Abstract
This study introduces a novel approach to zinc sintering, showcasing the remarkable potential of the cold sintering process. This innovative technique enables the compaction of zinc at close to room temperatures through the synergistic application of uniaxial pressure and a solvent. The cold sintering process stands out as an environmentally conscious production method, yielding substantial reductions in both energy consumption and harmful emissions. The present research demonstrates the sintering of zinc powder utilizing its oxide surface layer. Operating within temperatures close to room temperature and applying a pressure of 500 MPa, cold sintering occurred already after a holding time of 5 min. To increase the porosity, diverse NaCl concentrations (<60%) were employed and the size range of 1.25–1.6 mm was selected to improve the salt dissolution. To facilitate the sintering procedure, a strategic inclusion of diluted acetic acid (50%) and ethanol was made. This additive duo effectively facilitated the dispersion of acetic acid throughout the sample matrix. In the cold sintering process, processing parameters including solvent, pressure, temperature and holding time play critical roles in zinc densification.
1. Introduction
Metallic foams are a class of porous materials with significant potential for various applications due to their unique combination of properties. Compared to dense materials they have an additional degree of freedom in design—their porosity. They are broadly categorized into two main types based on their pore structure: open-cell foams—these foams have interconnected pores, which allow for the passage of fluids or gases [1]; and closed-cell foams—in this type, the pores are completely sealed from each other, this structure provides high compressive strength and excellent energy absorption [2].
Several methods have been developed to produce metallic foams, each offering distinct advantages in controlling the final foam’s structure and properties. These techniques can be grouped into three main categories: melt foaming—cost-effective but often results in foams with non-uniform pore structures [3,4]; powder metallurgy or the space holder method—this method offers precise control over the foam’s porosity and pore size; and vapor deposition and casting-based methods—they are less common methods that can produce highly specific and intricate foam structures for specialized applications [1].
Unlike other manufacturing techniques, the space holder method produces metallic foam with highly controllable and customizable structures with open pores [2]. This is crucial for optimizing properties like high energy absorption, low density, and enhanced strength-to-weight ratio [5], allowing one to also manufacture structurally graded porous materials. The ability to tailor the pore structure makes them ideal for applications in different areas of engineering like biomedical implants from metals like magnesium or titanium, automotive industries, which mainly employ Al foams, and aerospace industries, which mostly employ Al or Al-steel foams and aluminum-based heat exchangers from Al, Ni, or Cu or fuel cells W, Mo [5].
The space holder method typically involves four main steps: mixing—metal powder is mixed with a predetermined amount of space holder particles (e.g., salt, urea, ammonium bicarbonate, dolomite, titanium hydride (TiH2), polymer spheres, etc.); compaction—the mixture is pressed into a desired shape, ensuring a uniform distribution of the space holder; sintering—the compacted mixture is heated to a temperature below the metal’s melting point, causing the metal particles to sinter and form a solid matrix; and space holder removal—the space holder is mostly dissolved, also known as leaching, which is a critical step that defines the final porosity. This process is performed until the samples present constant mass and no more of the space holder can be removed [2].
Zinc foams, a specific type of metallic foam, have attracted significant research interest due to their unique properties and wide range of potential applications: batteries, catalysts or for the reduction of CO2 [6,7,8,9]. As a porous material, zinc foam combines the inherent characteristics of zinc—such as its low melting point (419.5 °C), good electrical conductivity, and corrosion resistance in certain environments—with the structural advantages of a cellular material [10]. The resulting structure, which typically consists of a network of interconnected pores, provides a high specific surface area, low density, and good energy absorption capabilities [11]. The resulting zinc foam exhibits mechanical properties that are highly dependent on its porosity, with higher porosity leading to a decrease in compressive strength and elastic modulus. This relationship can often be modeled by the Gibson-Ashby model for cellular solids [12].
An important possible application of the open pore zinc foams is that anodes in zinc air batteries can be extremely reversible with Coulombic efficiencies approaching 100%.
The porous 3D structure suppresses passivation by providing a large specific surface area and reduces local current density, thereby promoting the uniform deposition of zinc during its use, one of the important causes for zinc air batteries failure. The porous architecture offers ample space to accommodate dendrite growth, which prevents short circuits and enhances overall battery performance, leading to a higher capacity and longer cycle life [13].
Manufacturing zinc foams presents several key difficulties, primarily related to the material’s properties and the complexities of the foaming process. These challenges must be addressed to ensure the final product has a controlled pore structure and desirable mechanical integrity. The main challenges can be grouped in three main categories:
- -
- Difficulties caused by the low melting point and its oxidation tendency: Zinc’s low melting point makes it susceptible to collapse and deformation during melt foaming, and a rapid oxidation of the liquid zinc surface will also negatively impact the foam’s stability [14].
- -
- Difficulties in Controlling Pore Structure: The conventional melt-foaming method, while simple, often produces foams with a non-uniform pore size and distribution. Achieving a homogeneous structure is challenging [15].
- -
- Issues specific to Powder Metallurgy: When using the space holder method, a key powder metallurgy technique, several issues can arise. These include the deformation or crushing of the space holder particles during the compaction step, which leads to modified pores. Insufficient compaction can result in poor bonding between zinc particles, leading to weaker cell walls and a lack of structural integrity. In contrast, excessive compaction can cause the clogging of pores near the die walls, hindering pore interconnectivity [5]. Incomplete removal of the space holder may occur as follows: the final step of the space holder method, where the filler is removed, can be problematic. If the space holder is not completely leached out, residual particles can remain in the foam, acting as impurities that degrade its mechanical properties and corrosion resistance [16].
To overcome these limitations, cold sintering emerges as a highly promising alternative to current advanced ceramic synthesis like Spark Plasma Sintering [17,18]. This low-temperature process utilizes a transient liquid phase and pressure to promote bonding between metal particles at temperatures far below the melting point [19].
The cold sintering process is based on a bilateral uniaxial pressing of a powder mixture that is mixed with a solvent and is pressed at a moderate temperature (300 °C). To date, several approaches have been used for the cold sintering of over 100 materials, ceramics and composites [20]. Sintering at this low temperature is possible by changing the mechanism by which the atoms are transferred to the sintering neck. The densification of the powder is possible by using a chemically active transient liquid phase to dissolve the layer covering the powder surface and to facilitate the self-diffusion of metal atoms. This contrasts with classical sintering, where the creation of sintering necks occurs by transporting material into the particle contact area through accelerated diffusion due to the increase in sintering temperature to T ~ 0.7–0.8 of the melting temperature of the main component in the mixture.
The current understanding of the CSP of ceramics explains densification being driven by three stages: particle rearrangement, dissolution and precipitation [21]. Cold sintering largely depends on the correct choice of solvents. Chemistry has a very important role in the choice of solvents as well as in the mechanism of cold sintering. So far, three cold sintering mechanisms have been identified:
- Based on mechano-chemical effects.
- Purely chemical effects between the solvent and the surface of the powder material.
- Pressure-assisted sintering in the presence of a liquid phase, which is similar to classical sintering with the liquid phase, but due to the high pressure applied, this effect occurs at considerably higher temperatures.
The cold sintering process is governed by three primary factors. First, the material parameters are dictated by the starting powder. Second, the choice of solvent plays a critical role, as the most suitable solvent must facilitate chemical interactions with the powder under the processing conditions required to achieve densification.
A good example is zinc oxide with an aqueous solution of acetic acid [21]. In this case, acetic acid interacts with the surface of the ZnO particles, leading to the formation of hydrated zinc acetate species at the contact points. These dissolved species are then transported along chemical gradients around the particle contacts, before precipitating and crystallizing on lower-stress surfaces as they decompose.
The mechanical strength of the cold sintered ZnO was 65 MPa in the case of using acetic acid as a transient liquid media [22]. This value was considerably increased (by 40%) by changing the used liquid media from acetic acid to formic acid [22,23]. These differences were found to be caused not by the transient liquid, but they were associated with inhomogeneous pressure application and the hindered liquid phase evaporation [24].
Another factor that influences the ceramics strength is their fracture toughness. The resistance to crack propagation is influenced by the microstructure as well as by the fracture energy of the material [25]. For this, a lot of effort was centered on identifying the effect of the particle size [26,27], grain boundaries and the nature of the secondary phases. For the ZnO, only crystalline grain boundaries were reported with no secondary phases [28,29,30].
Cold sintering (CSP) of Zn powders offers a simple and efficient process for fabricating Zn metal foam by using the dissolution/reprecipitation mechanism to transfer material to the contact points of the particles and thus create the sintering neck between them, ensuring the necessary mechanical properties for use in the intended applications. Aqueous acetic acid solutions have been shown to cold sinter Zn powders in the production of conductive inks [31]; however, limited attention is focused on the development of porous Zn material by cold (chemical) sintering [32].
The novelty of this paper is the successful integration of Cold Sintering Process (CSP) with a millimeter-scale NaCl space holder to fabricate open-cell zinc (Zn) foams. This novel coupling of CSP with the Sintering–Dissolution Process (SDP), allows for the creation of porous materials with both high porosity and high specific surface area. To our knowledge, this approach has never been used for the cold sintering of zinc powder in previous studies. This research underscores the potential of cold sintering as a transformative technology for the future of materials science and engineering, with a particular emphasis on its role in the processing of zinc-based materials.
2. Materials and Methods
The starting materials selected for this work consist of fine, spherical zinc powder with the diameter of <10 µm as shown in Figure 1a. The main idea of using fine particles was to increase their surface area and, with it, to increase the surface oxide quantity that can be cold sintered.

Figure 1.
The starting powders: (a) spherical Zn powder and (b) angular NaCl particles.
The NaCl particles were added as space holder in order to increase the porosity (calculated for a final porosity of 40 and 60%) since they are cheap and inert at these conditions and simple to eliminate. The angular NaCl particles had a particle size range of 1.25–1.6 mm (Figure 1b). We opted for this high size range to accelerate the dissolution of the salt particles [33]. The use of the higher-size pore former results in larger pores and creates wider, more open pathways within the metallic structure. This allows the leaching solvent (water in the present case) easier and faster access to the pore’ former material and facilitates the rapid egress of the dissolved components, reducing the overall dissolution time and complexity compared to navigating smaller, more tortuous channels.
Open cell foams from spherical zinc powder (Figure 1a) were successfully fabricated by the combination of Cold Sintering and the Sintering and Dissolution Process (SDP), using NaCl as space holder (up to 60 wt%). Cold sintering was carried out at 25 °C (RT), 250 °C and 300 °C, at a pressure of 500 MPa, with a holding time of 5 min to obtain a sample with the highest possible porosity and high compressive strength.
The powder mix was placed in a hardened steel cylindrical mold, followed by ∼6 wt% (0.06 g liquid to each gram of powder mixture Zn + NaCl) of a mix of acetic acid, water, and ethyl alcohol (in a 1:1:2 volume ratio) to facilitate the cold sintering.
This solution dissolves the zinc oxide present on the surface of the particles. As the solvent evaporates, zinc oxide reprecipitates at the contact points between zinc particles, promoting the formation of sintering necks. The addition of alcohol improves the dispersion of acetic acid throughout the sample.
After cooling, the samples were left to rest for 24 h and then subjected to the salt dissolution in flowing water for 24 h (until reaching constant mass). The dried samples (room temperature 24 h, placed on absorbing paper tissues in a low-humidity atmosphere—RH 20–30%) were measured to determine the diameter and height using a caliper with a precision of 0.1 mm, weighted on a balance with a precision of 0.001 g. After that, the density of the samples was calculated as the ratio of the weight divided by the calculated volume. The total porosity was calculated [34] using Equation (1) (the zinc density was taken as ρZn = 7.14 g/cm3):
The open porosity and pore size distribution was measured by mercury intrusion porosimetry (MIP) on a Pascal 140 device (Thermo Electron, Waltham, MA, USA). The closed porosity was calculated as the difference of the previous two.
Thermogravimetric and differential thermal analyses (TG–DTA) were carried out in air from room temperature up to 600 °C at a constant heating rate of 10 °C min−1. The TG–DTA system was coupled to a quadrupole mass spectrometer (QMS 200, Residual Gas Analyzer, Stanford Research Systems-RGA—Sunnyvale, CA, USA) for evolved gas analysis. Gas transfer was achieved using a stainless-steel capillary, 120 cm in length with an inner diameter of 0.075 mm. To avoid condensation of water vapor and volatile decomposition products, the capillary was maintained at approximately 100 °C throughout the measurements. The mass spectrometer operated at an ionization energy of 70 eV under standard acceleration voltage conditions.
The degree of sintering was estimated on several SEM images (taken on a Jeol 5600-LV microscope—Akishima, Tokyo, Japan) in secondary electron configuration coupled with an energy-dispersive X-ray (EDX) spectrometer UltimMAX65 (Oxford Instruments, Aztec software, version 4.2, High Wycombe, UK) and calculated as the ratio between the sintering neck and the corresponding particle dimensions [35] as shown in Figure 2.

Figure 2.
The measurements for the sintering degree estimation.
The degree of sintering (Sd) was calculated using Equation (2),
where X is the thickness of the sintering neck and D is the particle diameter. To obtain a reasonable image of the sintering degree, for each sample, at least 250 measurements were performed. The given value is the average value of these measurements.
The thermal decomposition behavior of the powder was analyzed using thermogravimetric analysis (TG). The analysis was conducted from ambient temperature up to 600 °C, with a heating rate of 10 K/min, in air, within an open system. Infrared spectra for the samples were obtained using a Bruker Tensor 27 FTIR spectrometer (Ettlingen, Germany) operated in Attenuated Total Reflection (ATR) mode.
X-ray diffractions were taken at room temperature on an Inel Equinox 3000 diffractometer (Inel, Artenay, France), operating with Co Kα radiation. The diffraction patterns were recorded in the 2θ = 20–100° interval. The diffractometer works in reflection mode and is equipped with a Curved Position Sensitive (CPS) detector which covers 90° at the same time. The acquisition time was 15 min. The detector’s minimum resolution is 0.07° FWHM (Full Width at Half Maximum) and the angle reproducibility is ±0.001° 2θ.
The mechanical behavior of the cold sintered foams under compression were measured according to ISO 13314:2011 [36] with a test speed of 0.2 mm/min, on cylindrical samples with a diameter of ~11.5 mm and a D/H ratio ~1 on three specimens for each type of sample. A test speed of 0.2 mm/min was used.
3. Results and Discussions
Cold sintering (CSP) of Zn powders offers a simple and effective process to manufacture Zn metal foam by utilizing the dissolution/reprecipitation mechanism to transfer material to the particle contact points and thus create the sintering necks between them, ensuring the needed mechanical properties. As an alternative to the classical sintering process, the present cold sintering uses aqueous acetic acid solutions to dissolve the passivation layer covering the metal surface and facilitate the self-exchange of metal atoms.
3.1. The Cold Sintering Process
To demonstrate the reaction mechanism, the products of the reaction of zinc powder with acetic acid were subjected to an FTIR spectroscopy analysis. TG-MS measurements were conducted on the dried zinc acetate hydrate product, after H2 evolution from the initial Zn-acetic acid reaction had already occurred at room temperature. Following this analysis, the vibration modes corresponding to the spectrum of zinc acetate and acetic acid used as solvent were observed. It is observed that the amount of acetic acid used is more than necessary to form zinc acetate.
Figure 3a presents the FTIR spectra of Zn powder, pure acetic acid, and the reacted compound, along with a reference spectrum of zinc acetate (SDBS No.: 2965). As expected, metallic Zn is essentially IR-inactive, showing no characteristic absorption features. Pure acetic acid exhibits a strong C=O stretching band near ~1715 cm−1 and C–O stretching modes below 1300 cm−1. In contrast, the spectrum of the reacted compound shows the disappearance of the sharp acid C=O band and the appearance of new absorption features in the 1600–1400 cm−1 region, corresponding to the asymmetric and symmetric stretching vibrations of the carboxylate (COO−) group. The separation (Δν) between these bands is approximately 150–170 cm−1, consistent with bidentate or bridging coordination of acetate ligands to the Zn2+ center [35]. These spectral changes confirm the conversion of acetic acid into zinc acetate.
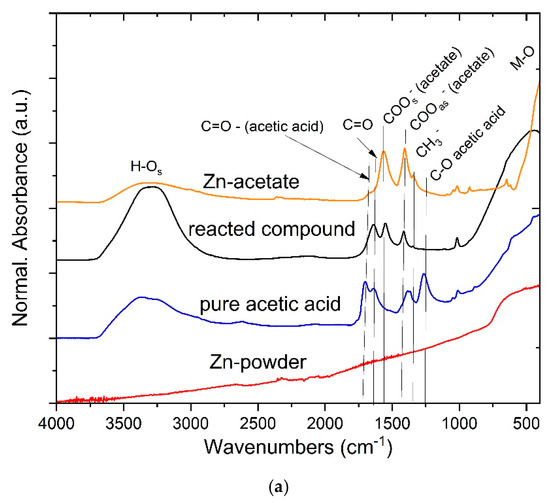
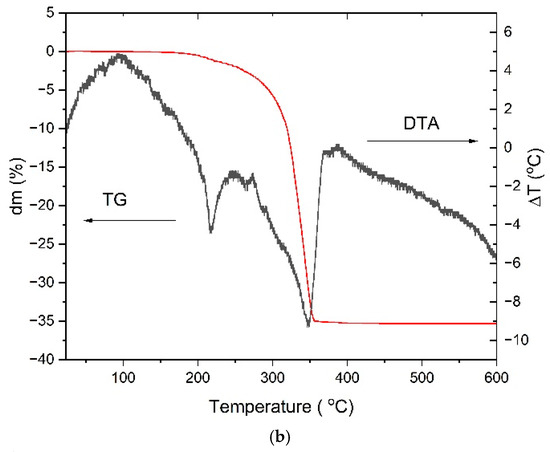
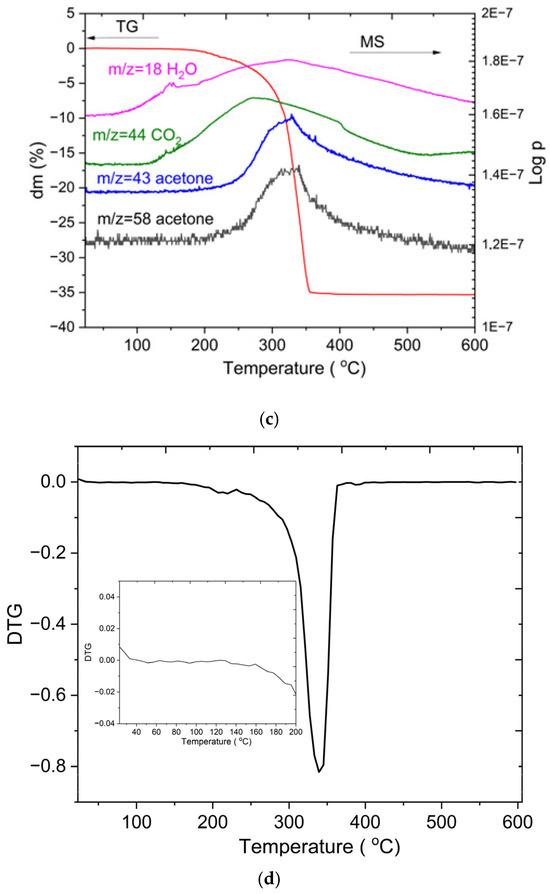
Figure 3.
FTIR spectra of Zn powder, pure acetic acid, and the reacted product (a), TG-DTA curve (b), TG-MS spectra for the acetate decomposition (c), the TG/DTG curve (d).
Figure 3b–d presents the thermogravimetric (TG/DTG), differential thermal analysis (DTA), and TG-mass spectrometry (MS) results of zinc acetate recorded in air at a heating rate of 10 °C/min. The TG and DTG data reveal two distinct features. The small mass change observed below ~100 °C, highlighted in the inset of the DTG curve, is attributed to the removal of adsorbed moisture or surface-bound species rather than structural dehydration of zinc acetate. The major thermal event occurs between 250 °C and 350 °C, corresponding to a significant mass loss of about 35%, which is assigned to the decomposition of zinc acetate and oxidation of organic fragments to ZnO. This process is accompanied by a strong exothermic peak in the DTA curve, consistent with the breakdown of acetate groups and formation of ZnO. Mass spectrometry of the evolved gases confirms the release of volatile organic species, including fragments attributable to acetone (m/z = 43, 58), as well as CO2 (m/z = 44) and H2O (m/z = 18). The strong correlation between the TG and MS data in this temperature range demonstrates the accuracy of the measurement and the rapid response (1 s) of the residual gas analyzer (RGA). The formation of acetone during the thermal decomposition of acetate ligands has been widely reported in the literature, further supporting our MS observations [37]. Above 350 °C, no additional mass loss or MS signals are detected, confirming the complete thermal conversion of zinc acetate to ZnO.
Although the zinc acetate is not evident in the Xray diffraction patterns (Figure 4), some data can be found in a deeper analysis of XRD pattern. Obviously, the zinc acetate cannot be observed in the XRD pattern due the fact that it forms a thin layer that cannot be detected by XRD in this configuration of apparatus. It must also be mentioned that the most intense peak of the zinc acetate can be found at lower diffraction angle as compared to the measured ones and the apparatus does not allow this. In Figure 4, ICDD-PDF file references are given: 04-0831 for Zn and 36-1451 for ZnO. At 25 °C, ZnO cannot be detected in the diffraction pattern; the ultrathin native oxide layer is below XRD detection limit. As previously mentioned, zinc acetate peaks are not observed, and ZnO peaks are not visible since the decomposition of zinc acetate has not started yet.
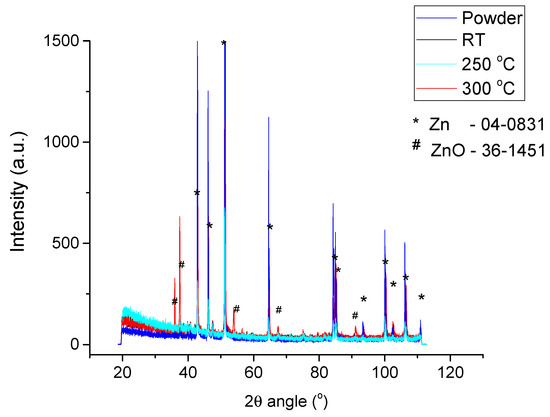
Figure 4.
Xray diffraction patterns before and after cold sintering at different. Temperatures (25 °C, 250 °C and 300 °C).
By increasing the temperature at 250 °C, the ZnO peaks are noticed in the diffraction patterns, and this suggests that the zinc acetate decomposition started at this temperature. This result comes to complement the DTA-TG measurements that indicated similar temperatures. Further increase in the temperature at 300 °C lead to the increase in the amount of ZnO, and this is confirmed by the increase in the intensity of the ZnO peaks as compared to the ones of Zn. Normally, the increase in the ZnO surface layer on the Zn particles leads to the increase of the ZnO amount observed in the x-ray pattern.
Based on FTIR, TG, MS, and XRD analyses, the transformation of zinc powder in acetic acid proceeds via a two-step process: first, the formation of zinc acetate, followed by its thermal decomposition into zinc oxide, accompanied by the evolution of acetone, water, carbon dioxide, and hydrogen, as summarized in the reaction system below (Equations (3) and (4)). The two water molecules were determined from TG analysis.
Step 1: Formation of zinc acetate
Zn (s) + 2 CH3COOH (aq) → Zn(CH3COO)2⋅xH2O (aq) + H2(g)
Step 2: Thermal decomposition of zinc acetate
Zn(CH3COO)2⋅2H2O → ZnO (s) + CH3COCH3(g) + H2O (g) + CO2(g)
3.2. The Porous Structure
The SEM analysis presents a well-sintered sample with some salt crystals remaining after the drying of the samples. This observation is a common defect observed in the SDP, unless the porosity and pore size is high, allowing an easy leaching [2]. Some areas present almost continuous structure (Figure 5a) mostly when the particle size is low and a more evident, powder metallurgy style surface with evident particles when the particles have higher diameters.

Figure 5.
Typical optical microscopy SEM images of the cold sintered Zn foams (a), with the corresponding EDS maps for Zn (b) and Cl (c) before leaching. The leached sample (d)—arrows mark the interconnection of pores with the corresponding EDS maps for Zn (e) and Cl (f) (60% porosity 300 °C/5 min).
In Figure 6, typical SEM images are presented for cold sintered samples at different temperatures; to calculate the degree of sintering, SEM images were used. On particle pairs of similar diameters, we calculated the ratio between the particle diameter and the sintering neck present on them [35].
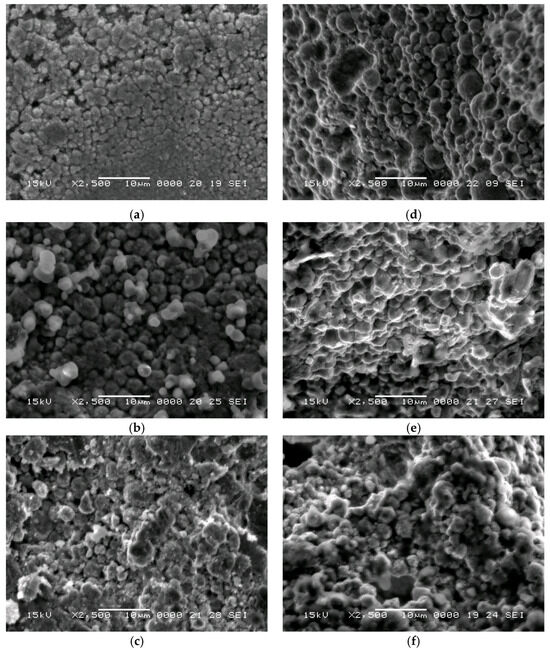
Figure 6.
SEM images presenting samples cell walls, cold sintered at different temperatures: (a) RT; (b) 250 °C; (c) 300 °C (40% porosity); (d) RT, (e) 250 °C, (f) 300 °C (60% porosity).
We did not notice a significant difference in the sintering degree with temperature (Figure 7). In the case of room temperature, we obtained 0.65. Upon increasing the temperature to 250 °C, it marginally increased to 0.66, and upon increasing the temperature to 300 °C, the sintering degree also increased slightly to 0.72.
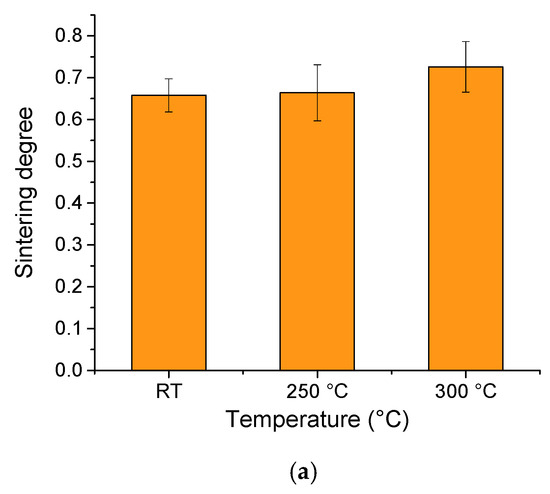
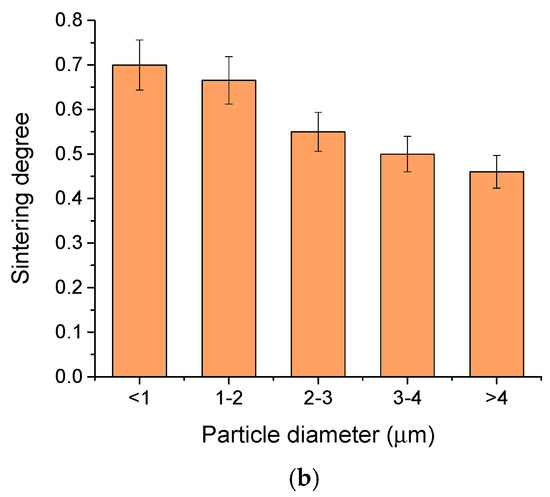
Figure 7.
The sintering degree with temperature (a) and with particle size (b).
A decrease in the sintering degree was observed as the particle diameter increases (Figure 7b), similar with findings in the case of the classical sintering [35]. As the particle size decreases, the specific surface area increases, improving the sintering. In all cases, we observed a sintering degree over 0.6 which indicates good sintering in every sample.
As in all porous structures, the total porosity is composed of an open porosity fraction and a closed one. As the total porosity increases, the fraction of open pores increases too. In the case of our samples, most pores are open and interconnected; otherwise, the pore former will be stuck inside, being unable to dissolve during the dissolution step. Table 1 presents the results of our measurements.

Table 1.
The total, open, and closed porosity values.
Inside the porous structures, the pores have a bottle-type shape. The pore former after dissolution creates pores having identical volume, however they are linked together by narrow channels (Figure 8b). Mercury intrusion porosimetry measures these channels, and not the full macropore formed by the NaCl space holder, since the raw data used during this measurement technique is the pressure and the impregnated volume of mercury for each pressure step. The presence of these channels increases the time needed for the NaCl dissolution since the water flow inside the porous structure is limited. This is evidenced also by the mercury porosimetry shown in Figure 8 and summarized in Table 2. The apparent pore size distribution is between 4 and 100 µm. The apparent increase in the high fraction of pores (90 µm and 100 µm) at higher sintering temperatures is linked to the transformation of the Zn-acetate to ZnO accompanied by a rearrangement that opens up the channels.
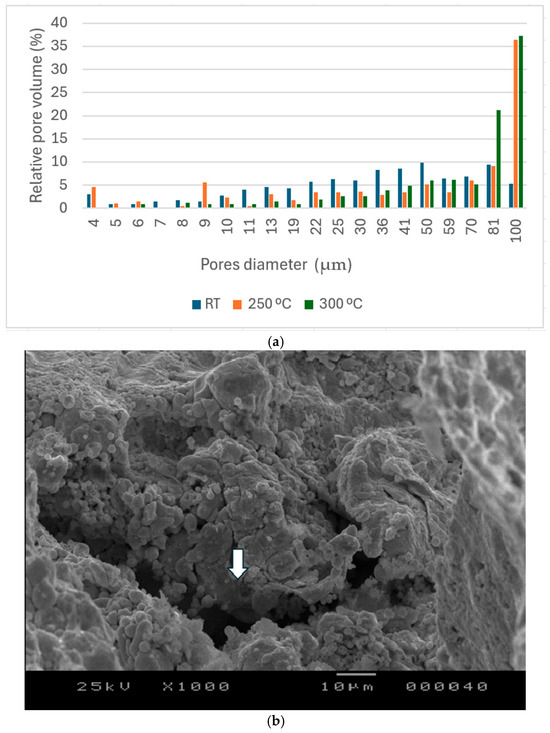
Figure 8.
The pore size distribution in samples (p = 60%) sintered at different temperatures (a), and SEM images presenting narrow channels between the macropores (b).
Table 2.
D10, D50 D90 for the pores size distribution.
3.3. Compression Tests
The first choice in evaluating the mechanical properties of metallic foams is the compression test. In the present case, we choose to evaluate samples having two porosities in the range typical for foams manufactured by powder metallurgy, namely 40% and 60% cold sintered at room temperature, 250 °C and 300 °C.
The appearance of the curves is specific to fragile metal foams (Figure 9), and all the characteristic points of the compressibility curves of metal foams are clearly visible. The compressive strength of the metal foam (σc) is the stress corresponding to the first local maximum in the compression curve. The plateau stress (σpl) is the arithmetic mean of the stresses for deformations in the deformation range of 20–40%; the stress values are calculated on deformation ranges smaller than 0.1%. The densification strain (σspl) is the corresponding strain corresponding to the end point of the plateau, considered as the point on the characteristic curve at which the stress is 1.3 times higher than the plateau stress. The results are presented in Table 3. The densification is at least 40%, not extremely high for a foam. However, considering that a significant amount of fragile zinc oxide forms at the particle interfaces and that zinc has a hexagonal structure with limited deformability, the deformation properties of the foams are reasonable.
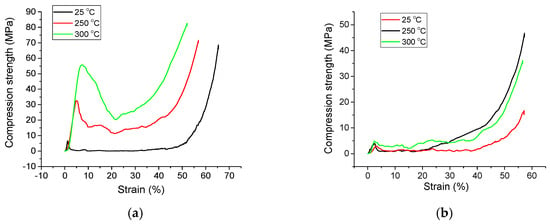
Figure 9.
The compression curves for samples with P = 40% (a) and P = 60% (b).
Table 3.
Compression properties of selected samples.
From the compression strength point of view, there are two main factors that modify the properties in a wide range. These are the porosity and the temperature, since in all cases, the sintering was found to be good. From the porosity’s point, the higher it is, the lower the mechanical properties, since there is less material to withstand the applied pressure. The compression strength of the higher porosity foams was further decreased by the brittle nature of the material where the lack of deformation enhances the weak spots, and so, the final compression strength is low. In the case of the temperature, the zinc oxide has the best compression strength of the three materials present in the samples (zinc acetate, metallic zinc and zinc oxide). The higher the fraction of the converted zinc acetate to zinc oxide, the higher the compression strength will be. As the DTA-TG-MS curve shows only after 200 °C, the decomposition of the acetate intensifies. Compared to other Zn foams obtained by liquid infiltration [12], our foams have higher mechanical properties although they also have higher densities (plateau stress of <2 MPa for a relative density of 0.2). Foams made by powder metallurgy [37] had slightly higher mechanical properties when sintered at temperatures over 400 °C (compression strength of 6 MPa at a porosity of 74%). Significant scattering of the data is observed in the cases of foams obtained by PM compared to liquid-based ones. To reduce this scattering effect, one should increase by at least an order of magnitude the sample size/pores size ratio.
4. Conclusions
We manufactured Zn metallic foams by combining cold sintering and the space holder method starting from fine Zn powder.
The FT-IR measurements confirmed the fact that during the cold sintering, the samples of zinc particles partially react with the acetic acid and form zinc acetate. Thermal analysis confirms complete conversion of zinc acetate to ZnO between 250 and 350 °C, with ~35% mass loss accompanied by release of acetone, CO2, and H2O. No further decomposition occurs above 350 °C, indicating complete transformation.
The SEM images presented a good sintering between the particles; the sintering degree is over 0.6, suggesting an advanced stage of sintering.
The compression curves indicate brittle foam behavior with densification strain exceeding 40%. Compression strength is primarily governed by porosity and sintering temperature, ranging from 56 MPa (40% porosity, 300 °C) to under 5 MPa (60% porosity). Higher porosity reduces mechanical properties due to less load-bearing material, amplified by the brittle foam structure. Increased sintering temperature enhances strength through conversion of zinc acetate to zinc oxide, which exhibits superior mechanical properties compared to zinc acetate and metallic zinc.
Author Contributions
Conceptualization, G.T.; Methodology, M.N. and T.F.M.; Formal analysis, G.T., N.A.S., T.F.M. and I.V.-S.; Investigation, M.N., G.T., N.A.S. and G.-A.R.; Writing—review & editing, I.V.-S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in the study are included in the article; further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ashby, M.F.; Evans, A.G.; Fleck, N.A.; Gibson, L.J.; Hutchinson, J.W.; Wadley, H.N.G. Metal Foams: A Design Guide; Butterworth-Heinemann: Woburn, MA, USA, 2000; pp. 1–254. [Google Scholar]
- Banhart, J. Metal Foams—From Fundamental Research to Applications. In Frontiers in the Design of Materials; Raj, B., Ranganathan, S., Bhanu Sankara Rao, K., Matthew, M.D., Shankar, P., Eds.; Universities Press (India) Limited: Telangana, India, 2007. [Google Scholar]
- Yu, C.-J.; Eifert, H.H.; Banhart, J.; Baumeister, J. Metal foaming by a powder metallurgy method: Production, properties and applications. Mater. Res. Innov. 1998, 2, 181–188. [Google Scholar] [CrossRef]
- Hassan, A.; Alnaser, I.A. A Review of Different Manufacturing Methods of Metallic Foams. ACS Omega 2024, 9, 6280–6295. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Bhatnagar, N. A survey of fabrication and application of metallic foams (1925–2017). J. Porous Mater. 2017, 25, 537–554. [Google Scholar] [CrossRef]
- Salimi, M.; Khoiee, S.M.M.; Alamdari, E.K.; Rezaei, M.; Karbasi, M. Nano Porous Zinc Synthesis on Soft Polyurethane Foam Using Conductive Ink and Electroplating Method. Metals 2022, 12, 1945. [Google Scholar] [CrossRef]
- Ge, Q.; Liu, X.; Qiao, A.; Mu, Y. Compressive Properties and Degradable Behavior of Biodegradable Porous Zinc Fabricated with the Protein Foaming Method. J. Funct. Biomater. 2022, 13, 151. [Google Scholar] [CrossRef]
- Luo, W.; Zhang, J.; Li, M.; Züttel, A. Boosting CO Production in Electrocatalytic CO2 Reduction on Highly Porous Zn Catalysts. ACS Catal. 2019, 9, 3783–3791. [Google Scholar] [CrossRef]
- Wang, X.; Wang, D.; Zhao, M.; Han, F. A novel approach to fabricate Zn coating on Mg foam through a modified thermal evaporation technique. J. Mater. Sci. Technol. 2018, 34, 1558–1563. [Google Scholar] [CrossRef]
- Gracheva, A.; Polozov, I.; Popovich, A. Additive Manufacturing of Biodegradable Metallic Implants by Selective Laser Melting: Current Research Status and Application Perspectives. Metals 2025, 15, 754. [Google Scholar] [CrossRef]
- Kádár, C.; Gorejová, R.; Kubelka, P.; Oriňaková, R.; Orbulov, I.N. Mechanical and Degradation Behavior of Zinc-Based Biodegradable Metal Foams. Adv. Eng. Mater. 2024, 26, 2301496. [Google Scholar] [CrossRef]
- Gibson, L.J.; Ashby, M.F. Cellular Solids: Structure and Properties; Cambridge University Press: Cambridge, UK, 1999; pp. 1–510. [Google Scholar]
- Ma, L.; Schroeder, M.A.; Borodin, O.; Pollard, T.P.; Ding, M.S.; Wang, C.; Xu, K. Realizing high zinc reversibility in rechargeable batteries. Nat. Energy 2020, 5, 743–749. [Google Scholar] [CrossRef]
- Kovacik, J.; Simancik, F. Comparison of zinc and aluminium foam behaviour. Kovove Mater. 2004, 42, 79–90. [Google Scholar]
- Cruz-Ramírez, A.; Contreras-Hernández, I.; Colin-García, E.; Plascencia-Barrera, G.; Pérez-Labra, M.; Gutiérrez-Pérez, V.H.; García-Hernández, M. Performance Assessment on the Manufacturing of Zn-22Al-2Cu Alloy Foams Using Barite by Melt Route. Crystals 2024, 14, 872. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, X.; Qiao, A.; Mu, Y. Mechanical Properties and In Vitro Corrosion of Biodegradable Open-Cell Zn Alloy Foams. J. Mater. Eng. Perform. 2022, 32, 5221–5236. [Google Scholar] [CrossRef]
- Shichalin, O.; Ivanov, N.; Seroshtan, A.; Nadaraia, K.; Simonenko, T.; Rogachev, K.; Marmaza, P.; Zaikova, A.; Sin’KOva, M.; Ikhtonov, G.; et al. ZnFe2O4 controlled synthesis: Key to improving properties of functional ceramic materials for energy storage applications. J. Phys. Chem. Solids 2025, 205, 112804. [Google Scholar] [CrossRef]
- Shichalin, O.; Ivanov, N.; Seroshtan, A.; Nadaraia, K.; Simonenko, T.; Gurin, M.; Kornakova, Z.; Shchitovskaya, E.; Barkhudarov, K.; Tsygankov, D.; et al. Spark plasma sintering of Ti2AlC/TiC MAX-phase based composite ceramic materials and study of their electrochemical characteristics. Ceram. Int. 2024, 50, 53120–53128. [Google Scholar] [CrossRef]
- Guo, J.; Guo, H.; Baker, A.L.; Lanagan, M.T.; Kupp, E.R.; Messing, G.L.; Randall, C.A. Cold Sintering: A Paradigm Shift for Processing and Integration of Ceramics. Angew. Chem. Int. Ed. Engl. 2016, 55, 11457–11461. [Google Scholar] [CrossRef] [PubMed]
- Ndayishimiye, A.; Sengul, M.Y.; Sada, T.; Dursun, S.; Bang, S.H.; Grady, Z.A.; Tsuji, K.; Funahashi, S.; van Duin, A.C.; Randall, C.A. Roadmap for densification in cold sintering: Chemical pathways. Open Ceram. 2020, 2, 100019. [Google Scholar] [CrossRef]
- Jabr, A.; Ribul, E.; Salamon, D.; Bermejo, R. Understanding the lower fracture resistance of cold sintered ceramics. J. Eur. Ceram. Soc. 2024, 45, 116968. [Google Scholar] [CrossRef]
- Lowum, S.; Floyd, R.; Bermejo, R.; Maria, J.-P. Mechanical strength of cold-sintered zinc oxide under biaxial bending. J. Mater. Sci. 2018, 54, 4518–4522. [Google Scholar] [CrossRef]
- Jabr, A.; Fanghanel, J.; Fan, Z.; Bermejo, R.; Randall, C. The effect of liquid phase chemistry on the densification and strength of cold sintered ZnO. J. Eur. Ceram. Soc. 2022, 43, 1531–1541. [Google Scholar] [CrossRef]
- Mamaghani, K.R.; Parvin, N. The measurement and improvement of tensile strength in cold-sintered zinc oxide. Mater. Chem. Phys. 2025, 339, 130753. [Google Scholar] [CrossRef]
- Lube, T. Fracture Toughness Measurement. In Encyclopedia of Materials: Technical Ceramics and Glasses; Elsevier: Amsterdam, The Netherlands, 2021; Volume 1, pp. 762–774. [Google Scholar] [CrossRef]
- Nur, K.; Mishra, T.P.; da Silva, J.G.P.; Gonzalez-Julian, J.; Bram, M.; Guillon, O. Influence of powder characteristics on cold sintering of nano-sized ZnO with density above 99%. J. Eur. Ceram. Soc. 2021, 41, 2648–2662. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Nakanishi, M.; Hashimoto, S. Effect of particle size distribution on cold sintering of amorphous silica. J. Eur. Ceram. Soc. 2025, 45, 117380. [Google Scholar] [CrossRef]
- Kang, X.; Floyd, R.; Lowum, S.; Cabral, M.; Dickey, E.; Maria, J. Mechanism studies of hydrothermal cold sintering of zinc oxide at near room temperature. J. Am. Ceram. Soc. 2019, 102, 4459–4469. [Google Scholar] [CrossRef]
- Funahashi, S.; Guo, J.; Guo, H.; Wang, K.; Baker, A.L.; Shiratsuyu, K.; Randall, C.A. Demonstration of the cold sintering process study for the densification grain growth of ZnOceramics. J. Am. Ceram. Soc. 2017, 100, 546–553. [Google Scholar] [CrossRef]
- de Beauvoir, T.H.; Estournès, C. Room temperature ZnO ageing after Cold Sintering Process: Grain boundaries evolution characterization by in situ electrochemical impedance spectroscopy. J. Eur. Ceram. Soc. 2024, 44, 4797–4803. [Google Scholar] [CrossRef]
- Majee, S.; Karlsson, M.C.F.; Wojcik, P.J.; Sawatdee, A.; Mulla, M.Y.; Alvi, N.U.H.; Dyreklev, P.; Beni, V.; Nilsson, D. Low temperature chemical sintering of inkjet-printed Zn nanoparticles for highly conductive flexible electronic components. npj Flex. Electron. 2021, 5, 14. [Google Scholar] [CrossRef]
- Jayasayee, K.; Clark, S.; King, C.; Dahl, P.I.; Tolchard, J.R.; Juel, M. Cold Sintering as a Cost-Effective Process to Manufacture Porous Zinc Electrodes for Rechargeable Zinc-Air Batteries. Processes 2020, 8, 592. [Google Scholar] [CrossRef]
- Thalmaier, G.; Sechel, N.A.; Vida-Simiti, I. Heat Transfer Enhancement of Paraffin Phase Change Composite Material Using Recycled Aluminum Sawing Chips. JOM 2019, 71, 1049–1055. [Google Scholar] [CrossRef]
- Sechel, A.N.; Prică, C.-V.; Marinca, T.F.; Popa, F.; Baglaevschi, L.-M.; Thalmaier, G.; Vida-Simiti, I. Characterization of Invar Syntactic Foams Obtained by Spark Plasma Sintering. Appl. Sci. 2025, 15, 2932. [Google Scholar] [CrossRef]
- Vida-Simiti, I.; Jumate, N.; Thalmaier, G.; Sechel, N.; Moldovan, V. Study of gradual porous metallic membranes obtained by powder sedimentation. J. Porous Mater. 2010, 19, 21–27. [Google Scholar] [CrossRef]
- ISO 13314:2011; Mechanical Testing of Metals—Ductility Testing—Compression Test for Porous and Cellular Metals. International Organization for Standardization: Geneva, Switzerland, 2011.
- Sadighikia, S.; Abdolhosseinzadeh, S.; Asgharzadeh, H. Production of high porosity Zn foams by powder metallurgy method. Powder Met. 2014, 58, 61–66. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).