34]. Most importantly, 3T3-L1 cells and human fat cells are similar in their response to cytokines and growth factors [36,37] and they show comparable protein secretion [38,39]; moreover, the proteins secreted by 3T3-L1 cells are frequently tested on human cell models [40,41]. 3T3-L1 cells were kept in Dulbecco’s modified Eagle medium (DMEM) with 4.5 g/L glucose, with the addition of 1% (v/v) antibiotics (penicillin/streptomycin solution) and 10% (v/v) fetal calf serum (FCS, Merck Life Science S.r.l., Milan, Italy). When 3T3-L1 cells reached confluence, to induce differentiation into mature adipocytes, 5 µg/mL insulin (Merck Life Science S.r.l., Milan, Italy), 1 µM dexamethasone (Merck Life Science S.r.l., Milan, Italy) and 0.5 mM 3-isobutyl-1-methylxanthine (IBMX) (Merck Life Science S.r.l., Milan, Italy) were added to the culture medium. After 2 days, the medium was changed to DMEM 4.5 g/L glucose, containing 10% fetal bovine serum (FBS) and 1 µg/mL insulin. Cells were maintained in this medium for 8 days, and we changed it every 3 days [34,35]. This protocol has been largely validated in many studies by Oil Red-O staining of differentiated adipocytes [34,36,42].
Both differentiated and undifferentiated 3T3-L1 cells were stimulated with Chemically Defined Lipid Concentrate (lipid mix, 11905-031, Gibco, Thermo Fisher Scientific, Monza, Italy) diluted 1:50 for 12 and 48 h, and differentiated adipocytes were kept for 48 h in mild hypoxic condition (5% O2). Mild hypoxia approximates the low tissue oxygen levels observed in vivo in obese rodents and humans, and stimulates adipogenesis and lipogenesis, supporting the role of hypoxia in the development of body metabolic impairments [43]. At the end of the 48 h treatments, the supernatant of differentiated adipocytes (conditioned medium, CM) was removed, centrifuged (14,000× g, 20 min) to eliminate any cellular debris, and tested for activity. The CM was then subjected to an acidification procedure necessary to activate the latent form of TGFβ, following a previously tested protocol [34,44]. Acidification of CM to pH 3.0 was carried out by dropwise addition of 5 N HCI. After incubation on ice for 30 min, the pH of CM was restored to 7.2 by dropwise addition of 5 N NaOH [44]. This protocol allows the activation of the cytokine by a chemical method without requiring the partial enzymatic proteolysis occurring in vivo.
The human pancreatic cancer cells PANC-1 and CAPAN-2 were grown in DMEM culture medium supplemented with 10% fetal bovine serum and 1% antibiotics (penicillin/streptomycin (Merck Life Science S.r.l., Milan, Italy). Both cellular lines were derived from primary tumors and are representative of different levels of tumor aggressiveness [29]. PANC-1 and CAPAN-2 cells were stimulated with the conditioned medium diluted 1:3 with fresh growth medium or with 0.5 ng/mL TGFβ1 (PeproTech, Rocky Hill, NJ, USA) for 8 days.
The experimental flowchart is summarized in Figure S1 (Supplementary Materials).
2.2. Measurement of the Mitochondrial Membrane Potential (ΔΨm)
The JC-1 dye (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide) was used to determine ΔΨm by flow cytometry, as previously reported [29]. JC-1 stains mitochondria in living cells in a membrane potential-dependent manner; the increase in the red/green fluorescence intensity ratio (FL2/FL1 channels of flow cytometer) measures the enhanced mitochondrial activity. After treatment, cells were loaded with the dye (2 µg/mL, Biotium, Hayward, CA, USA) and analyzed as described [26].
2.3. Evaluation of Mitochondrial ATP Levels
Undifferentiated and differentiated 3T3-L1 cells were seeded into six-well plates. At the end of the treatment, the cells were harvested, subjected to mechanical lysis in lysis buffer without detergents, and the mitochondria were separated by differential centrifugation. After a brief centrifugation at 12,000× g for 20 s to collect the nuclei, the supernatant was centrifuged at 12,000× g for 30 min to obtain the mitochondria. Then, the pellet was resuspended in PBS and it was analyzed for mitochondrial ATP content with the ATP Bioluminescent Assay kit (FL-AA, Merck Life Science S.r.l., Milan, Italy). ATP was measured as previously reported [26]. Briefly, we quantified ATP as relative light units (RLU), which were then converted into nmol ATP/mg mitochondrial proteins; data were expressed in comparison to control values as percentage values. Experiments were repeated three times in triplicate [26].
2.4. Western Blotting Analysis
At the end of the treatments, the subcellular fractionation of total, mitochondrial and cytoplasmic proteins extracted from 3T3-L1 pre-adipocytes and adipocytes was carried out and samples were subjected to Western blotting analysis as previously described [45]. Details of the Western blotting procedure are reported in Supplementary Materials. The blots were incubated with the following antibodies: mouse monoclonal anti-cytochrome C (556433, BD Pharmingen, Milan, Italy) and mouse monoclonal anti-VDR (sc-13133, Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA). The proteins of interest and the loading controls were detected on the same membranes. The mouse anti-actin antibody (sc-8432, Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), and the mouse anti-voltage-dependent anion channel (VDAC) antibody (anti-porin 31HL, Calbiochem, Merck Life Science S.r.l., Milan, Italy), were used as loading controls of, respectively, the total and mitochondrial proteins. After incubation with a peroxidase-conjugated anti-mouse secondary antibody (Bio-Rad Laboratories, Hercules, CA, USA), the proteins of interest were detected and quantified by ChemiDocTM MP System (Bio-Rad Laboratories, Hercules, CA, USA). VDR antibody detected a doublet and VDR was identified as the lower band, as demonstrated by the VDR silencing experiments previously published [45,46,47].
2.5. ELISA Analysis
The TGFβ1 release was quantified in 50 µL cellular supernatants of treated 3T3-L1 cells, using the human ELISA TGFβ1 assay (R&D, Minneapolis, MN, USA), according to the manufacturer’s instructions. Data are expressed as ng/mL. Experiments were performed in triplicate and were repeated three times.
2.6. Real-Time Polymerase Chain Reaction (qRT-PCR)
At the end of the treatments, total RNA was extracted as previously described [48]. One μg of total RNA was reverse transcribed into cDNA [49]. Quantitative PCR was carried out in a final volume of 20 μL using the iTaqTM Universal SYBR® Green Supermix (Bio-Rad, Hercules, CA, USA) with specific primers to amplify the following human genes: E-cadherin [26], fibronectin [50], cytochrome c oxidase subunit 2 (COX II), mitochondrial adenosine 5′-triphosphate (ATP) synthase F0 subunit 6 (MT-ATP6, ATP synthase subunit beta (ATP5B) [48]. Ribosomal subunit protein (S14) [49] was used as an internal control. The real-time PCR parameters were previously reported [40]. The 2-ΔΔCT method was applied and the quantification of PCR gene products was expressed in arbitrary units. PCR efficiency and high-linearity amplification plots (r > 0.976) of the analyzed transcripts were similar. Melt curve analysis confirmed the specificity of PCRs.
2.7. Wound Healing Assay
PANC-1 e CAPAN-2 cells were seeded in a 24-well plate, and were treated for 8 days. On the seventh day, confluent cells were starved overnight. A wound line was generated with a sterile pipette tip, followed by treatments with TGFβ (0.5 ng/mL) and conditioned medium (1:3 dilution) for 24 h. At the beginning (T0) and at the end (T24) of the incubations time, the wound was photographed at 20× magnification under bright field illumination. The wound area was measured in its central part by ImageJ software (ImageJ version 1.54, Sun Microsystems Inc., Palo Alto, CA, USA). The percentage of wound closure was calculated as follows: 100 − [(area at t24/area at t0) × 100] [26].
2.8. Cell Proliferation Assay
PANC-1 and CAPAN-2 cells were seeded in 96-multiwell plates and were incubated with conditioned medium (CM) and with 0.5 ng/mL TGFβ. After 5 days, the plates were fixed with 11% glutaraldehyde for 15 min, and then cells were stained with 0.1% crystal violet solution for 20 min. After solubilization, the absorbance of crystal violet was recorded at 595 nm [26]. Data were collected as the average of three independent experiments for each experimental condition.
2.9. Statistical Analysis
The data are expressed as the mean ± standard error (SEM) of three independent experiments. The statistical analysis of the data was carried out by ANOVA with Tukey’s post hoc correction from GraphPad Prism 5.0 (GraphPad Software, Inc., San Diego, CA, USA). Significance was set for values with p < 0.05.
3. Results
3.1. Lipid Mix Impairs Mitochondrial Respiration and ATP Production in 3T3-L1 Adipocytes, Without Affecting Mitochondrial Integrity
The first aim of this work was setting an in vitro experimental model of fat tissue cells that could be stimulated to reproduce a pathological condition of mitochondrial dysfunction. 3T3-L1 cells can be differentiated into adipocytes using different hormonal stimuli [51]. After differentiation, cells transform a fibroblastoid phenotype into a typical adipocyte appearance, and fill their cytoplasm with lipid droplets (Figure S2 in Supplementary Materials). We selected this cell culture model as it is widely used and standardized to study the effects of molecules on adipogenesis, inflammation and metabolism [51,52,53]. Moreover, the amino acid sequences for mature mouse and human TGFβ are 99–100% identical [54]; therefore, if the cytokine was secreted in 3T3-L1 growth medium, its biological effects could be further investigated in human cells. To experimentally induce mitochondrial dysmetabolism, we used a lipid mix able to generate a metabolic stress by uncoupling the respiratory chain from oxidative phosphorylation. In fact, high concentrations of long-chain free fatty acids can cause uncoupling by a mechanism of fatty acid cycling [55,56]. We analyzed the mitochondrial metabolic alterations occurring in 3T3-L1 pre-adipocytes and differentiated adipocytes in response to lipid mix treatment. First, the flow cytometric analysis with the JC-1 dye measured the mitochondrial membrane potential, proportional to mitochondrial respiratory activity, and was carried out only in undifferentiated 3T3-L1 cells treated for 48 h, because in differentiated adipocytes the abundance of lipid deposits interfered with the measurement. We observed a decrease in mitochondrial membrane potential when 3T3-L1 cells were treated with lipid mix, compared to untreated control (Figure 1A). This result suggested a possible condition of mitochondrial stress. Next, we found a decrease in ATP levels in the pre-adipocytes and fully differentiated adipocytes treated with lipid mix, compared to the respective untreated controls (Figure 1B). In addition, we detected a slight difference in ATP production between the short 12 h treatment and the longer 48 h treatment with the lipid mix, suggesting a partial protective restoration of ATP levels exerted by cells to avoid cytotoxicity. The results of both these analyses confirmed the reduced mitochondrial activity, demonstrating that a concentrated mixture of lipids could alter mitochondrial respiration, thus proposing a new model in vitro of adipocytes exposed to metabolic stress. Notwithstanding the energy loss, the integrity of mitochondrial compartment was not compromised by the metabolic derangement, as demonstrated by the analysis of intracellular cytochrome C localization. In both models of 3T3-L1 treated with the lipid mix for 48 h, the Western blot analysis revealed that the expression of cytochrome c was unchanged both in the mitochondrial and in the cytoplasmic compartment, demonstrating that the lipid mix did not induce the export of this soluble mitochondrial protein (Figure 1C). Similarly, the total expression of cytochrome C did not change. Original blots are shown in Figure S3 (Supplementary Materials). Altogether, these data suggest that although mitochondrial metabolism was altered by uncoupling agents, mitochondrial structures were not damaged and cells were protected from apoptosis.
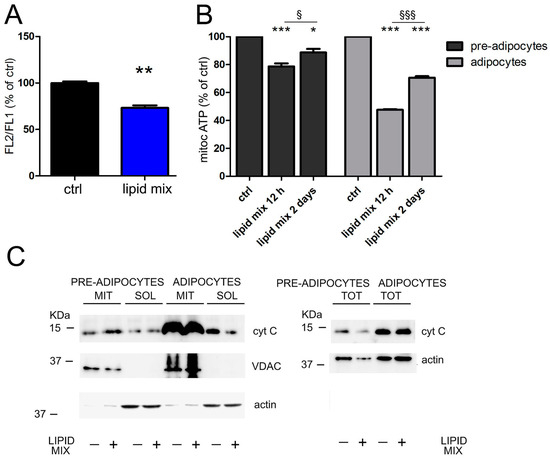
Figure 1.
Lipid mix impairs mitochondrial respiration. (A) 3T3-L1 pre-adipocytes were incubated for 48 h with the lipid mix and the mitochondrial membrane potential was evaluated by flow cytometric measurement of the JC-1 dye. The FL2/FL1 ratio was expressed as a percentage of the value obtained for untreated cells. (B) Pre-adipocytes and 3T3-L1 differentiated adipocytes were incubated with the lipid mix for 12 h and for 48 h and mitochondrial ATP was evaluated relative to the control. (C) After 48 h of treatment with the lipid mix and in untreated cells, the expression of cytochrome C (cyt C) was evaluated by Western blot analysis of total (TOT), mitochondrial (MIT) and cytoplasmic (SOL) extracts. Actin and VDAC were used as loading controls for total, cytoplasmic and mitochondrial extracts. Data are shown as means ± SEM of three independent experiments. * p < 0.05; ** p < 0.01 and *** p < 0.001 vs. ctrl.; § p < 0.05; §§§ p < 0.001.
3.2. Differentiated 3T3-L1 Adipocytes Subjected to Metabolic Stress Produce TGFβ
After finding an effective approach to induce metabolic stress, we decided to verify the production of TGFβ in our dysmetabolic cellular model. We treated differentiated adipocytes with lipid mix for 48 h; moreover, we also tested the effect of hypoxia, a well-known promoter of mitochondrial dysfunction, to compare the effect exerted by these two stressors on TGFβ production. The secreted cytokine was measured by ELISA assay in the supernatant of treated adipocytes (henceforth referred to as conditioned medium) and found to be highly increased compared to the undetectable levels of control cells. In fact, Figure 2A shows the 8-fold increase of the levels of TGFβ secreted in the adipocyte medium under hypoxia conditions (from 0.053 ng/mL to 0.402 ng/mL) and the 19-fold increase of TGFβ in the medium of cells treated with the concentrated lipid mix (from 0.053 ng/mL to 1.010 ng/mL). Next, to verify whether TGFβ was secreted by adipocytes in its active form, we investigated its efficacy in inducing vitamin D receptor (VDR) expression in pancreatic cancer cells. This property was previously demonstrated in two pancreatic cancer cell lines [29] and in this model was exploited as a readout of TGFβ activity. As shown in Figure 2B, the conditioned medium obtained from adipocytes exposed to treatments did not increase VDR expression in human pancreatic cancer cells, when it was used unprocessed. The increased adipocyte secretion of TGFβ not accompanied by biological activity led us to suspect the secretion of a latent form of the cytokine, as it is well known that the extracellular activation of latent TGFβ must be a major regulatory step controlling TGFβ effects [57]. Whereas in vivo the mechanisms activating the pro-form are numerous and not yet fully elucidated (for instance, the acidosis-dependent upregulation of thrombospondin-1 [58] and the cell-associated plasmin protease activity [59]), in vitro it is relatively easy to induce conformational changes of latent TGFβ, leading to the release of mature and active TGFβ. For instance, purified latent TGFβ can be activated by acidic pH [59], which is a mechanism occurring also in vivo when a similar pH is generated by osteoclasts [57]. We decided to carry out the procedure of subjecting the conditioned medium to strong acidification and subsequent neutralization, which was able to activate TGFβ, and finally we could detect the expression of VDR as the readout of TGFβ activity. Indeed, after the activation of the conditioned medium by acidification, we detected the induced expression of VDR when pancreatic cancer cells were treated with medium from hypoxic adipocytes and most strongly with medium from adipocytes exposed to lipid mix, as shown in Figure 2C. Original blots are displayed in Figure S4 (Supplementary Materials). We could conclude that dysmetabolic adipocytes secreted a latent form of TGFβ, which was modified and activated by acidification. This in vitro procedure of TGFβ activation was previously reported [44,60].
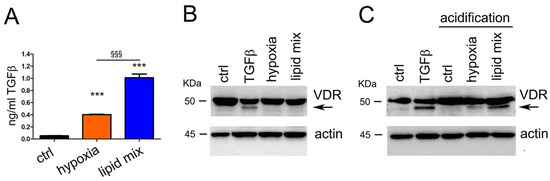
Figure 2.
Hypoxia and lipid mix stimulate the production of TGFβ in its latent form. (A) After 48 h of treatment, the amount of TGFβ secreted by differentiated adipocytes was measured in conditioned growth medium by ELISA method. (B,C) The induction of vitamin D receptor (VDR) expression was used as a readout of TGFβ activity. The expression of VDR was evaluated by Western blot analysis of total lysates from PANC-1 cells treated for 48 h with adipocyte-conditioned growth medium or with 10 ng/mL TGFβ. The band corresponding to VDR is indicated by the arrow. Actin expression was evaluated as loading control. Data are shown as means ± SEM of three independent experiments. *** p < 0.001 vs. ctrl.; §§§ p < 0.001.
Since in the 3T3-L1 model we characterized the cellular response to the lipid mix, and because this stimulus is the most powerful inducer of TGFβ secretion, in subsequent experiments we further investigated only this metabolic stress trigger.
3.3. Pancreatic Cancer Cells Exposed to TGFβ Secreted by Dysmetabolic Adipocytes Increase Their Respiratory Activity
The data obtained in our previous work revealed the metabolic impact of TGFβ on human pancreatic cancer cells and in particular the dose-dependent sensitivity of mitochondrial activity to cytokine treatment [29]. In fact, we showed that long-term exposure of PANC-1 and CAPAN-2 cells to a low, non-toxic dose of TGFβ (2 ng/mL) produced a strong increase in mitochondrial respiratory activity [33]. Based on these previous observations, we decided to use the conditioned medium obtained from adipocytes exposed to lipid mix to evaluate the metabolic modulation exerted by the inflammatory cytokine on the same pancreatic tumor lines. PANC-1 and CAPAN-2 cell lines were treated with conditioned medium (CM) diluted three times with fresh medium to ensure the nutrients necessary for growth. The final TGFβ concentration was therefore about 0,35 ng/mL, close to the lowest doses of the cytokine used in other experimental settings [61,62]. First, we carried out a flow cytometric analysis with JC-1 dye to measure mitochondrial respiratory activity and this investigation revealed some interesting metabolic changes, as shown in Figure 3A. In fact, when PANC-1 and CAPAN-2 cells were exposed to prolonged (8 days) treatment with the conditioned medium, we observed an increase in mitochondrial membrane potential similar to the one observed following prolonged treatment with commercial TGFβ. To further investigate the metabolic effect of the cytokine produced by adipocytes subjected to metabolic stress, we evaluated its transcriptional effects on mitochondrial respiration. In PANC-1 cells, the 8-day treatment with the conditioned medium resulted in a strong transcriptional induction of some subunits of respiratory chain and ATP synthase (COX-II, MT-ATP6 and ATP-5B) compared to control, whereas the prolonged treatment with commercial TGFβ was less effective (Figure 3B). These results were confirmed by the analysis of the second pancreatic cancer cell line CAPAN-2, as shown in Figure 3C. The results of these experiments demonstrated the metabolic efficacy of TGFβ secreted in the conditioned medium; interestingly, the effect on the transcription of respiratory elements is greater than that exerted by similar dosage of synthetic TGFβ.
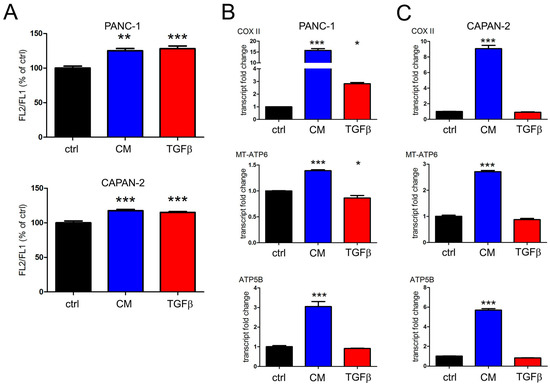
Figure 3.
The adipocyte-conditioned medium increases respiratory activity of two pancreatic cancer cell lines. PANC-1 and CAPAN-2 cells were incubated for 8 days with the conditioned medium (CM) or 0.5 ng/mL TGFβ. (A) Mitochondrial membrane potential was evaluated by JC-1 flow cytometric assay. The FL2/FL1 ratio was expressed as a percentage of the value obtained for untreated cells (ctrl). (B) The transcription of subunits of respiratory chain (COX II) and ATP synthase (MT-ATP6 and ATP5B) was evaluated by RT-PCR in PANC-1 cells and (C) in CAPAN-2 cells. Data are shown as means ± SEM of three independent experiments. * p < 0.05; ** p < 0.01 and *** p < 0.001 vs. ctrl.
3.4. The Conditioned Medium from Adipocytes Exposed to Metabolic Stress Favors the Progression of Tumor Cells from a Proliferative to a Migratory State
The last part of our study investigated two distinctive features of TGFβ-induced tumor progression, namely the nuclear effects of the cytokine on epithelial–mesenchymal transition (EMT) and cellular migration. We sought to confirm whether the prolonged exposure to the cytokine secreted from dysmetabolic adipocytes could enhance EMT and invasion of pancreatic cancer, as demonstrated in our previous in vitro study on TGFβ [29].
We incubated PANC-1 and CAPAN-2 cells for 8 days with both the conditioned medium and 0.5 ng/mL of synthetic TGFβ, to equalize the dosage of TGFβ used in treatments. The morphological changes indicative of EMT were observed by light microscopy (Figure S5 in Supplementary Materials) and suggested further analysis of EMT markers by RT-PCR. In addition to the stretched appearance of pancreatic cancer cells, we confirmed the ongoing transition by measuring the transcriptional suppression of E-cadherin and the increase of fibronectin mRNA after the treatment of both cell types with conditioned medium. The transcriptional effects exerted by the similar dosage of synthetic TGFβ were less intense. These results are shown in the first two panels of Figure 4A (PANC-1 cells) and Figure 4B (CAPAN-2 cells). Furthermore, we carried out the wound healing assay to test the migration of PANC-1 and CAPAN-2 cells after prolonged treatment. In both PANC-1 and CAPAN-2 cells, the wound closure rate following treatment with conditioned medium from dysmetabolic adipocytes was approximately 4 times higher than in the control and more effective than prolonged treatment with synthetic TGFβ. A representative image of these experiments is displayed in Figure 4C, and the wound closure is quantified in the third panel of Figure 4A,B. Based on these data, in this study we confirmed the pro-invasive effect of prolonged treatment with low doses of TGFβ and most interestingly, we demonstrated that the effect can be induced by the TGFβ produced by dysmetabolic fat cells.
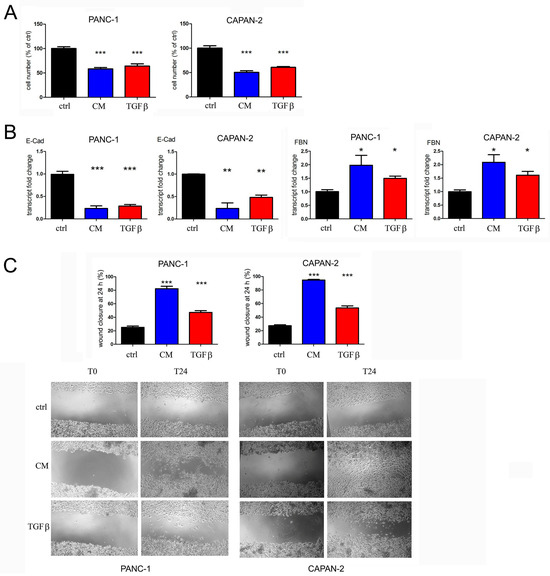
Figure 4.
The adipocyte-conditioned medium exerts antiproliferative effects and enhances the epithelial–mesenchymal transition and the migration of PANC-1 and CAPAN-2 cells. The cells were left untreated (control, ctrl) or were incubated with the conditioned medium (CM) or 0.5 ng/mL TGFβ. (A) After 5 days of treatment, cell proliferation was measured by crystal violet staining, and the values are expressed as percentages of the untreated control. (B) The transcription of E-cadherin (E-Cad) and fibronectin (FBN) was analyzed by RT-PCR. (C) Cell motility was evaluated as wound closure by wound healing assay. Representative images of wound healing assay are shown for both cell lines. Data are shown as means ± SEM of three independent experiments. * p < 0.05; ** p < 0.01 and *** p < 0.001 vs. ctrl.
4. Discussion
TGFβ signaling has been extensively described, but there is no clear explanation of the causes that trigger massive cytokine production in cancer and several inflammatory diseases. Adipose tissue is considered a site of TGFβ production and visceral fat is an important risk factor for the development of several cancers [12,13,14,15]. Obesity in humans is associated with increased expression and release of TGFβ, although its low level of secretion by human adipose tissue has so far prevented the measurement of the active form of the cytokine and the investigation of factors influencing TGFβ conversion of the latent form to the active one in most human studies [10].
Visceral fat accumulation can be originated by adipose tissue dysfunction, associated with changes in the cellular composition, increased lipid storage and secretion of proinflammatory, atherogenic and diabetogenic factors [63]. In fact, obese patients display hypertrophic, hypoxic and inflamed adipose tissue [53]. Interestingly, the improvement of the mitochondrial function in adipocytes can prevent adipose tissue inflammation, fibrosis and insulin resistance [1]. This suggests that adipose tissue malfunction and metabolic disorders are linked to mitochondrial impairment, defective oxidative phosphorylation (OXPHOS) and altered adipose-derived secreted factors [1]. Indeed, several studies on obese mice models reported the reduction of mitochondrial mass, high reactive oxygen species (ROS) production and low cellular ATP levels of adipose tissue [64,65,66]. However, it remains unclear whether the altered mitochondrial activity is a direct consequence or the cause of maladaptation in adipose tissue.
A positive correlation between obesity and PDAC risk has been definitely established [15]. Preclinical studies employing PDAC mouse models have convincingly demonstrated that obesity accelerates pancreatic cancer development [15]. Many in vivo studies investigating the link between dysfunctional adipose tissue and pancreatic cancer have demonstrated that a high-fat diet, which promotes adipocyte metabolic perturbation, favors inflammation and pancreatic cancer [67,68,69]; however, the direct correlation between adipocyte metabolic stress and pancreatic cancer progression has not been fully investigated yet.
Based on this evidence, in this proof-of-concept study we searched novel metabolic triggers of TGFβ production in adipocytes and we investigated the link between dysmetabolism, inflammation and cancer. For this purpose, we proposed a simplified in vitro model to verify whether the deranged energy metabolism in adipocytes can trigger the secretion of the proinflammatory TGFβ and then we tested the effects of this production on a model of pancreatic cancer. Although human preadipocytes and adipose stem cells are excellent models for studying adipogenesis and obesity-related metabolic alterations as well as donor differences, in this study the aim of demonstrating the induction of TGFβ secretion by metabolic stress could be achieved using the simplest model of 3T3-L1 adipocytes, which are widely used because of the highly standardized protocols of differentiation and treatments. Future studies will develop a more complex human model to confirm this initial observation and to investigate the relevance of the fat tissue microenvironment. In this study, we identified two different causes of metabolic failure that can induce TGFβ production in our adipose cell model, namely hypoxia and a mix of concentrated fatty acids.
We investigated the response to hypoxia because this known stimulus of cellular stress is associated with the hypertrophic and dysmetabolic phenotype of adipocytes. Moreover, in vitro studies have shown that experimentally induced hypoxia in adipocytes stimulates the production of inflammatory mediators and that the accumulation of hypoxia-inducible factor 1α (HIF-1α) leads to activation of transcription of pro-fibrotic genes [6,35]. In addition, many studies have reported the interdependence of HIF-1α and TGFβ signaling [70,71], and one study demonstrated that hypoxic hepatocytes increased the ratio of active TGFβ to total TGFβ in the culture medium [72]. In this study, for the first time we demonstrated that hypoxia is a metabolic factor capable of stimulating TGFβ production in differentiated adipocytes.
As a further cause of metabolic stress, we took into consideration the factors that can alter the mitochondrial function, and we focused on the uncoupling properties of fatty acids. The transfer of fatty acids through the mitochondrial membrane can proceed by a flip-flop mechanisms or can be due to a uniport mediated by anion carriers; in both cases, the consequent release of protons into the mitochondrial matrix (acidification) causes a decrease in the protonmotive force, driving uncoupling from ATP synthesis, and the uncoupling can be observed experimentally upon the fatty acid addition to mitochondria [55,56]. Uncoupling, like hypoxia, can be considered a stress stimulus that lowers energy levels due to impairment of mitochondrial respiration. Accordingly, the concentrated lipid mix used in this study induced a deregulation of the mitochondrial bioenergetic system of adipocytes, reproducing a condition of metabolic failure. Interestingly, both hypoxia and the fatty acid-driven uncoupling induced TGFβ synthesis and secretion. This evidence is a novel and exciting discovery because the influence of mitochondrial dysmetabolism on cytokine production had never been investigated before and sheds a new light on the association between obesity, dysmetabolism and inflammation.
In this proof-of-concept study, we established an adipose cell model that could be stressed and forced to produce TGFβ; however, in vivo, the interaction with the microenvironment is necessary to control TGFβ activity. For instance, the activation of TGFβ may be regulated locally at the tumor site by proteinases such as plasmin secreted by tumor and stromal cells [59], moreover it has been reported that the interaction with integrins [73] and the extracellular tumoral acidosis [58] are necessary to TGFβ maturation. Since our simplified cellular model missed the microenvironment responsible for TGFβ activation, it was necessary to evaluate the activity of the secreted cytokine. We chose to analyze the VDR expression as the readout of TGFβ activity, based on our previously published observations that synthetic TGFβ induces VDR and modulates mitochondrial activity of human pancreatic cancer cells [29]. Interestingly, we realized that in our experimental model TGFβ was secreted in its latent form, and a transient acidification was necessary to activate the cytokine. These results are in agreement with the evidence that the tumoral acidic environment is fundamental for the activation of latent TGFβ, possibly together with other activating mechanisms, and can lead to an enhanced response to the cytokine [60].
The novelty of this study lies in the observation that mitochondrial dysfunction and hypoxia can induce TGFβ secretion. Some hypotheses can be formulated to explain the influence of mitochondrial failure and hypoxic stress on TGFβ gene transcription. We believe that oxidative stress generated by ROS could be the main mediator of TGFβ modulation. ROS are produced by the aberrant function of mitochondrial complexes during hypoxic and metabolic uncoupling stress, and can control TGFβ expression [57,74] and hypoxia-inducible factor HIF-1α activity [75], which is known to mediate the TGFβ-dependent pathways [76]. Indeed, the increased levels of ROS can induce the transcription of TGF-β and stimulate its release and activation [77]. In turn, ROS production is also enhanced by HIF-1α stabilized in hypoxic condition and by TGFβ, in a feed-forward loop that supports cancer progression [78]. Future studies will investigate the molecular mechanisms involved in TGFβ induction.
In the second part of this study, the conditioned medium that contained a fair amount of TGFβ was used on human pancreatic cancer cells to verify that the cytokine secreted by dysmetabolic adipocytes exerted the metabolic and pro-cancerogenic effects previously characterized when commercial TGFβ was studied [29]. Indeed, after this treatment the tumor cells increased the transcription of the elements of mitochondrial respiration, and EMT and cellular migration were enhanced. This investigation can be considered a novel proof-of-concept study in which we tested an in vitro model that could mimic the causes and the effects of an inflammatory condition occurring in vivo; for example, in the tumor microenvironment. In fact, the metabolic failure that could affect every tissue could trigger TGFβ synthesis and secretion, particularly in adipocytes; in response to metabolic impairment, the cytokine would stimulate adipocytes and neighbor cells by autocrine and paracrine activity, to reinforce energy metabolism, contain the dysmetabolic energy loss and sustain cell survival, in a positive feedback loop. Unfortunately, the pro-tumorigenic activity of TGFβ would also support cancer progression and spreading. This model is depicted in Figure 5.
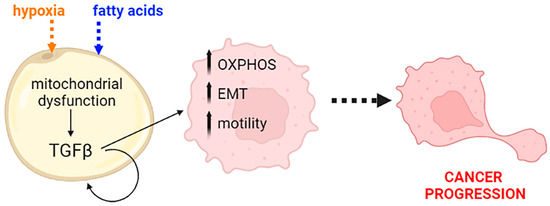
Figure 5.
A schematic representation of the role of TGFβ as a response to mitochondrial dysfunction. The cytokine produced by dysmetabolic adipocytes exerts its stimulatory activity on pancreatic transformed cells and enhances cancer progression by increasing energy availability, reprogramming the cells toward the epithelial–mesenchymal transition (EMT) and inducing migration. OXPHOS: oxidative phosphorylation. Created with BioRender.com.
Some works in the literature [79,80] and our recent article [29] report that TGFβ acts as a stimulant of mitochondrial metabolism and subsequent energy production. From this evidence and the results obtained in this study, we believe that the synthesis of this cytokine can be considered an adaptive response to signals of dysmetabolism or mitochondrial deregulation, which lead to an energy deficiency; in these conditions, TGFβ stimulates the cells to overcome these difficulties by increasing the activity of the respiratory chain and oxidative phosphorylation.
This study lays the foundations for a further investigation of the role of inflamed adipose tissue as a predisposing factor to carcinogenesis. The signaling of TGFβ is a crucial regulator of intestinal homeostasis and inflammation; therefore, dysregulation of this pathway could be associated with inflammatory carcinogenesis of the gastrointestinal tract [81]. In the study of Hertzer et al. carried out in the KrasG12D conditional mouse model of pancreatic cancer, a high-fat diet generated an inflammatory environment at the level of peri-pancreatic fat, suggesting that an initial inflammation of adipose tissue may extend to nearby organs, through the secretion of inflammatory cytokines [69]. Measuring cytokine levels at the tumor site in vivo is complex and often the tumor cells themselves are resistant to its activity; it is plausible that the final effect of the cytokine in vivo could be comparable to that observed in vitro at low doses. Dimeloe et al. have quantified the amounts of TGFβ in 11 tumor effusions and reported a concentration range between 0.5 and 2 ng/mL [82]. Similarly to previous studies in which the cytokine was used at low doses of 1 ng/mL [61,62], in our experimental setting, the conditioned medium of the dysmetabolic adipocytes was used on pancreatic cancer at a concentration of TGFβ corresponding to 0.35 ng/mL. Although from this study we can draw some conclusions on TGFβ secretion and activity, we cannot exclude the presence of other inflammatory factors in the conditioned medium, which would reinforce the metabolic and pro-tumoral effects of TGFβ measured in our model. In fact, in the assays carried out in pancreatic cancer cell lines, we observed that the conditioned medium was more effective than the treatment with recombinant TGFβ. Further research is necessary to explore in more detail the cellular response to metabolic stress.
Based on the presented evidence, this proof-of-concept study has the merit of setting a simplified in vitro model of adipocytes sensitive to metabolic stress, together with a human cancer model that can be tested to verify the consequences of chronic inflammation. In fact, the prolonged (8 days) exposure to these low levels of the cytokine(s) was very efficient in potentiating the mitochondrial activity that favored tumor progression, evaluated as epithelial–mesenchymal transition and cell migration. Therefore, the approach of studying in vitro the long-term effects of low doses of TGFβ secreted by dysmetabolic adipocytes has the advantage of evaluating the results of a persistent stimulation similar to what is originated in vivo by the chronic inflammation of adipose tissue.
Considering the extensive fibro-inflammatory stroma surrounding pancreatic cancer, and the abundant infiltrating fat mass, the investigation of the mechanisms triggering TGFβ secretion could support new therapeutic and preventive approaches to fight pancreatic cancer.
5. Conclusions
This proof-of-concept research unravels the importance of metabolic stress as a strong inducer of TGFβ production. For the first time, we set an in vitro model of differentiated adipocytes that responded to mitochondrial metabolic failure triggered by hypoxia or by a lipid mix by secreting the inflammatory cytokine; moreover, we verified that the pro-metastatic activity of the cytokine was associated with enhanced respiratory activity, tumor progression and migration of a pancreatic cancer model.
Because we tested a combined model that could mimic the causes and the effects of an inflammatory condition occurring in vivo, this work establishes a new paradigm for the metabolic role of TGFβ and paves the way to further investigation on inflamed adipose tissue as a predisposing factor to carcinogenesis.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/app15084300/s1, Supplementary Methods: Western blotting analysis; Figure S1: The experimental flowchart; Figure S2: Picture of proliferating pre-adipocytes (A) and differentiated adipocytes (B) taken with a digital camera under bright field illumination; Figure S3: Western blotting image source for adipocyte cells shown in Figure 1C; Figure S4: Western blotting image source for PANC-1 cells shown in Figure 2B,C; Figure S5: morphological changes of pancreatic cancer cells.
Author Contributions
Conceptualization, F.S.; methodology, F.S. and I.G.; validation, F.S., A.A. and L.B.; formal analysis, F.S.; investigation, A.A., L.B., E.V. and I.G.; resources, I.G. and L.B.; data curation, F.S.; writing—original draft preparation, A.A. and F.S.; writing—review and editing, A.A., L.B., I.G., E.V. and F.S.; visualization, F.S.; supervision, F.S.; project administration, L.B.; funding acquisition, F.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Ministero dell’Istruzione Università e Ricerca.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chouchani, E.T.; Kajimura, S. Metabolic Adaptation and Maladaptation in Adipose Tissue. Nat. Metab. 2019, 1, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, S.; Jokinen, R.; Rissanen, A.; Pietiläinen, K.H. White Adipose Tissue Mitochondrial Metabolism in Health and in Obesity. Obes. Rev. 2020, 21, e12958. [Google Scholar] [CrossRef] [PubMed]
- Heilbronn, L.K.; Gan, S.K.; Turner, N.; Campbell, L.V.; Chisholm, D.J. Markers of Mitochondrial Biogenesis and Metabolism Are Lower in Overweight and Obese Insulin-Resistant Subjects. J. Clin. Endocrinol. Metab. 2007, 92, 1467–1473. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Lanza, I.R.; Swain, J.M.; Sarr, M.G.; Nair, K.S.; Jensen, M.D. Adipocyte Mitochondrial Function Is Reduced in Human Obesity Independent of Fat Cell Size. J. Clin. Endocrinol. Metab. 2014, 99, E209–E216. [Google Scholar] [CrossRef]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in Inflammation and Metabolic Disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef]
- Trayhurn, P.; Wood, I.S. Adipokines: Inflammation and the Pleiotropic Role of White Adipose Tissue. Br. J. Nutr. 2004, 92, 347–355. [Google Scholar] [CrossRef]
- Lee, M.-J. Transforming Growth Factor Beta Superfamily Regulation of Adipose Tissue Biology in Obesity. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2018, 1864, 1160–1171. [Google Scholar] [CrossRef]
- Alessi, M.C.; Bastelica, D.; Morange, P.; Berthet, B.; Leduc, I.; Verdier, M.; Geel, O.; Juhan-Vague, I. Plasminogen Activator Inhibitor 1, Transforming Growth Factor-Beta1, and BMI Are Closely Associated in Human Adipose Tissue during Morbid Obesity. Diabetes 2000, 49, 1374–1380. [Google Scholar] [CrossRef]
- Yadav, H.; Quijano, C.; Kamaraju, A.K.; Gavrilova, O.; Malek, R.; Chen, W.; Zerfas, P.; Zhigang, D.; Wright, E.C.; Stuelten, C.; et al. Protection from Obesity and Diabetes by Blockade of TGF-β/Smad3 Signaling. Cell Metab. 2011, 14, 67–79. [Google Scholar] [CrossRef]
- Fain, J.N.; Tichansky, D.S.; Madan, A.K. Transforming Growth Factor Β1 Release by Human Adipose Tissue Is Enhanced in Obesity. Metabolism 2005, 54, 1546–1551. [Google Scholar] [CrossRef]
- Samad, F.; Yamamoto, K.; Pandey, M.; David, J. Loskutoff Elevated Expression of Transforming Growth Factor–b in Adipose Tissue from Obese Mice. Mol. Med. 1997, 3, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Lysaght, J.; van der Stok, E.P.; Allott, E.H.; Casey, R.; Donohoe, C.L.; Howard, J.M.; McGarrigle, S.A.; Ravi, N.; Reynolds, J.V.; Pidgeon, G.P. Pro-Inflammatory and Tumour Proliferative Properties of Excess Visceral Adipose Tissue. Cancer Lett. 2011, 312, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Vongsuvanh, R.; George, J.; Qiao, L.; van der Poorten, D. Visceral Adiposity in Gastrointestinal and Hepatic Carcinogenesis. Cancer Lett. 2013, 330, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Brocco, D.; Florio, R.; De Lellis, L.; Veschi, S.; Grassadonia, A.; Tinari, N.; Cama, A. The Role of Dysfunctional Adipose Tissue in Pancreatic Cancer: A Molecular Perspective. Cancers 2020, 12, 1849. [Google Scholar] [CrossRef]
- Eibl, G.; Rozengurt, E. Obesity and Pancreatic Cancer: Insight into Mechanisms. Cancers 2021, 13, 5067. [Google Scholar] [CrossRef]
- Xu, M.; Jung, X.; Hines, O.J.; Eibl, G.; Chen, Y. Obesity and Pancreatic Cancer: Overview of Epidemiology and Potential Prevention by Weight Loss. Pancreas 2018, 47, 158–162. [Google Scholar] [CrossRef]
- Li, D. Body Mass Index and Risk, Age of Onset, and Survival in Patients with Pancreatic Cancer. JAMA 2009, 301, 2553. [Google Scholar] [CrossRef]
- Park, W.; Chawla, A.; O’Reilly, E.M. Pancreatic Cancer: A Review. JAMA 2021, 326, 851–862. [Google Scholar] [CrossRef]
- Kendall, R.T.; Feghali-Bostwick, C.A. Fibroblasts in Fibrosis: Novel Roles and Mediators. Front. Pharmacol. 2014, 5, 123. [Google Scholar] [CrossRef]
- Chung, J.Y.-F.; Chan, M.K.-K.; Li, J.S.-F.; Chan, A.S.-W.; Tang, P.C.-T.; Leung, K.-T.; To, K.-F.; Lan, H.-Y.; Tang, P.M.-K. TGF-β Signaling: From Tissue Fibrosis to Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 7575. [Google Scholar] [CrossRef]
- Liu, S.; Ren, J.; ten Dijke, P. Targeting TGFβ Signal Transduction for Cancer Therapy. Signal Transduct. Target. Ther. 2021, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-H.; Eibl, G. Obesity-Induced Adipose Tissue Inflammation as a Strong Promotional Factor for Pancreatic Ductal Adenocarcinoma. Cells 2019, 8, 673. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. Inflammation and Tumor Progression: Signaling Pathways and Targeted Intervention. Signal Transduct. Target. Ther. 2021, 6, 263. [Google Scholar] [CrossRef] [PubMed]
- Lovisa, S. Epithelial-to-Mesenchymal Transition in Fibrosis: Concepts and Targeting Strategies. Front. Pharmacol. 2021, 12, 737570. [Google Scholar] [CrossRef]
- Yang, L.; Roh, Y.S.; Song, J.; Zhang, B.; Liu, C.; Loomba, R.; Seki, E. Transforming Growth Factor Beta Signaling in Hepatocytes Participates in Steatohepatitis through Regulation of Cell Death and Lipid Metabolism in Mice. Hepatology 2014, 59, 483–495. [Google Scholar] [CrossRef]
- Ricca, C.; Aillon, A.; Viano, M.; Bergandi, L.; Aldieri, E.; Silvagno, F. Vitamin D Inhibits the Epithelial-Mesenchymal Transition by a Negative Feedback Regulation of TGF-β Activity. J. Steroid Biochem. Mol. Biol. 2019, 187, 97–105. [Google Scholar] [CrossRef]
- Soukupova, J.; Malfettone, A.; Bertran, E.; Hernández-Alvarez, M.I.; Peñuelas-Haro, I.; Dituri, F.; Giannelli, G.; Zorzano, A.; Fabregat, I. Epithelial–Mesenchymal Transition (EMT) Induced by TGF-β in Hepatocellular Carcinoma Cells Reprograms Lipid Metabolism. Int. J. Mol. Sci. 2021, 22, 5543. [Google Scholar] [CrossRef]
- Liu, Q.-Q.; Huo, H.-Y.; Ao, S.; Liu, T.; Yang, L.; Fei, Z.-Y.; Zhang, Z.-Q.; Ding, L.; Cui, Q.-H.; Lin, J.; et al. TGF-β1-induced Epithelial-mesenchymal Transition Increases Fatty Acid Oxidation and OXPHOS Activity via the p-AMPK Pathway in Breast Cancer Cells. Oncol. Rep. 2020, 44, 1206–1215. [Google Scholar] [CrossRef]
- Fiz, C.; Apprato, G.; Ricca, C.; Aillon, A.; Bergandi, L.; Silvagno, F. TGF Beta Induces Vitamin D Receptor and Modulates Mitochondrial Activity of Human Pancreatic Cancer Cells. Cancers 2021, 13, 2932. [Google Scholar] [CrossRef]
- Liu, H.; Chen, Y.-G. The Interplay Between TGF-β Signaling and Cell Metabolism. Front. Cell Dev. Biol. 2022, 10, 846723. [Google Scholar] [CrossRef]
- Sun, Q.; Fang, L.; Tang, X.; Lu, S.; Tamm, M.; Stolz, D.; Roth, M. TGF-β Upregulated Mitochondria Mass through the SMAD2/3→C/EBPβ→PRMT1 Signal Pathway in Primary Human Lung Fibroblasts. J. Immunol. 2019, 202, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Guido, C.; Whitaker-Menezes, D.; Capparelli, C.; Balliet, R.; Lin, Z.; Pestell, R.G.; Howell, A.; Aquila, S.; Andò, S.; Martinez-outschoorn, U.; et al. Metabolic reprogramming of cancer-associated fibroblasts by TGF-beta drives tumor growth: Connecting TGF-beta signaling with "Warburg-like" cancer metabolism and L-lactate production. Cell Cycle 2012, 11, 3019–3035. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.-S.; Lee, J.-H.; Hwang, S.-C.; Choi, K.S.; Yoon, G. TGF Β1 Induces Prolonged Mitochondrial ROS Generation through Decreased Complex IV Activity with Senescent Arrest in Mv1Lu Cells. Oncogene 2005, 24, 1895–1903. [Google Scholar] [CrossRef]
- Rahimi, N.; Tremblay, E.; McAdam, L.; Roberts, A.; Elliott, B. Autocrine Secretion of TGF-Β1 and TGF-Β2 by Pre-Adipocytes and Adipocytes: A Potent Negative Regulator of Adipocyte Differentiation and Proliferation of Mammary Carcinoma Cells. In Vitro Cell. Dev. Biol.-Anim. 1998, 34, 412–420. [Google Scholar] [CrossRef]
- Hosogai, N.; Fukuhara, A.; Oshima, K.; Miyata, Y.; Tanaka, S.; Segawa, K.; Furukawa, S.; Tochino, Y.; Komuro, R.; Matsuda, M.; et al. Adipose Tissue Hypoxia in Obesity and Its Impact on Adipocytokine Dysregulation. Diabetes 2007, 56, 901–911. [Google Scholar] [CrossRef]
- Luo, H.; Guo, Y.; Liu, Y.; Wang, Y.; Zheng, R.; Ban, Y.; Peng, L.; Yuan, Q.; Liu, W. Growth Differentiation Factor 11 Inhibits Adipogenic Differentiation by Activating TGF-Beta/Smad Signalling Pathway. Cell Prolif. 2019, 52, e12631. [Google Scholar] [CrossRef]
- Guerrero, J.; Tobar, N.; Cáceres, M.; Espinoza, L.; Escobar, P.; Dotor, J.; Smith, P.C.; Martínez, J. Soluble Factors Derived from Tumor Mammary Cell Lines Induce a Stromal Mammary Adipose Reversion in Human and Mice Adipose Cells. Possible Role of TGF-Β1 and TNF-α. Breast Cancer Res. Treat. 2010, 119, 497–508. [Google Scholar] [CrossRef]
- Peeraully, M.R.; Jenkins, J.R.; Trayhurn, P. NGF Gene Expression and Secretion in White Adipose Tissue: Regulation in 3T3-L1 Adipocytes by Hormones and Inflammatory Cytokines. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E331–E339. [Google Scholar] [CrossRef]
- Ding, Q.; Mracek, T.; Gonzalez-Muniesa, P.; Kos, K.; Wilding, J.; Trayhurn, P.; Bing, C. Identification of Macrophage Inhibitory Cytokine-1 in Adipose Tissue and Its Secretion as an Adipokine by Human Adipocytes. Endocrinology 2009, 150, 1688–1696. [Google Scholar] [CrossRef]
- Johnston, P.G.; Rondinone, C.M.; Voeller, D.; Allegra, C.J. Identification of a Protein Factor Secreted by 3T3-L1 Preadipocytes Inhibitory for the Human MCF-7 Breast Cancer Cell Line. Cancer Res. 1992, 52, 6860–6865. [Google Scholar]
- Yum, C.; Andolino, C.; Larrick, B.; Sheeley, M.P.; Teegarden, D. 1α,25-Dihydroxyvitamin D Downregulates Adipocyte Impact on Breast Cancer Cell Migration and Adipokine Release. Nutrients 2024, 16, 3153. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, I.; Suchý, T.; Strnadová, M.; Thor, D. Qualitative and Quantitative Analysis of Lipid Droplets in Mature 3T3-L1 Adipocytes Using Oil Red O. STAR Protoc. 2024, 5, 102977. [Google Scholar] [CrossRef] [PubMed]
- Weiszenstein, M.; Musutova, M.; Plihalova, A.; Westlake, K.; Elkalaf, M.; Koc, M.; Prochazka, A.; Pala, J.; Gulati, S.; Trnka, J.; et al. Adipogenesis, Lipogenesis and Lipolysis Is Stimulated by Mild but Not Severe Hypoxia in 3T3-L1 Cells. Biochem. Biophys. Res. Commun. 2016, 478, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Constam, D.B.; Philipp, J.; Malipiero, U.V.; ten Dijke, P.; Schachner, M.; Fontana, A. Differential Expression of Transforming Growth Factor-Beta 1, -Beta 2, and -Beta 3 by Glioblastoma Cells, Astrocytes, and Microglia. J. Immunol. 1992, 148, 1404–1410. [Google Scholar] [CrossRef]
- Silvagno, F.; Consiglio, M.; Foglizzo, V.; Destefanis, M.; Pescarmona, G. Mitochondrial Translocation of Vitamin D Receptor Is Mediated by the Permeability Transition Pore in Human Keratinocyte Cell Line. PLoS ONE 2013, 8, e54716. [Google Scholar] [CrossRef]
- Consiglio, M.; Destefanis, M.; Morena, D.; Foglizzo, V.; Forneris, M.; Pescarmona, G.; Silvagno, F. The Vitamin D Receptor Inhibits the Respiratory Chain, Contributing to the Metabolic Switch That Is Essential for Cancer Cell Proliferation. PLoS ONE 2014, 9, e115816. [Google Scholar] [CrossRef]
- Ricca, C.; Aillon, A.; Bergandi, L.; Alotto, D.; Castagnoli, C.; Silvagno, F. Vitamin D Receptor Is Necessary for Mitochondrial Function and Cell Health. Int. J. Mol. Sci. 2018, 19, 1672. [Google Scholar] [CrossRef]
- Bergandi, L.; Lucia, U.; Grisolia, G.; Granata, R.; Gesmundo, I.; Ponzetto, A.; Paolucci, E.; Borchiellini, R.; Ghigo, E.; Silvagno, F. The Extremely Low Frequency Electromagnetic Stimulation Selective for Cancer Cells Elicits Growth Arrest through a Metabolic Shift. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1389–1397. [Google Scholar] [CrossRef]
- Bergandi, L.; Flutto, T.; Valentini, S.; Thedy, L.; Pramotton, R.; Zenato, S.; Silvagno, F. Whey Derivatives and Galactooligosaccharides Stimulate the Wound Healing and the Function of Human Keratinocytes through the NF-kB and FOXO-1 Signaling Pathways. Nutrients 2022, 14, 2888. [Google Scholar] [CrossRef]
- Turini, S.; Bergandi, L.; Gazzano, E.; Prato, M.; Aldieri, E. Epithelial to Mesenchymal Transition in Human Mesothelial Cells Exposed to Asbestos Fibers: Role of TGF-β as Mediator of Malignant Mesothelioma Development or Metastasis via EMT Event. Int. J. Mol. Sci. 2019, 20, 150. [Google Scholar] [CrossRef]
- Morrison, S.; McGee, S.L. 3T3-L1 Adipocytes Display Phenotypic Characteristics of Multiple Adipocyte Lineages. Adipocyte 2015, 4, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Ducluzeau, P.-H.; Priou, M.; Weitheimer, M.; Flamment, M.; Duluc, L.; Iacobazi, F.; Soleti, R.; Simard, G.; Durand, A.; Rieusset, J.; et al. Dynamic Regulation of Mitochondrial Network and Oxidative Functions during 3T3-L1 Fat Cell Differentiation. J. Physiol. Biochem. 2011, 67, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, B.; Smith, U. Cytokines Promote Wnt Signaling and Inflammation and Impair the Normal Differentiation and Lipid Accumulation in 3T3-L1 Preadipocytes. J. Biol. Chem. 2006, 281, 9507–9516. [Google Scholar] [CrossRef] [PubMed]
- Lively, S.; Lam, D.; Wong, R.; Schlichter, L.C. Comparing Effects of Transforming Growth Factor Β1 on Microglia From Rat and Mouse: Transcriptional Profiles and Potassium Channels. Front. Cell. Neurosci. 2018, 12, 115. [Google Scholar] [CrossRef]
- Skulachev, V.P. Fatty Acid Circuit as a Physiological Mechanism of Uncoupling of Oxidative Phosphorylation. FEBS Lett. 1991, 294, 158–162. [Google Scholar] [CrossRef]
- Ježek, P.; Engstová, H.; Žáčková, M.; Vercesi, A.E.; Costa, A.D.T.; Arruda, P.; Garlid, K.D. Fatty Acid Cycling Mechanism and Mitochondrial Uncoupling Proteins. Biochim. Biophys. Acta BBA Bioenerg. 1998, 1365, 319–327. [Google Scholar] [CrossRef]
- Annes, J.P.; Munger, J.S.; Rifkin, D.B. Making Sense of Latent TGFβ Activation. J. Cell Sci. 2003, 116, 217–224. [Google Scholar] [CrossRef]
- Ribeiro, S.M.F.; Poczatek, M.; Schultz-Cherry, S.; Villain, M.; Murphy-Ullrich, J.E. The Activation Sequence of Thrombospondin-1 Interacts with the Latency-Associated Peptide to Regulate Activation of Latent Transforming Growth Factor-β. J. Biol. Chem. 1999, 274, 13586–13593. [Google Scholar] [CrossRef]
- Munger, J.S.; Harpel, J.G.; Gleizes, P.-E.; Mazzieri, R.; Nunes, I.; Rifkin, D.B. Latent Transforming Growth Factor-β: Structural Features and Mechanisms of Activation. Kidney Int. 1997, 51, 1376–1382. [Google Scholar] [CrossRef]
- Jullien, P.; Berg, T.M.; Lawrence, D.A. Acidic Cellular Environments: Activation of Latent Tgf-β and Sensitization of Cellular Responses to Tgf-β and Egf. Int. J. Cancer 1989, 43, 886–891. [Google Scholar] [CrossRef]
- Guo, Q. Changes in Mitochondrial Function during EMT Induced by TGFβ-1 in Pancreatic Cancer. Oncol. Lett. 2017, 13, 1575–1580. [Google Scholar] [CrossRef] [PubMed]
- Katsuno, Y.; Meyer, D.S.; Zhang, Z.; Shokat, K.M.; Akhurst, R.J.; Miyazono, K.; Derynck, R. Chronic TGF-β Exposure Drives Stabilized EMT, Tumor Stemness, and Cancer Drug Resistance with Vulnerability to Bitopic mTOR Inhibition. Sci. Signal. 2019, 12, eaau8544. [Google Scholar] [CrossRef] [PubMed]
- Blüher, M. Adipose Tissue Dysfunction in Obesity. Exp. Clin. Endocrinol. Diabetes 2009, 117, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Schöttl, T.; Kappler, L.; Fromme, T.; Klingenspor, M. Limited OXPHOS Capacity in White Adipocytes Is a Hallmark of Obesity in Laboratory Mice Irrespective of the Glucose Tolerance Status. Mol. Metab. 2015, 4, 631–642. [Google Scholar] [CrossRef]
- Wang, P.-W.; Kuo, H.-M.; Huang, H.-T.; Chang, A.Y.W.; Weng, S.-W.; Tai, M.-H.; Chuang, J.-H.; Chen, I.-Y.; Huang, S.-C.; Lin, T.-K.; et al. Biphasic Response of Mitochondrial Biogenesis to Oxidative Stress in Visceral Fat of Diet-Induced Obesity Mice. Antioxid. Redox Signal. 2014, 20, 2572–2588. [Google Scholar] [CrossRef]
- Schöttl, T.; Pachl, F.; Giesbertz, P.; Daniel, H.; Kuster, B.; Fromme, T.; Klingenspor, M. Proteomic and Metabolite Profiling Reveals Profound Structural and Metabolic Reorganization of Adipocyte Mitochondria in Obesity. Obesity 2020, 28, 590–600. [Google Scholar] [CrossRef]
- Dawson, D.W.; Hertzer, K.; Moro, A.; Donald, G.; Chang, H.-H.; Go, V.L.; Pandol, S.J.; Lugea, A.; Gukovskaya, A.S.; Li, G.; et al. High-Fat, High-Calorie Diet Promotes Early Pancreatic Neoplasia in the Conditional KrasG12D Mouse Model. Cancer Prev. Res. 2013, 6, 1064–1073. [Google Scholar] [CrossRef]
- Khasawneh, J.; Schulz, M.D.; Walch, A.; Rozman, J.; Hrabe de Angelis, M.; Klingenspor, M.; Buck, A.; Schwaiger, M.; Saur, D.; Schmid, R.M.; et al. Inflammation and Mitochondrial Fatty Acid Beta-Oxidation Link Obesity to Early Tumor Promotion. Proc. Natl. Acad. Sci. USA 2009, 106, 3354–3359. [Google Scholar] [CrossRef]
- Hertzer, K.M.; Xu, M.; Moro, A.; Dawson, D.W.; Du, L.; Li, G.; Chang, H.-H.; Stark, A.P.; Jung, X.; Hines, O.J.; et al. Robust Early Inflammation of the Peripancreatic Visceral Adipose Tissue During Diet-Induced Obesity in the KrasG12D Model of Pancreatic Cancer. Pancreas 2016, 45, 458–465. [Google Scholar] [CrossRef]
- Basu, R.K.; Hubchak, S.; Hayashida, T.; Runyan, C.E.; Schumacker, P.T.; Schnaper, H.W. Interdependence of HIF-1α and TGF-β/Smad3 Signaling in Normoxic and Hypoxic Renal Epithelial Cell Collagen Expression. Am. J. Physiol.-Ren. Physiol. 2011, 300, F898–F905. [Google Scholar] [CrossRef]
- Jun, E.K.; Zhang, Q.; Yoon, B.S.; Moon, J.-H.; Lee, G.; Park, G.; Kang, P.J.; Lee, J.H.; Kim, A.; You, S. Hypoxic Conditioned Medium from Human Amniotic Fluid-Derived Mesenchymal Stem Cells Accelerates Skin Wound Healing through TGF-β/SMAD2 and PI3K/Akt Pathways. Int. J. Mol. Sci. 2014, 15, 605–628. [Google Scholar] [CrossRef] [PubMed]
- Copple, B.L. Hypoxia Stimulates Hepatocyte Epithelial to Mesenchymal Transition by Hypoxia-Inducible Factor and Transforming Growth Factor-β-Dependent Mechanisms. Liver Int. 2010, 30, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Chen, R.; Lin, B.; Deng, R.; Liang, Y.; Zeng, J.; Ma, S.; Qiu, X. Cross-Talk between the TGF-β and Cell Adhesion Signaling Pathways in Cancer. Int. J. Med. Sci. 2024, 21, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Rivera, S.; Monclus, E.A.; Synenki, L.; Zirk, A.; Eisenbart, J.; Feghali-Bostwick, C.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial Reactive Oxygen Species Regulate Transforming Growth Factor-β Signaling. J. Biol. Chem. 2013, 288, 770–777. [Google Scholar] [CrossRef]
- Movafagh, S.; Crook, S.; Vo, K. Regulation of Hypoxia-Inducible Factor-1a by Reactive Oxygen Species: New Developments in an Old Debate. J. Cell. Biochem. 2015, 116, 696–703. [Google Scholar] [CrossRef]
- Tirpe, A.A.; Gulei, D.; Ciortea, S.M.; Crivii, C.; Berindan-Neagoe, I. Hypoxia: Overview on Hypoxia-Mediated Mechanisms with a Focus on the Role of HIF Genes. Int. J. Mol. Sci. 2019, 20, 6140. [Google Scholar] [CrossRef]
- Barcellos-Hoff, M.H.; Dix, T.A. Redox-Mediated Activation of Latent Transforming Growth Factor-Beta 1. Mol. Endocrinol. 1996, 10, 1077–1083. [Google Scholar] [CrossRef]
- Chung, J.; Huda, M.N.; Shin, Y.; Han, S.; Akter, S.; Kang, I.; Ha, J.; Choe, W.; Choi, T.G.; Kim, S.S. Correlation between Oxidative Stress and Transforming Growth Factor-Beta in Cancers. Int. J. Mol. Sci. 2021, 22, 13181. [Google Scholar] [CrossRef]
- Negmadjanov, U.; Godic, Z.; Rizvi, F.; Emelyanova, L.; Ross, G.; Richards, J.; Holmuhamedov, E.L.; Jahangir, A. TGF-Β1-Mediated Differentiation of Fibroblasts Is Associated with Increased Mitochondrial Content and Cellular Respiration. PLoS ONE 2015, 10, e0123046. [Google Scholar] [CrossRef]
- Jiang, L.; Xiao, L.; Sugiura, H.; Huang, X.; Ali, A.; Kuro-o, M.; Deberardinis, R.J.; Boothman, D.A. Metabolic Reprogramming during TGFβ1-Induced Epithelial-to-Mesenchymal Transition. Oncogene 2015, 34, 3908–3916. [Google Scholar] [CrossRef]
- Hong, S. Connection between Inflammation and Carcinogenesis in Gastrointestinal Tract: Focus on TGF-β Signaling. World J. Gastroenterol. 2010, 16, 2080. [Google Scholar] [CrossRef] [PubMed]
- Dimeloe, S.; Gubser, P.; Loeliger, J.; Frick, C.; Develioglu, L.; Fischer, M.; Marquardsen, F.; Bantug, G.R.; Thommen, D.; Lecoultre, Y.; et al. Tumor-Derived TGF-β Inhibits Mitochondrial Respiration to Suppress IFN-γ Production by Human CD4+ T Cells. Sci. Signal. 2019, 12, eaav3334. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).