
Increasing the Thermostability of Luciferase from Antarctic Krill by Rational Design for Biotechnological Applications


Abstract
Featured Application
Abstract
1. Introduction
2. Materials and Methods
2.1. Homology Modeling and Selection of Mutation Sites
2.2. Plasmid Construction, Expression, and Purification of Luciferase Mutants
2.3. Determination of Luciferase Activity
2.4. Thermostability Analysis
2.5. Michaelis–Menten Kinetics Determination
2.6. Circular Dichroism (CD) Spectroscopy Analysis
3. Results and Discussions
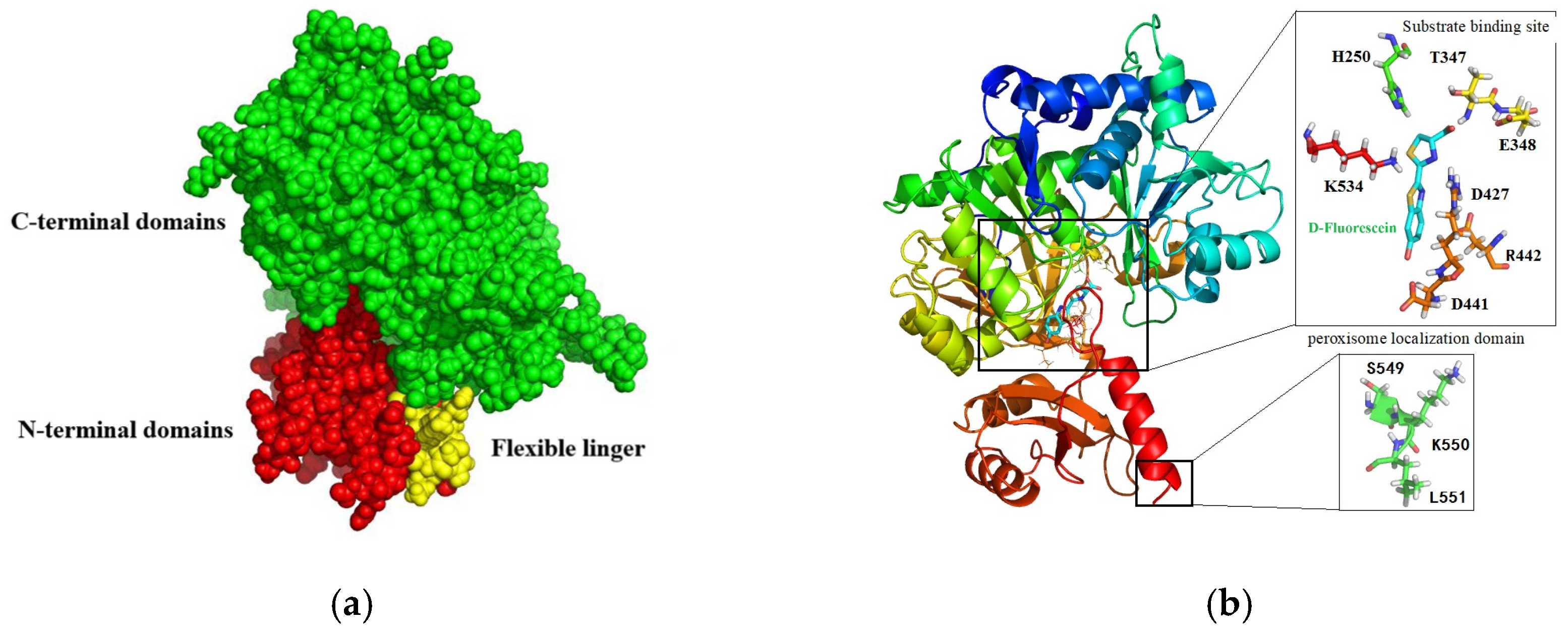
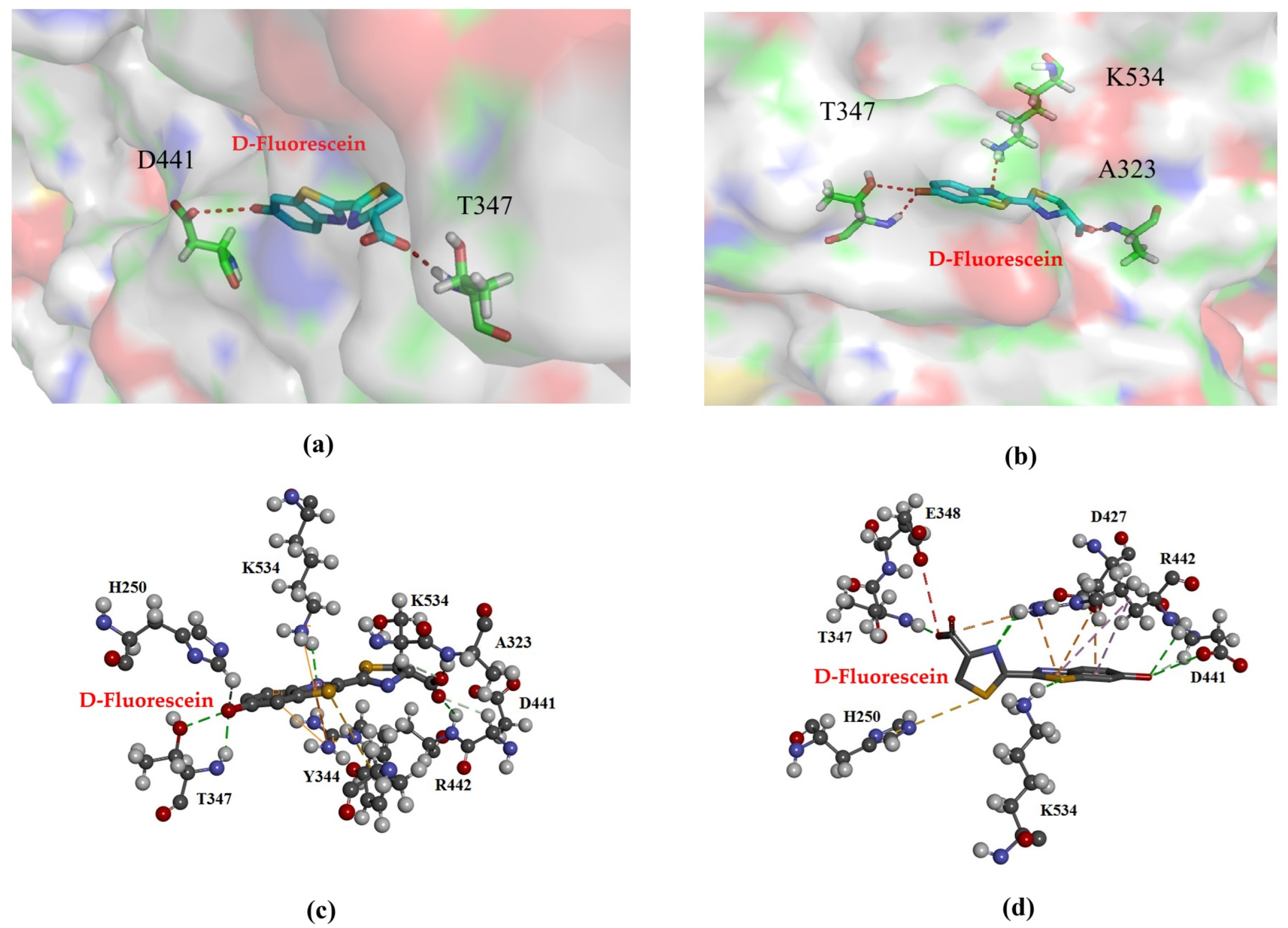
3.1. Homology Modeling
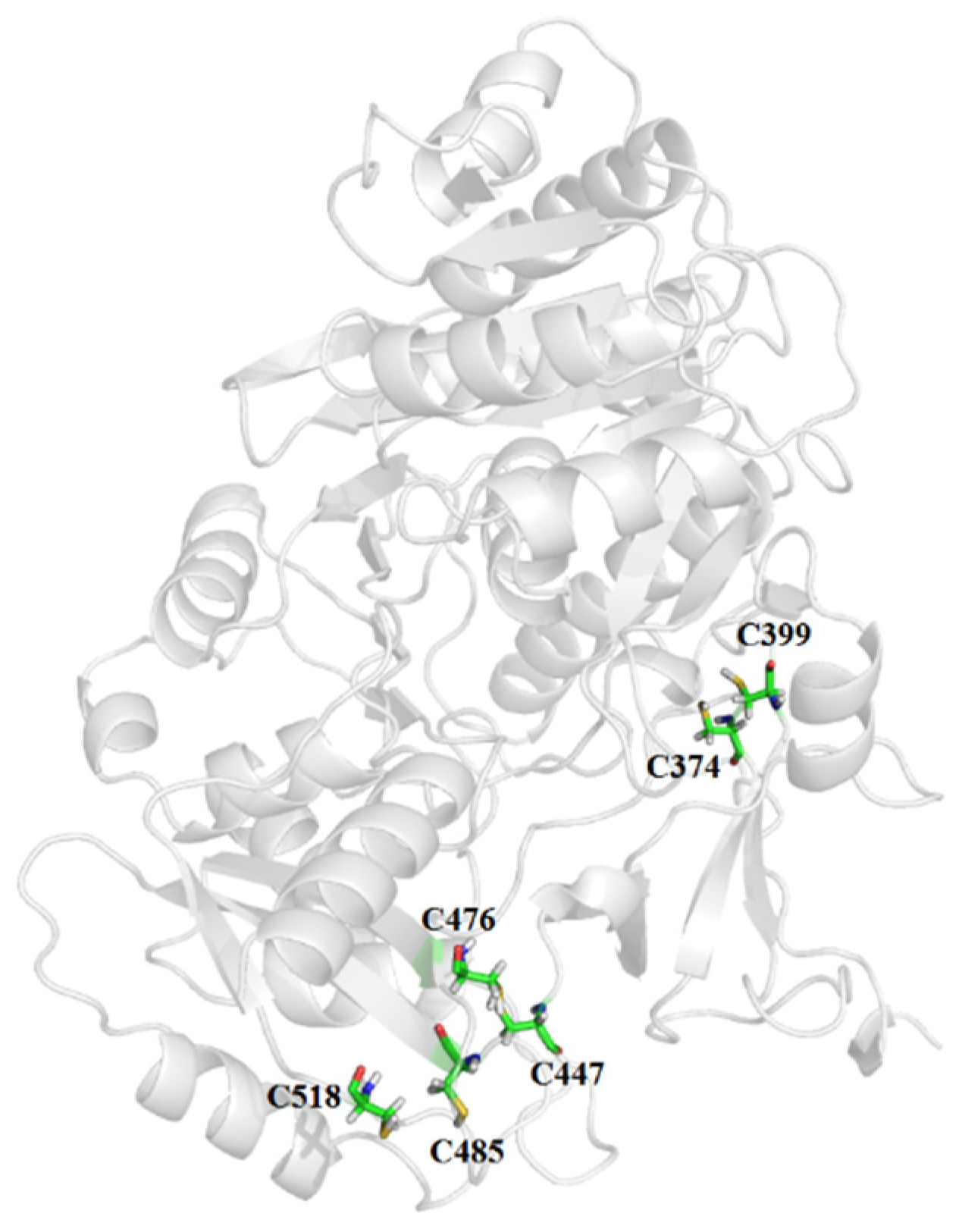
3.2. Construction of Mutants
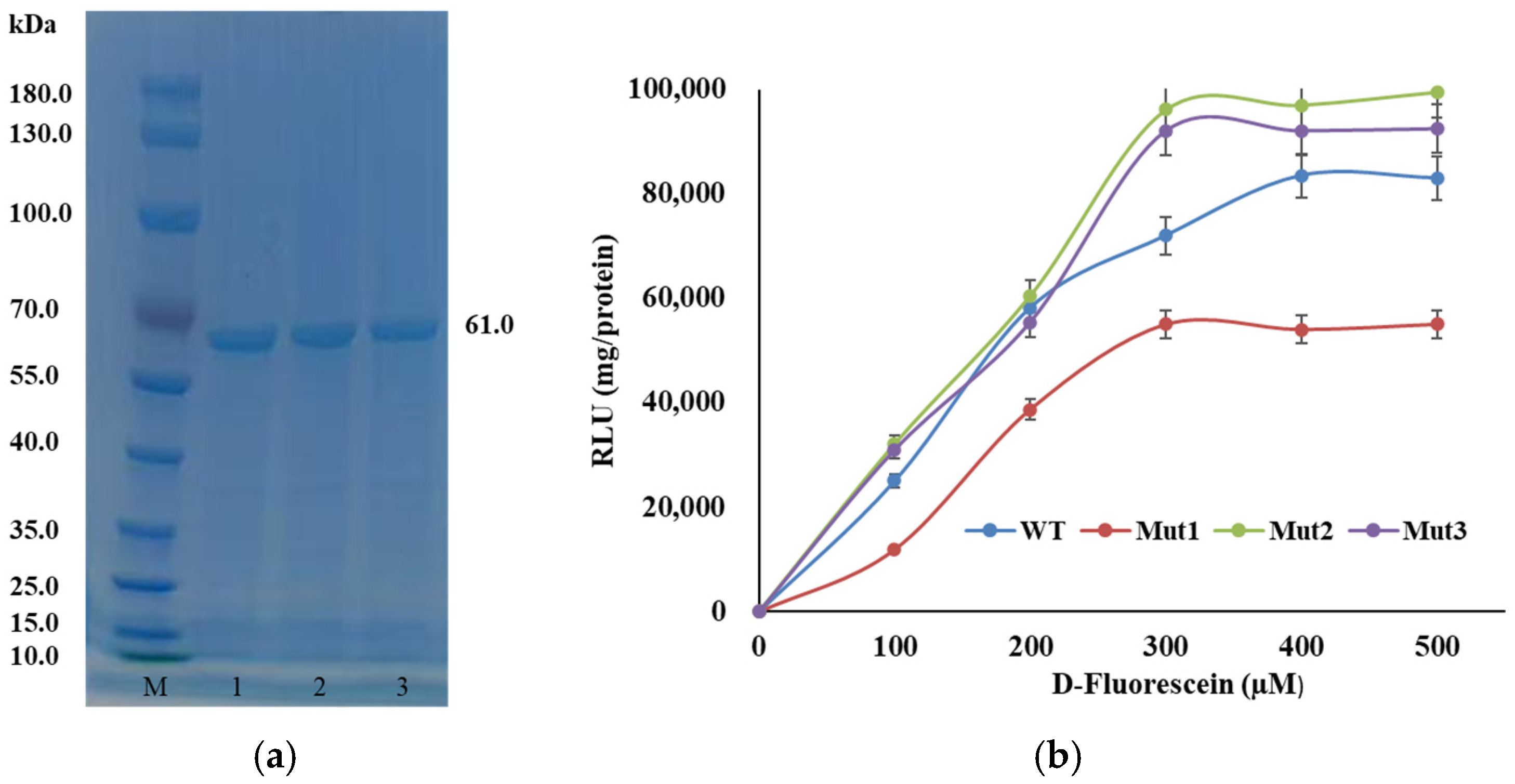
3.3. Plasmid Construction and Expression of Luciferase Mutants
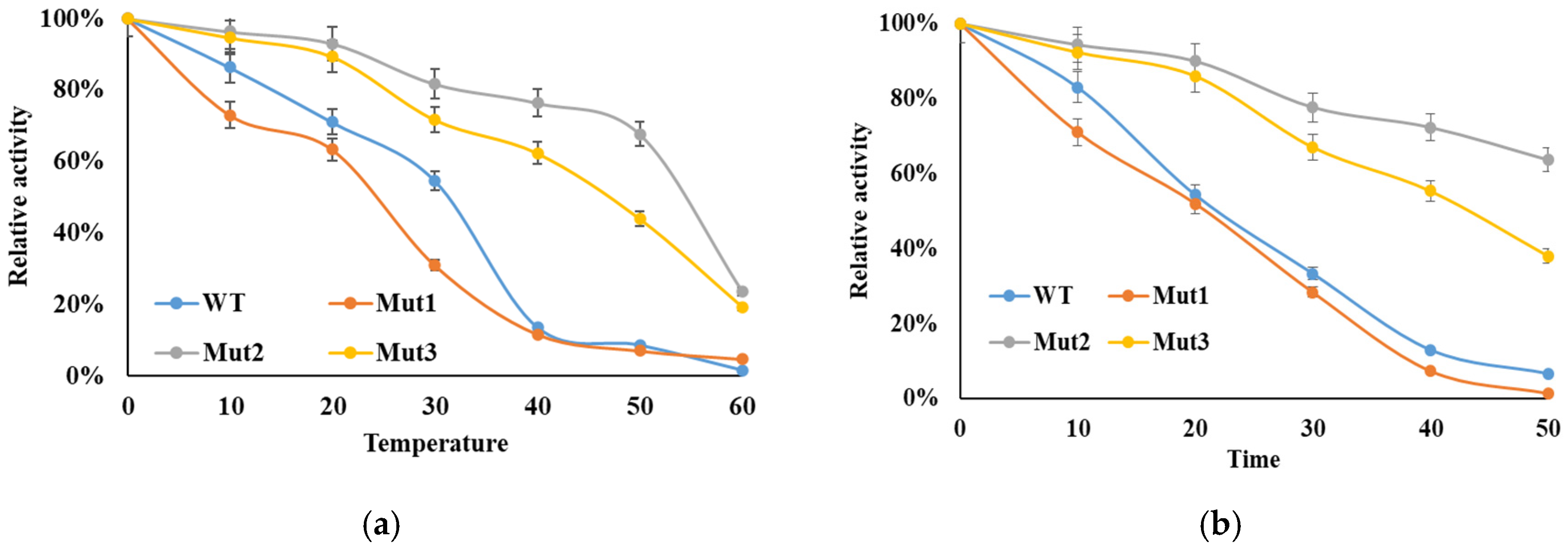
3.4. Enzymatic Thermostability
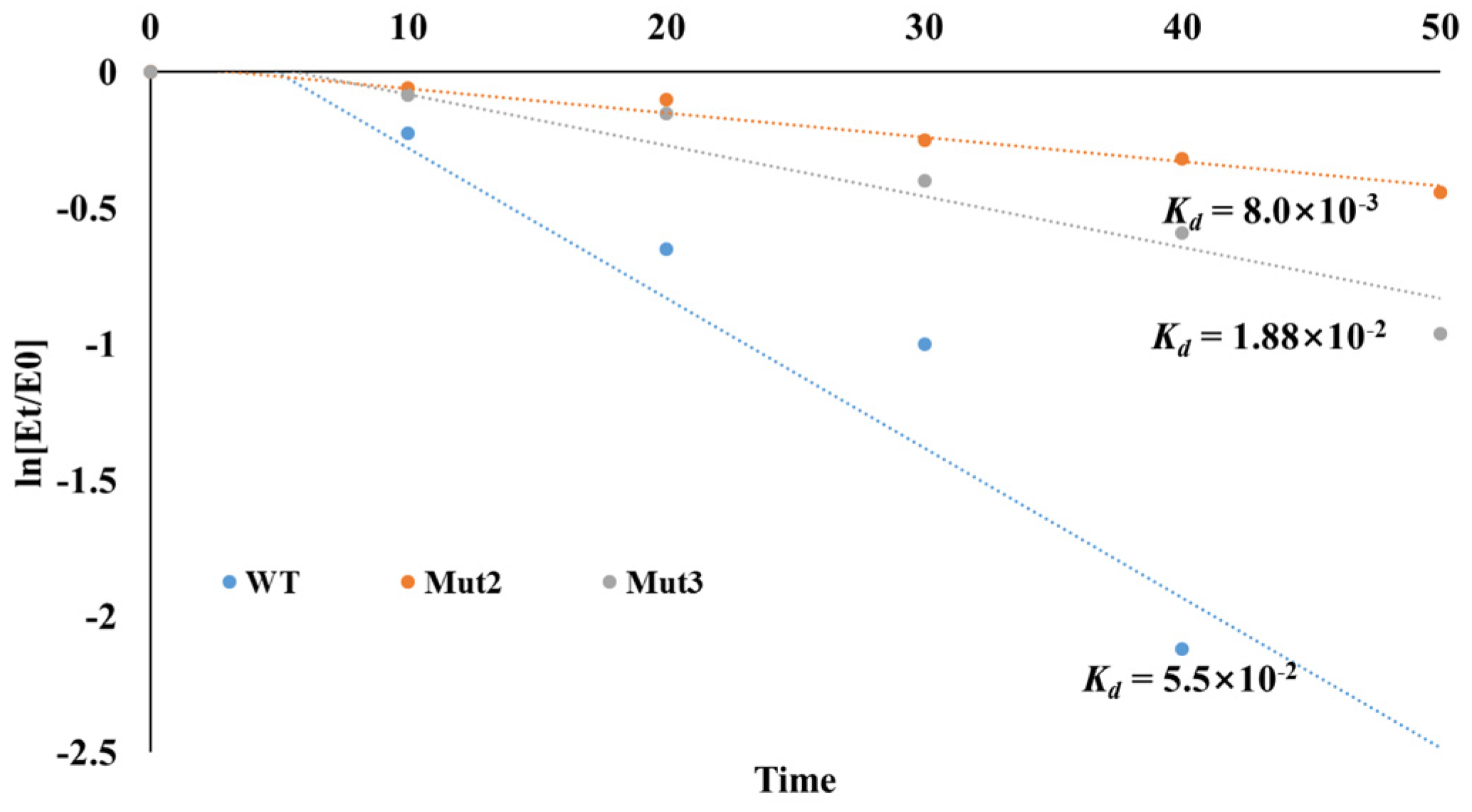
3.5. Thermal Deactivation Analysis
3.6. Kinetic Analysis
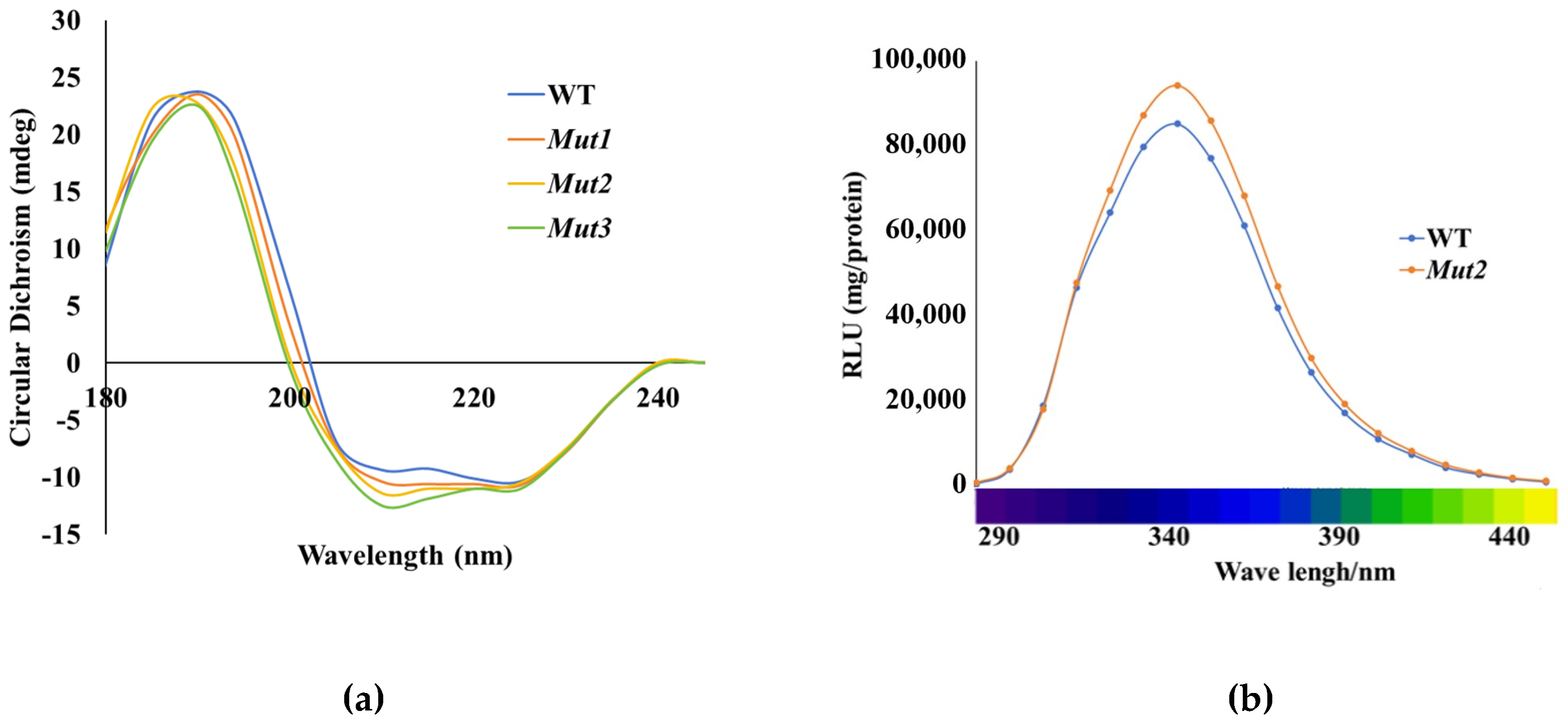
3.7. CD and Luminescence Spectra Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CD | Circular dichroism |
| LAK | Antarctic krill luciferase |
| IPTG | isopropyl-β-D-thiogalactopyranoside |
| WT | wild-type |
References
- Stefan, S.; Al-Handawi, M.B. Mechanically Assisted Bioluminescence with Natural Luciferase. Angew. Chem. Int. Ed. Engl. 2020, 59, 16485–16489. [Google Scholar]
- Kaskova, Z.M.; Tsarkova, A.S. 1001 lights: Luciferins, luciferases, their mechanisms of action and applications in chemical analysis, biology and medicine. Chem. Soc. Rev. 2016, 45, 6048–6077. [Google Scholar]
- Viviani, V.R. The origin, diversity, and structure function relationships of insect luciferases. CMLS Cell. Mol. Life Sci. 2002, 59, 1833–1850. [Google Scholar]
- Contag, C.H.; Spilman, S.D. Visualizing bioluminescent gene expression in 1iving mammals using reporter. Photochem. Photobiol. 1997, 66, 523–531. [Google Scholar] [PubMed]
- Niu, G.; Chen, X. Molecular imaging with activatable reporter systems. Theranostics 2012, 2, 413–423. [Google Scholar] [PubMed]
- Thore, A.; Ansehn, S. Detection of bacteriuria by luciferase assay of adenosine triphosphate. J. Clin. Microbiol. 1975, 1, 1–8. [Google Scholar] [PubMed]
- Alison, M.; Horsburgh, D.P. On-line microbial biosensing and fingerprinting of water pollutants. Biosens. Bioelectron. 2002, 17, 495–501. [Google Scholar]
- Yan, S.L.; Miao, S.N. ATP bioluminescence rapid detection of total viable count in soy sauce. Luminescence 2012, 27, 34–38. [Google Scholar]
- Deb, D.K.; Srivastava, K.K. Bioluminescent rapid vitro and high in Mycobacterium throughput aurum expressing screening of firefly luciferase antimycobacterial infected drugs for in macrophages. Biochem. Biophys. Res. Commun. 2000, 279, 457–461. [Google Scholar]
- Andrey, A.K.; Robert, A.D. Rapid and Sensitive Detection of Retrovirus Entry by Using a Novel Luciferase-Based Content-Mixing Assay. J. Virol. 2004, 78, 5124–5132. [Google Scholar]
- Kocher, B.; Piwnicaworms, D. Illuminating Cancer Systems with Genetically-Engineered Mouse Models and Coupled Luciferase Reporters In Vivo. Cancer Discov. 2013, 3, 616–628. [Google Scholar] [CrossRef]
- Koutsoudakis, G.; Kaul, A. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J. Virol. 2006, 80, 5308–5320. [Google Scholar] [CrossRef] [PubMed]
- Pietschmann, T.; Kaul, A. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. USA 2006, 103, 7408–7413. [Google Scholar] [CrossRef] [PubMed]
- Wood, K.V. The chemical mechanism and evolutionary development of beetle bioluminescence. Photochem. Photobiol. 1995, 62, 662–673. [Google Scholar] [CrossRef]
- Gui-Chun, L.; Ru, Z. Cloning and Characterization of Luciferase from the Chinese Firefly Lamprigera yunnana. Photochem. Photobiol. 2019, 95, 1186–1194. [Google Scholar]
- Tania, P.; Farhima, A. Firefly Luciferase Mutant with Enhanced Activity and Thermostability. ACS Omega 2018, 3, 2628–2633. [Google Scholar]
- Mitani, Y.; Futahashi, R. Tibetan Firefly Luciferase with Low Temperature Adaptation. Photochem. Photobiol. 2017, 93, 466–472. [Google Scholar] [CrossRef]
- Law, G.H.; Gandelman, O.A. Mutagenesis of solvent-exposed amino acids in Photinus pyralis luciferase improves thermostability and pH-tolerance. Biochem. J. 2006, 397, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Changwei, S.; Shuai, S. The enormous repetitive Antarctic krill genome reveals environmental adaptations and population insights. Cell 2023, 186, 1279–1294.e19. [Google Scholar]
- Imani, M.; Hosseinkhani, S. Design and introduction of a disulfide bridge in firefly luciferase: Increase of thermostability and decrease of pH sensitivity. Photochem. Photobiol. Sci. 2010, 9, 1167–1177. [Google Scholar] [CrossRef]
- Eijsink, V.G.H.; Bjørk, A. Rational engineering of enzyme stability. J. Biotechnol. 2004, 113, 105–120. [Google Scholar] [CrossRef]
- Tokurik, N.; Tawfik, D.S. Stability effects of mutations and protein evolvability. Curr. Opin. Struct. Biol. 2009, 19, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Ma, X. Protein cyclization enhanced thermostability and exopeptidase resistanceof green fluorescent protein. J. Microbiol. Biotechnol. 2010, 20, 460–466. [Google Scholar] [PubMed]
- Gilbert, H.F. Protein Disulfide Isomerase and Assisted Protein Folding. J. Biol. Chem. 1997, 272, 29399–29402. [Google Scholar] [CrossRef]
- Mamathambika, B.S.; Bardwell, J.C. Disulfide-linked protein folding pathways. Annu. Rev. Cell Dev. Biol. 2008, 24, 211–235. [Google Scholar] [CrossRef]
- Kaushik, M.; Sinha, P. Protein engineering and de novo designing of a biocatalyst. J. Mol. Recognit. 2016, 29, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Le, Q.A.T.; Joo, J.C. Development of thermostable Candida antarctica lipase b through novel in silico design of disulfide bridge. Biotechnol. Bioeng. 2012, 109, 867–876. [Google Scholar] [CrossRef]
- Hou, Q.Q.; Li, N. Design and regulation of the surface and interfacial behavior of protein molecules. Chin. J. Chem. Eng. 2020, 28, 2837–2847. [Google Scholar] [CrossRef]
- Li, C.; Ban, X. Rational design of disulfide bonds for enhancing the thermostability of the 1,4 α-glucan branching enzyme from Geobacillus thermoglucosidans STB02. J. Agric. Food Chem. 2020, 68, 13791–13797. [Google Scholar] [CrossRef]
- Vogt, G.; Woell, S. Protein thermal stability, hydrogen bonds, and ion pairs. J. Mol. Biol. 1997, 269, 631–643. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | AA Residue | Chi3 a | Energy (kcal/mol) | ΣB-Factor |
|---|---|---|---|---|
| Mut1 | F374C-G399C | 68.48 | 7.25 | 1.86 |
| Mut2 | M447C-G476G | −103.80 | 2.63 | 1.90 |
| Mut3 | A485C-T518C | −82.92 | 4.87 | 1.74 |
| Luciferase | Km (μM) | Kcat (s−1) |
|---|---|---|
| WT | 53.6 | 2.1 |
| Mut2 | 27.2 | 4.2 |
| Mut3 | 88.1 | 2.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Y.; Zheng, Y.; Ji, X.; Sheng, J. Increasing the Thermostability of Luciferase from Antarctic Krill by Rational Design for Biotechnological Applications. Appl. Sci. 2025, 15, 3563. https://doi.org/10.3390/app15073563
Ma Y, Zheng Y, Ji X, Sheng J. Increasing the Thermostability of Luciferase from Antarctic Krill by Rational Design for Biotechnological Applications. Applied Sciences. 2025; 15(7):3563. https://doi.org/10.3390/app15073563
Chicago/Turabian StyleMa, Yuqi, Yuan Zheng, Xiaofeng Ji, and Jun Sheng. 2025. "Increasing the Thermostability of Luciferase from Antarctic Krill by Rational Design for Biotechnological Applications" Applied Sciences 15, no. 7: 3563. https://doi.org/10.3390/app15073563
APA StyleMa, Y., Zheng, Y., Ji, X., & Sheng, J. (2025). Increasing the Thermostability of Luciferase from Antarctic Krill by Rational Design for Biotechnological Applications. Applied Sciences, 15(7), 3563. https://doi.org/10.3390/app15073563