

Unveiling State-of-Charge Effects on Elastic Properties of LiCoO2 via Deep Learning and Empirical Models

Abstract
1. Introduction
2. Method
2.1. System Preparation
2.2. Analytical Model
2.3. Deep Potential Model
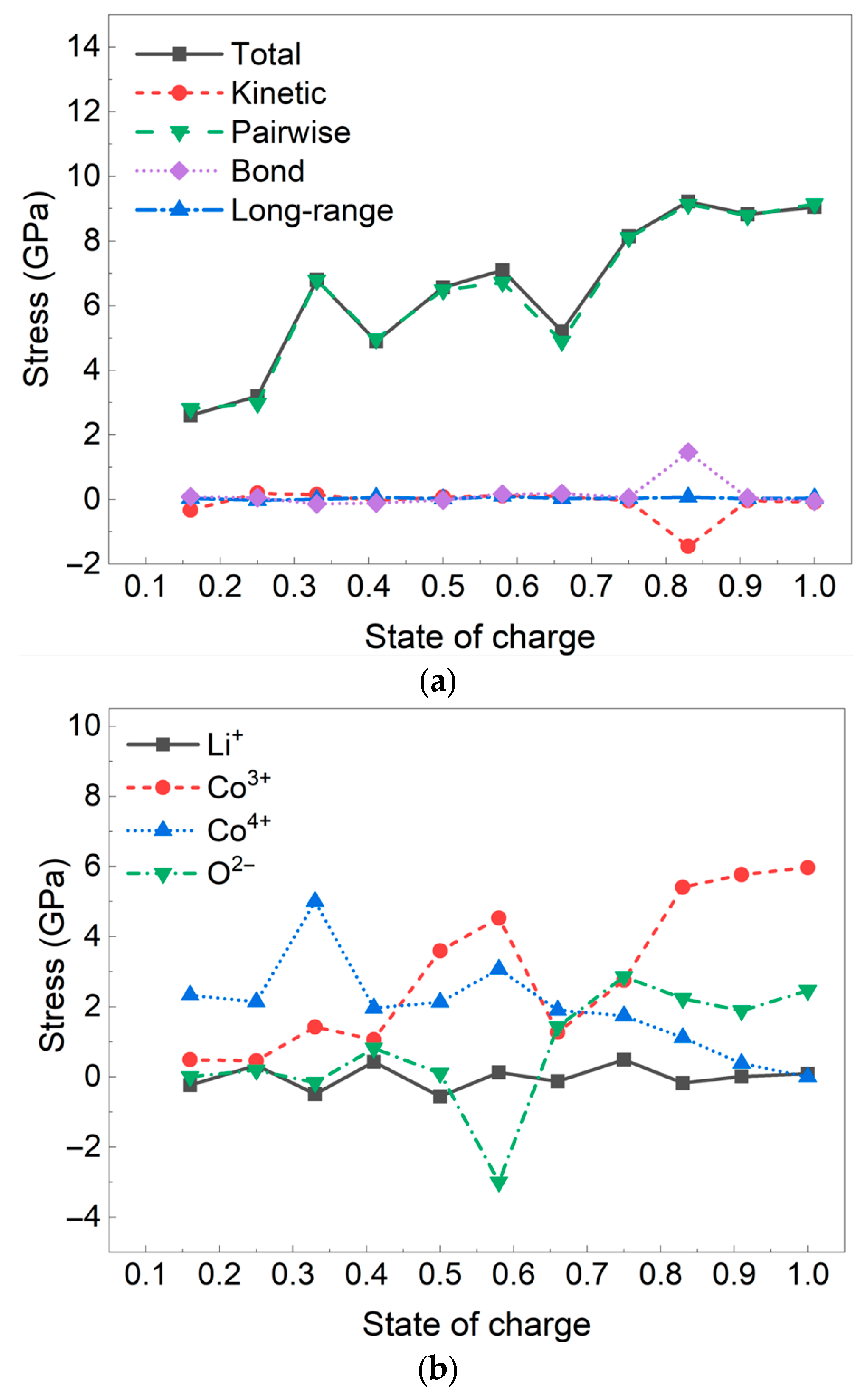
3. Results
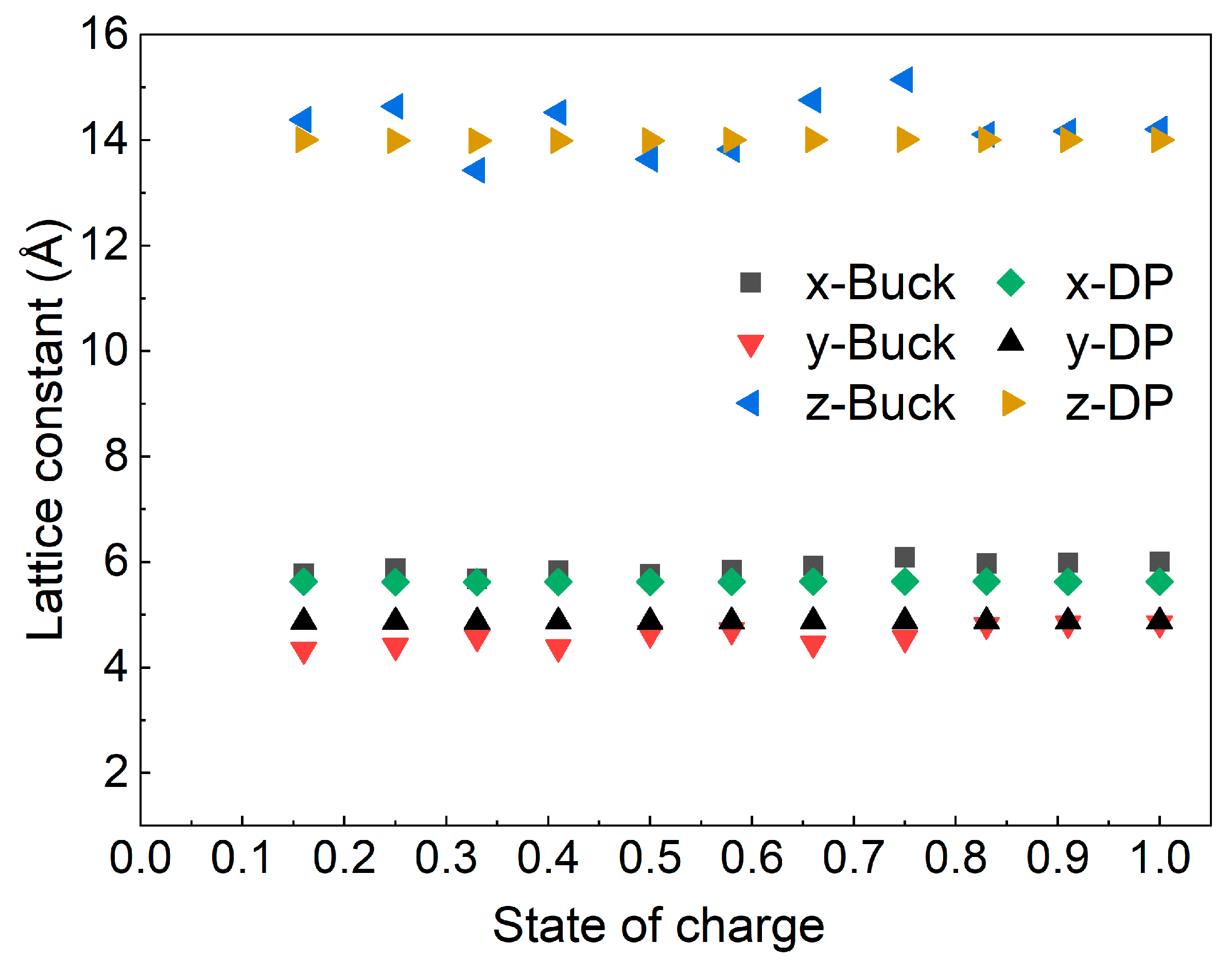
3.1. Validation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SOC | Buckingham Potential | Deep Potential | Previous Studies | |||||
|---|---|---|---|---|---|---|---|---|
| a (Å) | c (Å) | a (Å) | c (Å) | a (Å) | c (Å) | Reference | ||
| 1.00 | 6.01 | 14.21 | 5.62 | 14.00 | 5.631 | 14.054 | [36] | DFT |
| 5.629 | 14.052 | [37] | DFT | |||||
| 5.652 | 14.207 | [38] | DFT | |||||
| 5.680 | 14.160 | [42] | DFT | |||||
| 5.583 | 13.593 | [14] | MD | |||||
| 2.859 * | 14.02 | [43] | DFT | |||||
| 2.816 * | 14.04 | [44] | NI ** | |||||
| 2.840 * | 14.16 | [42] | DFT | |||||
| 0.91 | 5.99 | 14.17 | 5.62 | 14.00 | ||||
| 0.83 | 5.97 | 14.11 | 5.63 | 14.01 | ||||
| 0.75 | 6.09 | 15.15 | 5.63 | 14.00 | 2.811 * | 14.226 | [45] | NPD *** |
| 0.66 | 5.93 | 14.76 | 5.63 | 14.00 | 2.811 * | 14.286 | [45] | NPD *** |
| 0.58 | 5.85 | 13.83 | 5.62 | 14.00 | ||||
| 0.50 | 5.77 | 13.64 | 5.62 | 13.99 | 4.865 | 14.420 | [46] | XRD |
| 0.41 | 5.84 | 14.53 | 5.62 | 13.99 | ||||
| 0.33 | 5.68 | 13.43 | 5.62 | 13.98 | ||||
| 0.25 | 5.88 | 14.64 | 5.62 | 13.99 | ||||
| 0.16 | 5.78 | 14.39 | 5.63 | 14.01 | ||||
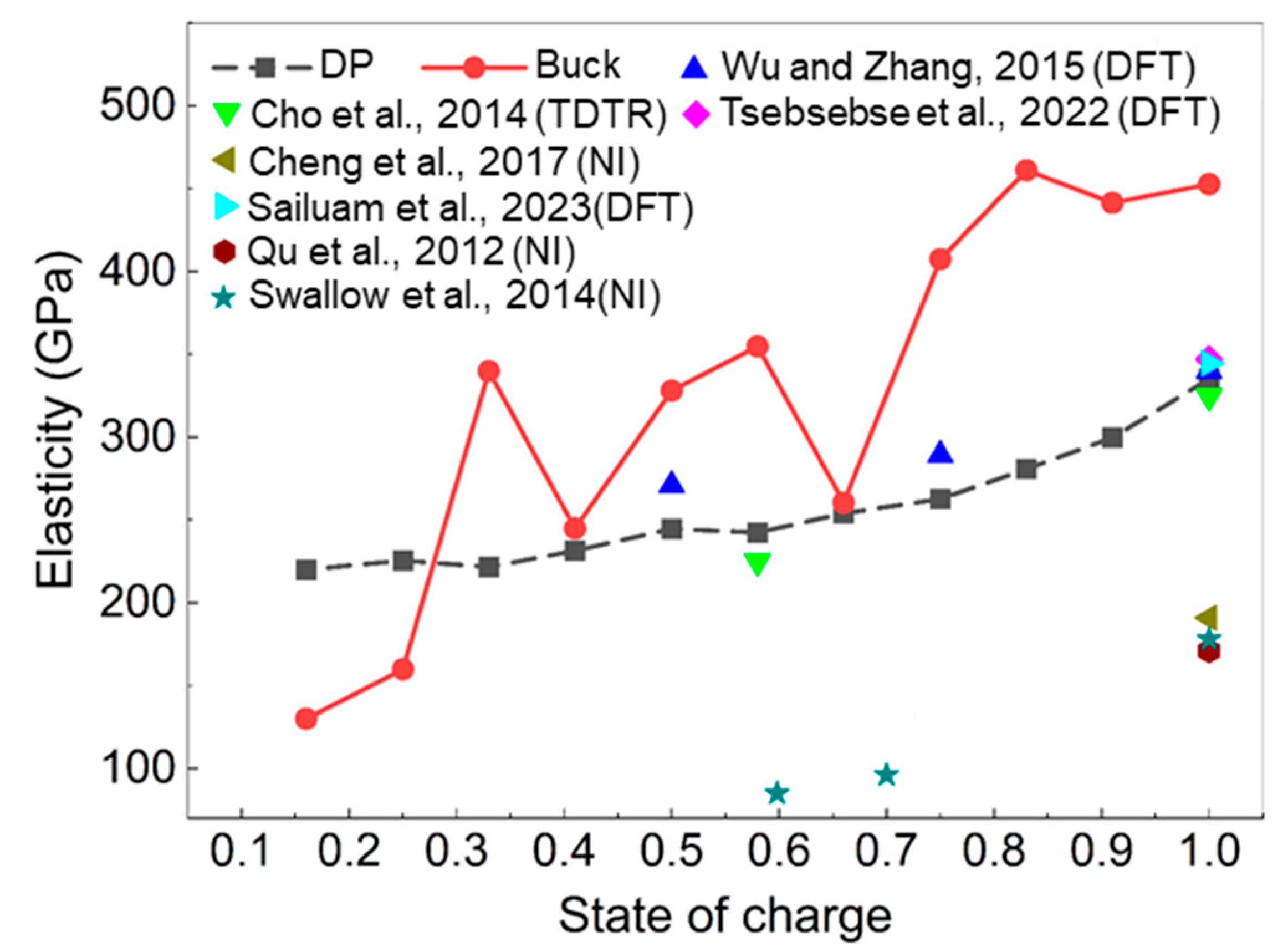
3.2. Elastic Modulus
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Castelvecchi, D. Electric cars and batteries: How will the world produce enough? Nature 2021, 596, 336–339. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Bao, Z.; Cui, Y.; Dufek, E.J.; Goodenough, J.B.; Khalifah, P.; Li, Q.; Liaw, B.Y.; Liu, P.; Manthiram, A.; et al. Pathways for practical high-energy long-cycling lithium metal batteries. Nat. Energy 2019, 4, 180–186. [Google Scholar] [CrossRef]
- Donders, M.E.; Arnoldbik, W.M.; Knoops, H.C.M.; Kessels, W.M.M.; Notten, P.H.L. Atomic Layer Deposition of LiCoO2 Thin-Film Electrodes for All-Solid-State Li-Ion Micro-Batteries. J. Electrochem. Soc. 2013, 160, A3066–A3071. [Google Scholar] [CrossRef]
- Akada, K.; Sudayama, T.; Asakura, D.; Kitaura, H.; Nagamura, N.; Horiba, K.; Oshima, M.; Hosono, E.; Harada, Y. Microscopic photoelectron analysis of single crystalline LiCoO2 particles during the charge-discharge in an all solid-state lithium ion battery. Sci. Rep. 2019, 9, 12452. [Google Scholar] [CrossRef]
- Müller, S.; Pietsch, P.; Brandt, B.-E.; Baade, P.; De Andrade, V.; De Carlo, F.; Wood, V. Quantification and modeling of mechanical degradation in lithium-ion batteries based on nanoscale imaging. Nat. Commun. 2018, 9, 2340. [Google Scholar] [CrossRef]
- Boyce, A.M.; Martínez-Pañeda, E.; Wade, A.; Zhang, Y.S.; Bailey, J.J.; Heenan, T.M.; Brett, D.J.; Shearing, P.R. Cracking predictions of lithium-ion battery electrodes by X-ray computed tomography and modelling. J. Power Sources 2022, 526, 231119. [Google Scholar] [CrossRef]
- Parks, H.C.W.; Boyce, A.M.; Wade, A.; Heenan, T.M.M.; Tan, C.; Martínez-Pañeda, E.; Shearing, P.R.; Brett, D.J.L.; Jervis, R. Direct observations of electrochemically induced intergranular cracking in polycrystalline NMC811 particles. J. Mater. Chem. A 2023, 11, 21322–21332. [Google Scholar] [CrossRef]
- Lee, S.; Park, S.S. Atomistic Simulation Study of Mixed-Metal Oxide (LiNi1/3Co1/3Mn1/3O2) Cathode Material for Lithium Ion Battery. J. Phys. Chem. C 2012, 116, 6484–6489. [Google Scholar] [CrossRef]
- Fisher, C.A.J.; Prieto, V.M.H.; Islam, M.S. Lithium Battery Materials LiMPO4 (M = Mn, Fe, Co, and Ni): Insights into Defect Association, Transport Mechanisms, and Doping Behavior. Chem. Mater. 2008, 20, 5907–5915. [Google Scholar] [CrossRef]
- Lee, S.; Park, J.; Sastry, A.M.; Lu, W. Molecular Dynamics Simulations of SOC-Dependent Elasticity of LixMn2O4 Spinels in Li-Ion Batteries. J. Electrochem. Soc. 2013, 160, A968. [Google Scholar] [CrossRef]
- Haq, I.U.; Lee, S. Molecular Dynamics Study of the Ni Content-Dependent Mechanical Properties of NMC Cathode Materials. Crystals 2025, 15, 272. [Google Scholar] [CrossRef]
- Fallahzadeh, R.; Farhadian, N. Molecular dynamics simulation of lithium ion diffusion in LiCoO2 cathode material. Solid State Ion. 2015, 280, 10–17. [Google Scholar] [CrossRef]
- Kong, F.; Longo, R.C.; Liang, C.; Nie, Y.; Zheng, Y.; Zhang, C.; Cho, K. Charge-transfer modified embedded atom method dynamic charge potential for Li–Co–O system. J. Phys. Condens. Matter 2017, 29, 475903. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Lee, K.-R.; Lee, B.-J. An interatomic potential for the Li-Co-O ternary system. Comput. Mater. Sci. 2018, 142, 47–58. [Google Scholar] [CrossRef]
- Han, J.; Zhang, L.; Car, R. Deep Potential: A General Representation of a Many-Body Potential Energy Surface. Commun. Comput. Phys. 2018, 23, 629–639. [Google Scholar] [CrossRef]
- Zhang, L.; Han, J.; Wang, H.; Car, R. Deep Potential Molecular Dynamics: A Scalable Model with the Accuracy of Quantum Mechanics. Phys. Rev. Lett. 2018, 120, 143001. [Google Scholar] [CrossRef]
- Bartók, A.P.; Kermode, J.; Bernstein, N.; Csányi, G. Machine Learning a General-Purpose Interatomic Potential for Silicon. Phys. Rev. X 2018, 8, 041048. [Google Scholar] [CrossRef]
- Lowe, J.S.; Siegel, D.J. Modeling the Interface between Lithium Metal and Its Native Oxide. ACS Appl. Mater. Interfaces 2020, 12, 46015–46026. [Google Scholar] [CrossRef]
- Li, Z.; Tan, X.; Fu, Z.; Liu, L.; Yang, J.-Y. Thermal transport across copper–water interfaces according to deep potential molecular dynamics. Phys. Chem. Chem. Phys. 2023, 25, 6746–6756. [Google Scholar] [CrossRef]
- Dahbi, M.; Saadoune, I.; Amarilla, J.M. LixNi0.7Co0.3O2 electrode material: Structural, physical and electrochemical investigations. Electrochim. Acta 2008, 53, 5266–5271. [Google Scholar] [CrossRef]
- Li, D.; Peng, Z.; Ren, H.; Guo, W.; Zhou, Y. Synthesis and characterization of LiNi1-xCoxO2 for lithium batteries by a novel method. Mater. Chem. Phys. 2008, 107, 171–176. [Google Scholar] [CrossRef]
- Akimoto, J.; Gotoh, Y.; Oosawa, Y. Synthesis and Structure Refinement of LiCoO2Single Crystals. J. Solid State Chem. 1998, 141, 298–302. [Google Scholar] [CrossRef]
- Ammundsen, B.; Rozière, J.; Islam, M.S. Atomistic Simulation Studies of Lithium and Proton Insertion in Spinel Lithium Manganates. J. Phys. Chem. B 1997, 101, 8156–8163. [Google Scholar] [CrossRef]
- Binks, D.J. Computational Modelling of Zinc Oxide and Related Oxide Ceramics. Ph.D. Thesis, University of Surrey, Surrey, UK, 1994. Available online: https://www.proquest.com/docview/301550144/abstract/8B6C340A95914E05PQ/1 (accessed on 1 July 2025).
- Catlow, C.R.A. Computer Modelling in Inorganic Crystallography; Academic Press: San Diego, CA, USA, 1997. [Google Scholar]
- Panchmatia, P.M.; Armstrong, A.R.; Bruce, P.G.; Islam, M.S. Lithium-ion diffusion mechanisms in the battery anode material Li1+xV1−xO2. Phys. Chem. Chem. Phys. 2014, 16, 21114–21118. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Dai, F.-Z.; Zhou, G.; Wang, X.; Xu, S. Unraveling the Dynamic Correlations between Transition Metal Migration and the Oxygen Dimer Formation in the Highly Delithiated LixCoO2 Cathode. J. Phys. Chem. Lett. 2023, 14, 3677–3684. [Google Scholar] [CrossRef]
- Rupp, M.; Tkatchenko, A.; Müller, K.-R.; von Lilienfeld, O.A. Fast and accurate modeling of molecular atomization energies with machine learning. Phys. Rev. Lett. 2012, 108, 058301. [Google Scholar] [CrossRef]
- Chmiela, S.; Tkatchenko, A.; Sauceda, H.E.; Poltavsky, I.; Schütt, K.T.; Müller, K.-R. Machine learning of accurate energy-conserving molecular force fields. Sci. Adv. 2017, 3, e1603015. [Google Scholar] [CrossRef]
- Dherin, B.; Avelin, B.; Karlsson, A.; Mazzawi, H.; Gonzalvo, J.; Munn, M. Training in reverse: How iteration order influences convergence and stability in deep learning’. arXiv 2025, arXiv:2502.01557. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.-Y.; Wang, H.; Car, R. Active learning of uniformly accurate interatomic potentials for materials simulation. Phys. Rev. Mater. 2019, 3, 023804. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, H.; Chen, W.; Zeng, J.; Zhang, L.; Wang, H. DP-GEN: A concurrent learning platform for the generation of reliable deep learning based potential energy models. Comput. Phys. Commun. 2020, 253, 107206. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, L.; Han, J. DeePMD-kit: A deep learning package for many-body potential energy representation and molecular dynamics. Comput. Phys. Commun. 2018, 228, 178–184. [Google Scholar] [CrossRef]
- Zeng, J.; Zhang, D.; Lu, D.; Mo, P.; Li, Z.; Chen, Y.; Rynik, M.; Huang, L.; Li, Z.; Shi, S.; et al. DeePMD-kit v2: A software package for deep potential models. J. Chem. Phys. 2023, 159, 054801. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Loa, I.; Kunc, K.; Syassen, K.; Amboage, M. Effect of pressure on the structural properties and Raman modes of LiCoO2. Phys. Rev. B 2005, 72, 224102. [Google Scholar] [CrossRef]
- Wang, M. Enthalpy of formation of LiNiO2, LiCoO2 and their solid solution, LiNi1−xCoxO2. Solid State Ion. 2004, 166, 167–173. [Google Scholar] [CrossRef]
- Kramer, D.; Ceder, G. Tailoring the Morphology of LiCoO2: A First Principles Study. Chem. Mater. 2009, 21, 3799–3809. [Google Scholar] [CrossRef]
- Tyagi, R.; Srinivasan, S. Molecular dynamics modeling of lithium ion intercalation induced change in the mechanical properties of LixMn2O4. J. Chem. Phys. 2020, 153, 164712. [Google Scholar] [CrossRef]
- Aykol, M.; Kim, S.; Wolverton, C. van der Waals Interactions in Layered Lithium Cobalt Oxides. J. Phys. Chem. C 2015, 119, 19053–19058. [Google Scholar] [CrossRef]
- Ji, J.; Lee, B.-J. Analyzing the effect of Li/Ni intermixing on Ni-rich layered cathode structures using atomistic simulation of the Li–Ni–Mn–Co–O quinary system. J. Power Sources 2022, 556, 232535. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, J. Ab initio study of anisotropic mechanical properties of LiCoO2 during lithium intercalation and deintercalation process. J. Appl. Phys. 2015, 118, 225101. [Google Scholar] [CrossRef]
- Sailuam, W.; Fongkaew, I.; Busayaporn, W.; Klinkla, R.; Phacheerak, K. Influence of pressure on elasticity, mechanical properties, and Li diffusion in battery electrode material LiCoO2: First-principles calculations. Results Phys. 2023, 52, 106788. [Google Scholar] [CrossRef]
- Qu, M.; Woodford, W.H.; Maloney, J.M.; Carter, W.C.; Chiang, Y.; Van Vliet, K.J. Nanomechanical Quantification of Elastic, Plastic, and Fracture Properties of LiCoO2. Adv. Energy Mater. 2012, 2, 940–944. [Google Scholar] [CrossRef]
- Hertz, J.T.; Huang, Q.; McQueen, T.; Klimczuk, T.; Bos, J.W.G.; Viciu, L.; Cava, R.J. Magnetism and structure of LixCoO2 and comparison to NaxCoO2. Phys. Rev. B 2008, 77, 075119. [Google Scholar] [CrossRef]
- Reimers, J.N.; Dahn, J.R. Electrochemical and in situ x-ray diffraction studies of lithium intercalation in LixCoO2. J. Electrochem. Soc. 1992, 139, 2091–2097. [Google Scholar] [CrossRef]
- Cho, J.; Losego, M.D.; Zhang, H.G.; Kim, H.; Zuo, J.; Petrov, I.; Cahill, D.G.; Braun, P.V. Electrochemically tunable thermal conductivity of lithium cobalt oxide. Nat. Commun. 2014, 5, 4035. [Google Scholar] [CrossRef]
- Swallow, J.G.; Woodford, W.H.; McGrogan, F.P.; Ferralis, N.; Chiang, Y.-M.; Van Vliet, K.J. Effect of Electrochemical Charging on Elastoplastic Properties and Fracture Toughness of LiX CoO2. J. Electrochem. Soc. 2014, 161, F3084–F3090. [Google Scholar] [CrossRef]
- Cheng, E.J.; Taylor, N.J.; Wolfenstine, J.; Sakamoto, J. Elastic properties of lithium cobalt oxide (LiCoO2). J. Asian Ceram. Soc. 2017, 5, 113–117. [Google Scholar] [CrossRef]
- Tsebesebe, N.; Kgatwane, K.; Ledwaba, R.; Ngoepe, P. Investigating the Structural and Electronic Properties of LiMO2 (M: Mn, Ni, Co) as Potential Cathode Materials: A DFT Study. J. Phys. Conf. Ser. 2022, 2298, 012010. [Google Scholar] [CrossRef]
- Morgan, L.M.; Islam, M.M.; Yang, H.; O’rEgan, K.; Patel, A.N.; Ghosh, A.; Kendrick, E.; Marinescu, M.; Offer, G.J.; Morgan, B.J.; et al. From Atoms to Cells: Multiscale Modeling of LiNixMnyCozO2 Cathodes for Li-Ion Batteries. ACS Energy Lett. 2022, 7, 108–122. [Google Scholar] [CrossRef]
- Guo, F.; Wu, X.; Liu, L.; Ye, J.; Wang, T.; Fu, L.; Wu, Y. Prediction of remaining useful life and state of health of lithium batteries based on time series feature and Savitzky-Golay filter combined with gated recurrent unit neural network. Energy 2023, 270, 126880. [Google Scholar] [CrossRef]
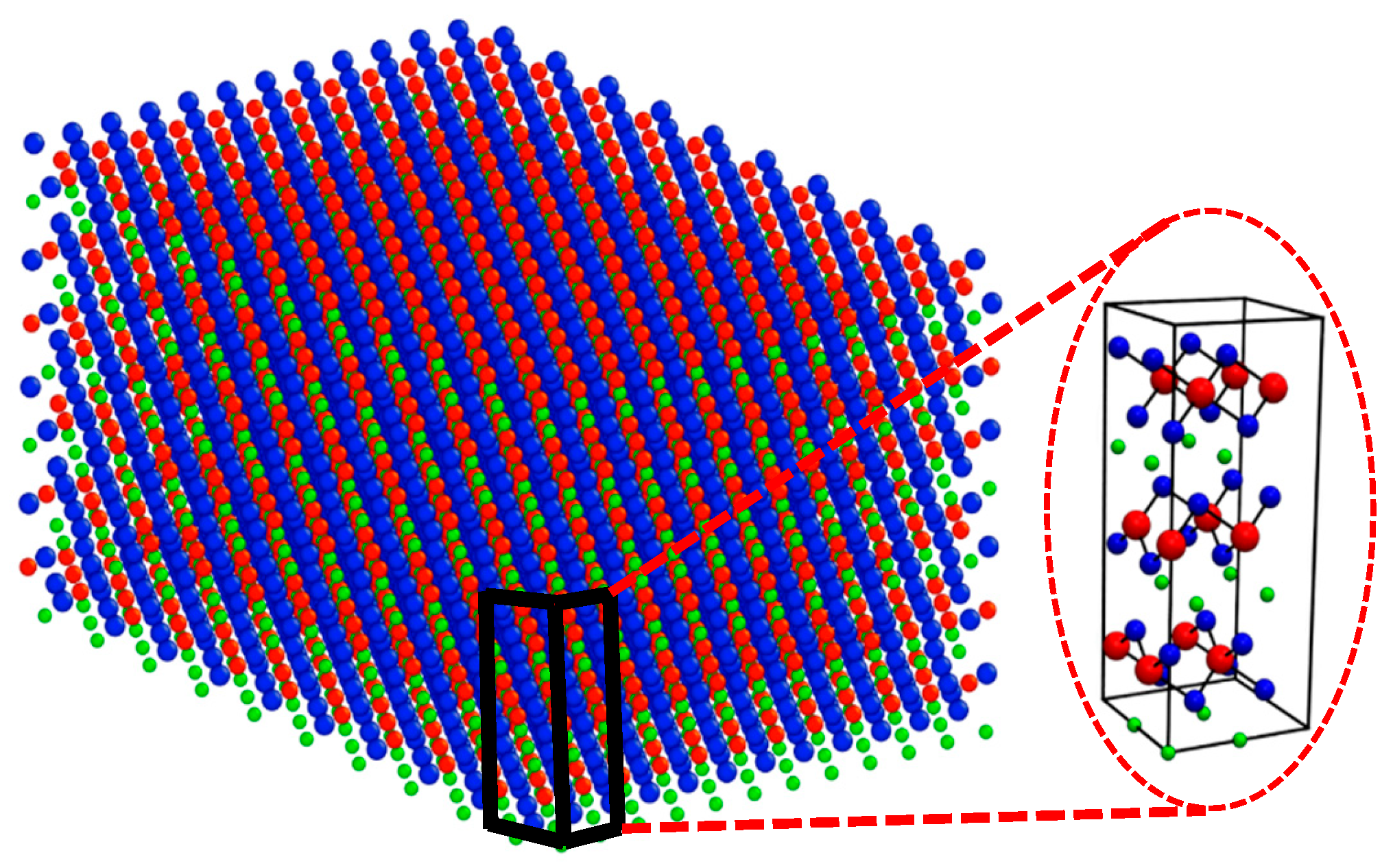
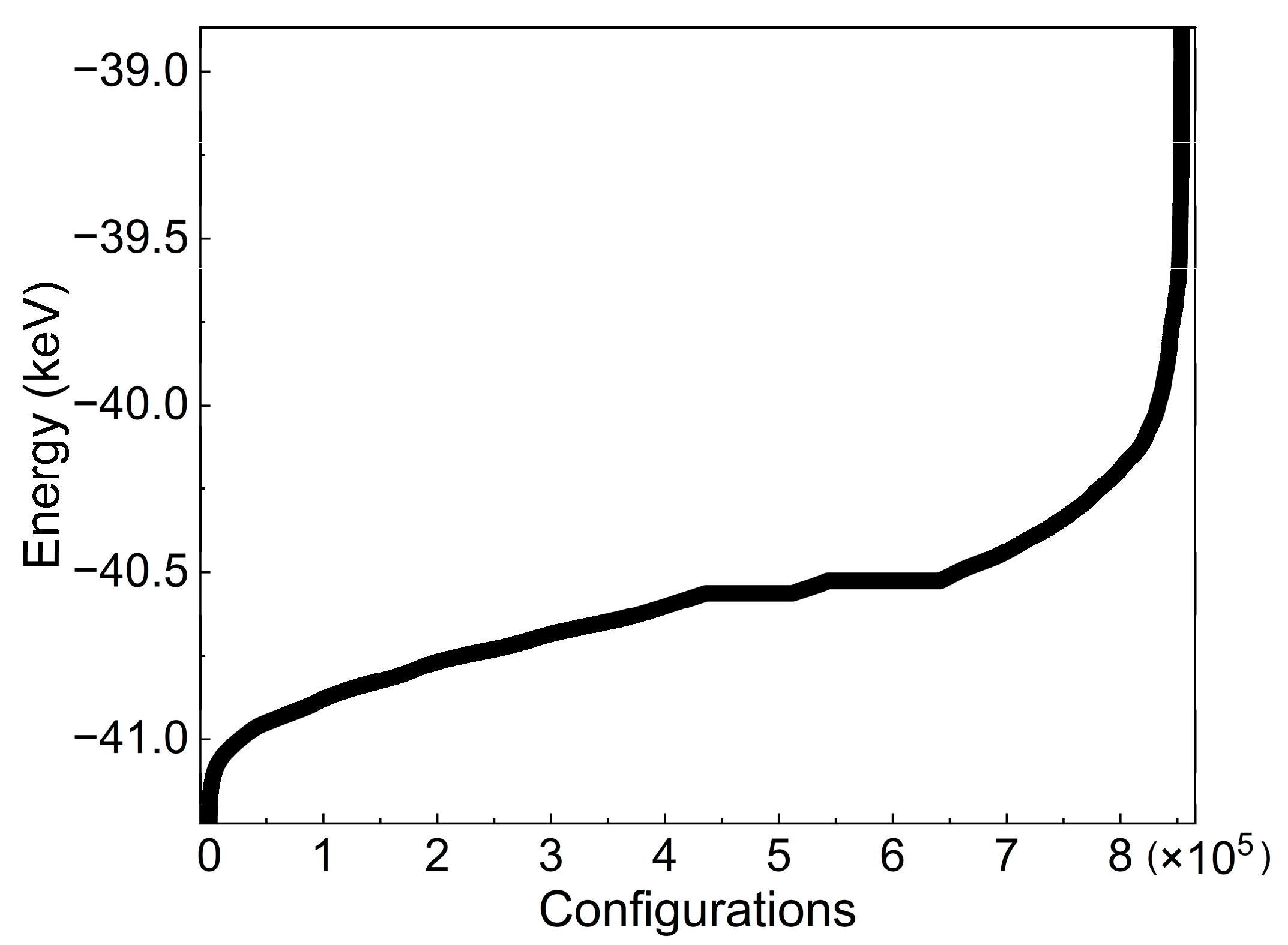
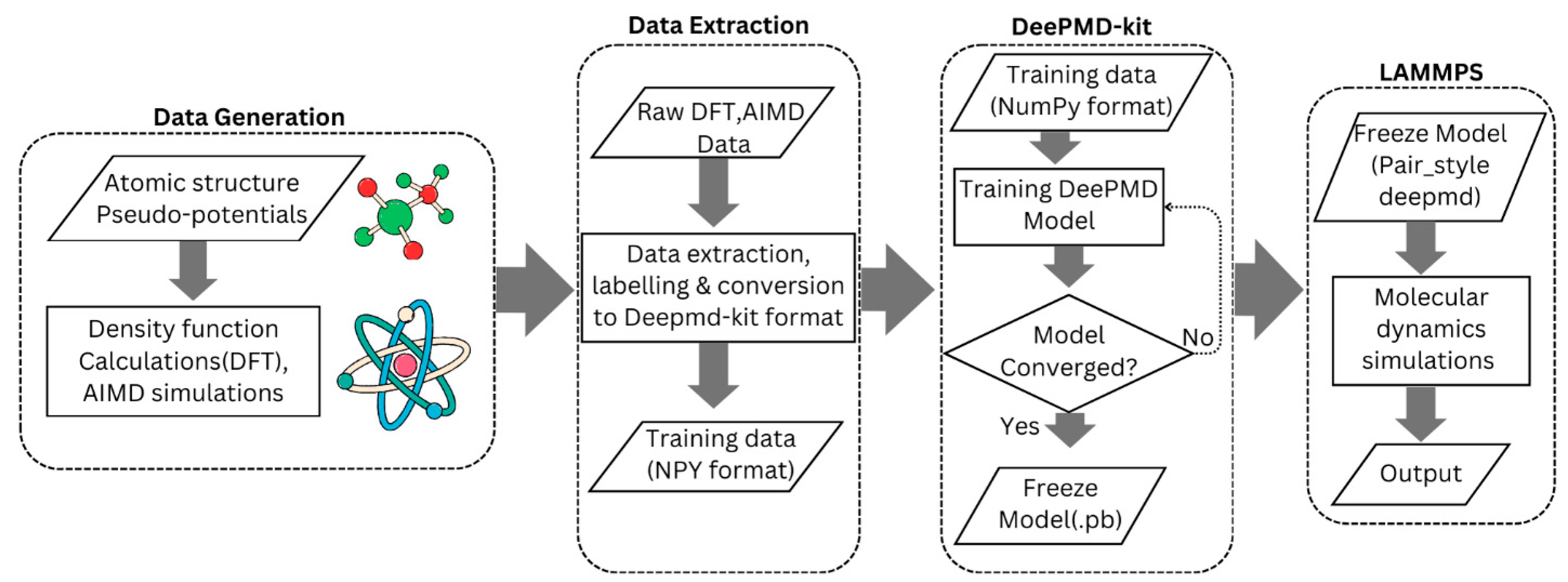

| SOC | Configurations |
|---|---|
| 1.00 | 1 |
| 0.91 | 144 |
| 0.83 | 4356 |
| 0.75 | 48,400 |
| 0.66 | 245,025 |
| 0.58 | 627,264 |
| 0.50 | 853,776 |
| 0.41 | 627,264 |
| 0.33 | 245,025 |
| 0.25 | 48,400 |
| 0.16 | 4356 |
| Interaction Pair | A (eV) | ρ (Å) | C (eVÅ6) | Reference |
|---|---|---|---|---|
| Li+–O2− | 632.1018 | 0.2906 | 0.00 | [8] |
| Co3+–O2− | 1329.82 | 0.3087 | 0.00 | [8] |
| Co4+–O2− | 1102.03 | 0.2984 | 0.00 | [24] |
| O2−–O2− | 22,764.3 | 0.149 | 65.0 | [8] |
| Ion | Y (e) | k (eVÅ−2) |
|---|---|---|
| Li+ | 1.0 | 9999.0 |
| Co3+ | 2.04 | 196.3 |
| Co4+ | 3.04 | 196.3 |
| O2− | –2.96 | 65.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haq, I.U.; Lee, S. Unveiling State-of-Charge Effects on Elastic Properties of LiCoO2 via Deep Learning and Empirical Models. Appl. Sci. 2025, 15, 7809. https://doi.org/10.3390/app15147809
Haq IU, Lee S. Unveiling State-of-Charge Effects on Elastic Properties of LiCoO2 via Deep Learning and Empirical Models. Applied Sciences. 2025; 15(14):7809. https://doi.org/10.3390/app15147809
Chicago/Turabian StyleHaq, Ijaz Ul, and Seungjun Lee. 2025. "Unveiling State-of-Charge Effects on Elastic Properties of LiCoO2 via Deep Learning and Empirical Models" Applied Sciences 15, no. 14: 7809. https://doi.org/10.3390/app15147809
APA StyleHaq, I. U., & Lee, S. (2025). Unveiling State-of-Charge Effects on Elastic Properties of LiCoO2 via Deep Learning and Empirical Models. Applied Sciences, 15(14), 7809. https://doi.org/10.3390/app15147809