
Stable Carbon Isotope Fractionation of Trichloroethylene Oxidized by Potassium Permanganate Under Different Environmental Conditions

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Experimental Procedure
2.3. Analysis Methods
3. Results and Discussion
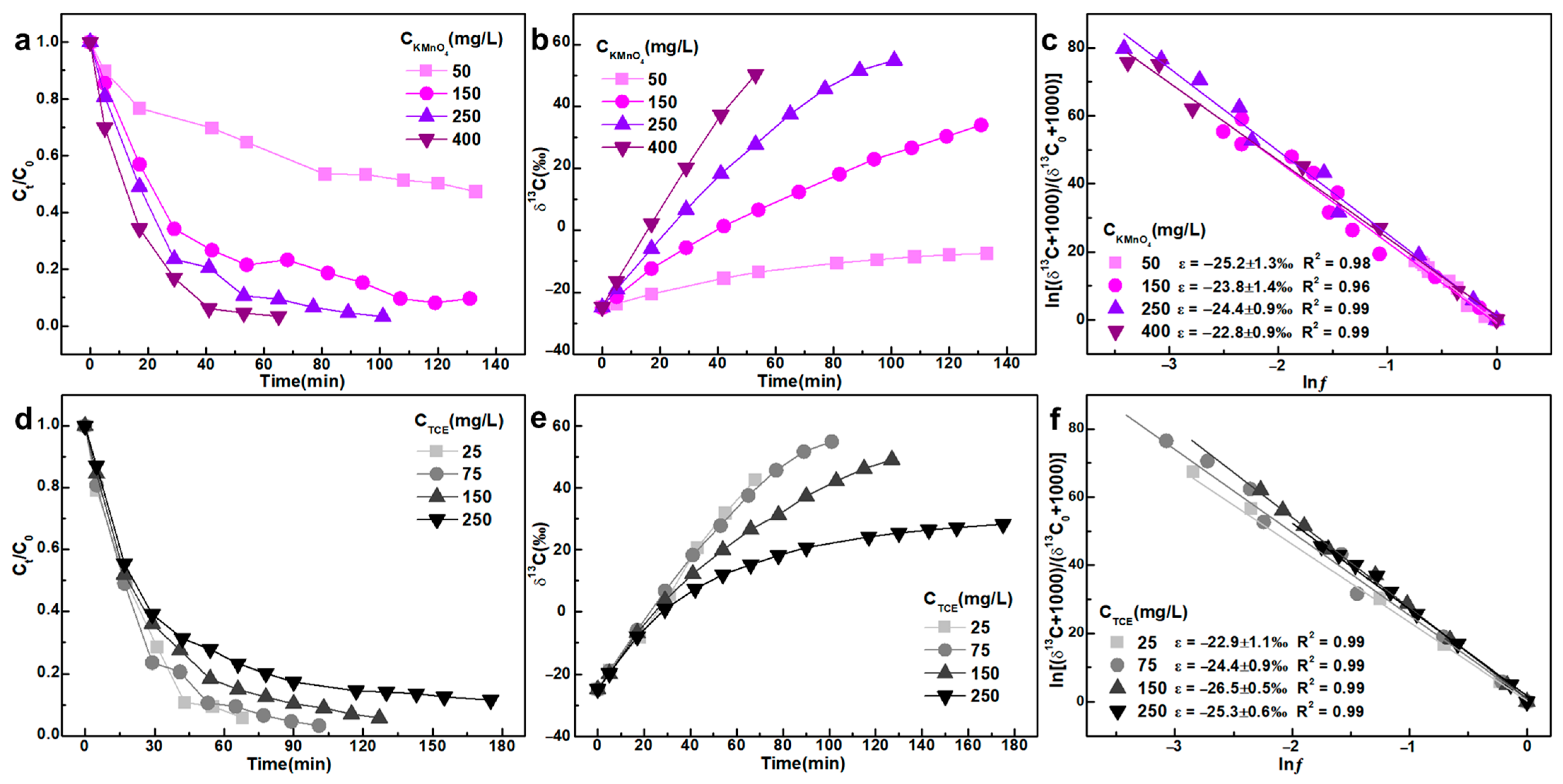
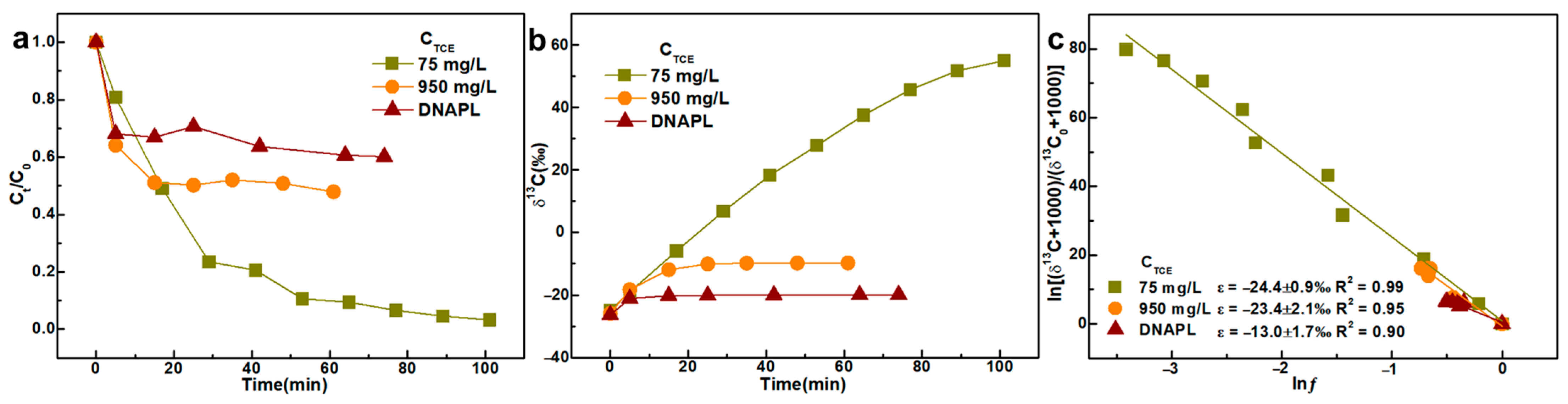
3.1. Initial Concentration of TCE and KMnO4
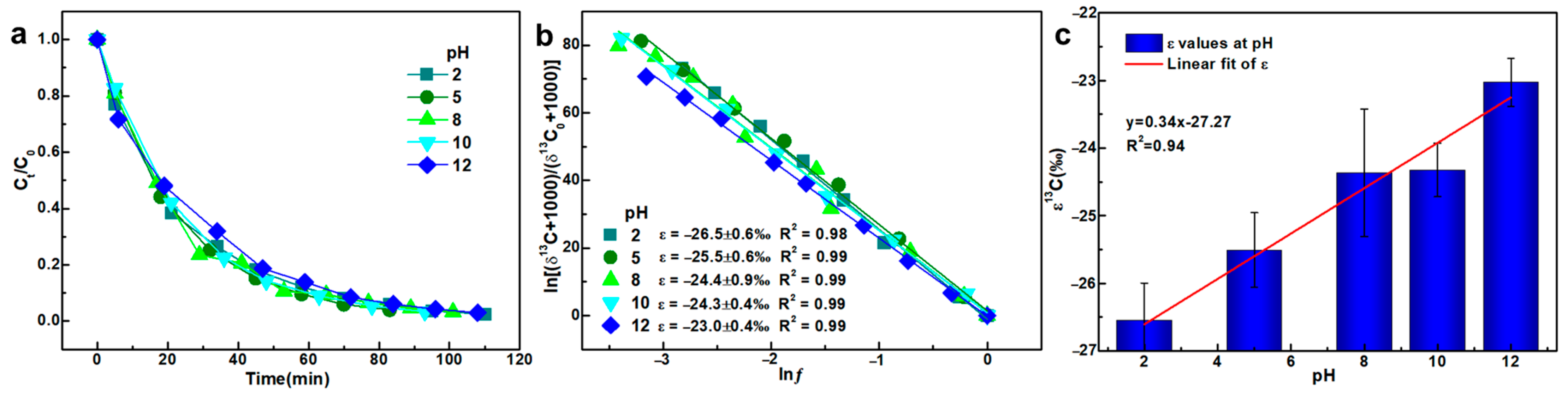
3.2. Initial pH
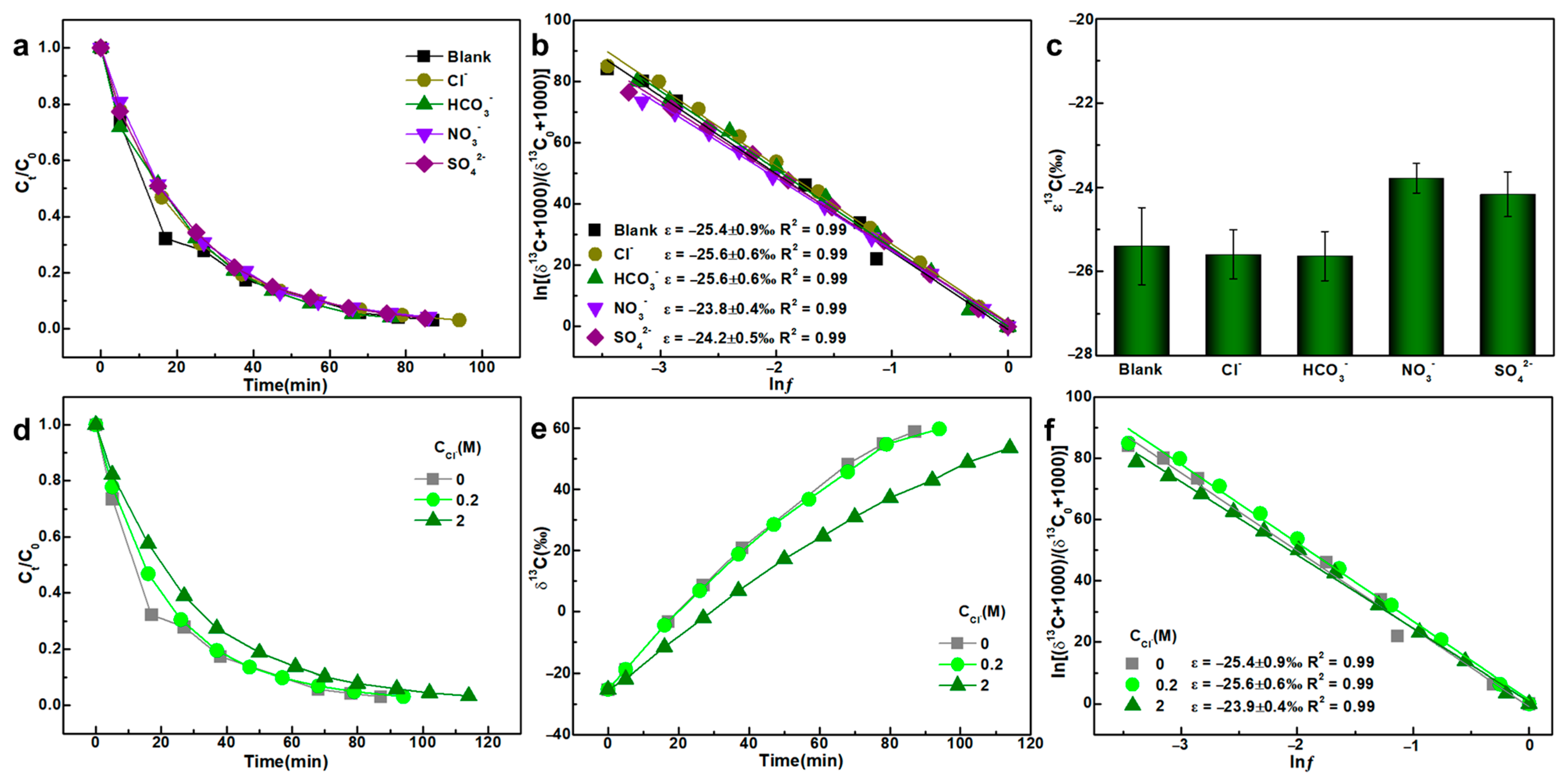
3.3. Anions
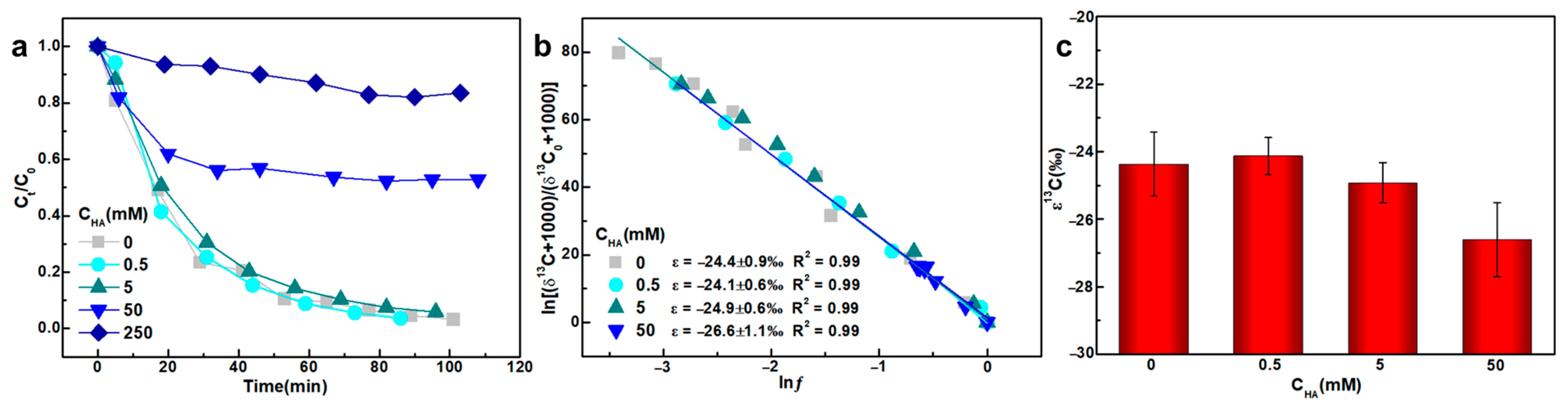
3.4. HA Concentrations
4. Conclusions
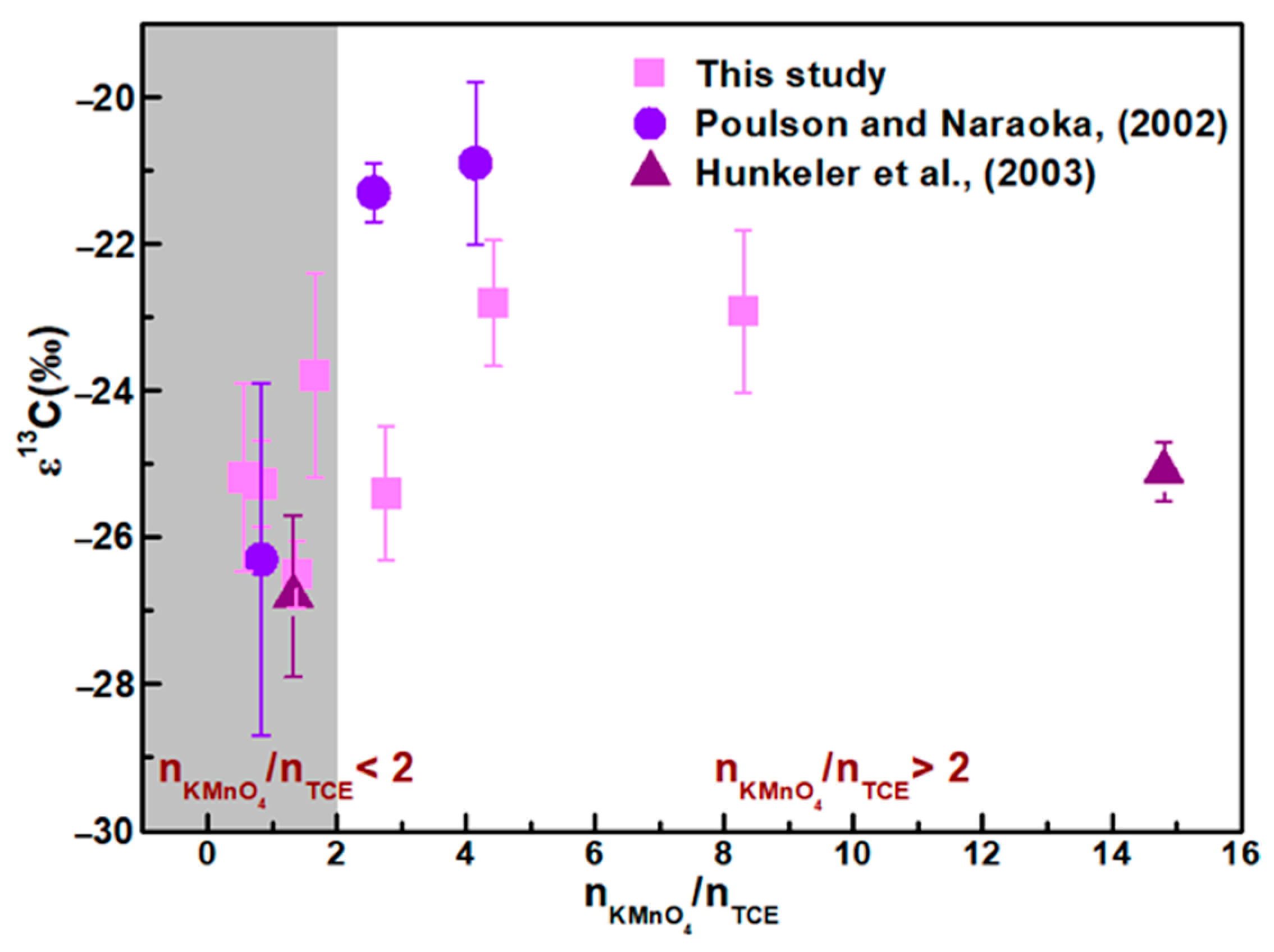
- The molar ratio n(KMnO4)/n(TCE) has a subtle yet definitive effect on ε values, which should not be overlooked during isotopic interpretation. We observed that ε values are higher when the molar ratio n(KMnO4)/n(TCE) exceeds 2, compared to ratios below 2. This effect may be attributed to insufficient KMnO4 (n(KMnO4)/n(TCE) < 2) being entirely consumed by the excess TCE, generating a substantial amount of MnO2 colloids that further react with residual TCE. Moreover, under severely limited conditions (n(KMnO4)/n(TCE) < 1), KMnO4 primarily cleaves the carbon-carbon double bond of TCE, resulting in the formation of carboxylic acid intermediates. These findings differ from earlier studies, which reported no influence of the n(KMnO4)/n(TCE) ratio on isotope fractionation.
- A linear relationship was observed between pH and the corresponding ε values, exhibiting a trend opposite to E0 as pH increased. This suggested that E0 and TCE degradation intermediates are likely key factors influencing the observed isotope effects during KMnO4 oxidation.
- The addition of SO42− and NO3− slightly influenced TCE carbon isotope fractionation, likely due to pH reductions during the reaction, which impart mild oxidizing characteristics to these anions under acidic conditions. Increasing initial Cl− concentration from 0 to 2 M also led to elevated ε values, potentially due to the salting-out effect that reduces TCE solubility in the aqueous phase.
- High concentration of HA (50–250 mM) significantly influenced carbon isotope fractionation during TCE degradation, likely due to competition between HA functional groups and TCE for KMnO4 consumption.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
Abbreviations
| CSIA | Compound-specific isotope analysis |
| ISCO | in situ chemical oxidation |
| DNAPL | dense non-aqueous phase liquid |
References
- McCarty, P.L. Groundwater Contamination by Chlorinated Solvents: History, Remediation Technologies and Strategies. In In Situ Remediation of Chlorinated Solvent Plumes; Stroo, H.F., Ward, C.H., Eds.; Springer: New York, NY, USA, 2010; pp. 1–28. [Google Scholar] [CrossRef]
- Chen, L.; Liu, Y.; Liu, F.; Jin, S. Treatment of co-mingled benzene, toluene and TCE in groundwater. J. Hazard. Mater. 2014, 275, 116–120. [Google Scholar] [CrossRef]
- Liang, S.; Wang, S.; Chang, Y.; Kao, C. Treatment of TCE-contaminated groundwater using in situ potassium permanganate oxidation: Effects and kinetics evaluation. Res. J. Biotechnol. 2015, 10, 20–24. [Google Scholar]
- Mercer, J.W.; Cohen, R.M. A review of immiscible fluids in the subsurface: Properties, models, characterization and remediation. J. Contam. Hydrol. 1990, 6, 107–163. [Google Scholar] [CrossRef]
- Kueper, B.H.; Abbott, W.; Farquhar, G. Experimental observations of multiphase flow in heterogeneous porous media. J. Contam. Hydrol. 1989, 5, 83–95. [Google Scholar] [CrossRef]
- Rivett, M.O.; Dearden, R.A.; Wealthall, G.P. Architecture, persistence and dissolution of a 20 to 45year old trichloroethene DNAPL source zone. J. Contam. Hydrol. 2014, 170, 95–115. [Google Scholar] [CrossRef]
- Rivett, M.O.; Turner, R.J.; Glibbery, P.; Cuthbert, M.O. The legacy of chlorinated solvents in the Birmingham aquifer, UK: Observations spanning three decades and the challenge of future urban groundwater development. J. Contam. Hydrol. 2012, 140–141, 107–123. [Google Scholar] [CrossRef]
- Rowe Barbara, L.; Toccalino Patricia, L.; Moran Michael, J.; Zogorski John, S.; Price Curtis, V. Occurrence and Potential Human-Health Relevance of Volatile Organic Compounds in Drinking Water from Domestic Wells in the United States. Environ. Health Perspect. 2007, 115, 1539–1546. [Google Scholar] [CrossRef] [PubMed]
- Abhilash, P.C.; Jamil, S.; Singh, N. Transgenic plants for enhanced biodegradation and phytoremediation of organic xenobiotics. Biotechnol. Adv. 2009, 27, 474–488. [Google Scholar] [CrossRef]
- Kumar Yadav, K.; Gupta, N.; Kumar, A.; Reece, L.M.; Singh, N.; Rezania, S.; Ahmad Khan, S. Mechanistic understanding and holistic approach of phytoremediation: A review on application and future prospects. Ecol. Eng. 2018, 120, 274–298. [Google Scholar] [CrossRef]
- Passatore, L.; Rossetti, S.; Juwarkar, A.A.; Massacci, A. Phytoremediation and bioremediation of polychlorinated biphenyls (PCBs): State of knowledge and research perspectives. J. Hazard. Mater. 2014, 278, 189–202. [Google Scholar] [CrossRef]
- Brusseau, M.L.; Guo, Z. Assessing contaminant-removal conditions and plume persistence through analysis of data from long-term pump-and-treat operations. J. Contam. Hydrol. 2014, 164, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Brusseau, M.L.; Fogg, G.E. Determining the long-term operational performance of pump and treat and the possibility of closure for a large TCE plume. J. Hazard. Mater. 2019, 365, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Davie, M.G.; Cheng, H.; Hopkins, G.D.; LeBron, C.A.; Reinhard, M. Implementing Heterogeneous Catalytic Dechlorination Technology for Remediating TCE-Contaminated Groundwater. Environ. Sci. Technol. 2008, 42, 8908–8915. [Google Scholar] [CrossRef] [PubMed]
- Kirschling, T.L.; Gregory, K.B.; Minkley, J.E.G.; Lowry, G.V.; Tilton, R.D. Impact of Nanoscale Zero Valent Iron on Geochemistry and Microbial Populations in Trichloroethylene Contaminated Aquifer Materials. Environ. Sci. Technol. 2010, 44, 3474–3480. [Google Scholar] [CrossRef]
- Huang, K.-C.; Hoag, G.E.; Chheda, P.; Woody, B.A.; Dobbs, G.M. Kinetic Study of Oxidation of Trichloroethylene by Potassium Permanganate. Environ. Eng. Sci. 1999, 16, 265–274. [Google Scholar] [CrossRef]
- Kao, C.M.; Huang, K.D.; Wang, J.Y.; Chen, T.Y.; Chien, H.Y. Application of potassium permanganate as an oxidant for in situ oxidation of trichloroethylene-contaminated groundwater: A laboratory and kinetics study. J. Hazard. Mater. 2008, 153, 919–927. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, A.; Gan, Y.; Li, X. Variability in carbon isotope fractionation of trichloroethene during degradation by persulfate activated with zero-valent iron: Effects of inorganic anions. Sci. Total Environ. 2016, 548–549, 1–5. [Google Scholar] [CrossRef]
- Gates Dianne, D.; Siegrist Robert, L. In-Situ Chemical Oxidation of Trichloroethylene Using Hydrogen Peroxide. J. Environ. Eng. 1995, 121, 639–644. [Google Scholar] [CrossRef]
- Liang, C.; Wang, Z.-S.; Mohanty, N. Influences of carbonate and chloride ions on persulfate oxidation of trichloroethylene at 20 °C. Sci. Total Environ. 2006, 370, 271–277. [Google Scholar] [CrossRef]
- Chen, L.; Hu, X.; Cai, T.; Yang, Y.; Zhao, R.; Liu, C.; Li, A.; Jiang, C. Degradation of Triclosan in soils by thermally activated persulfate under conditions representative of in situ chemical oxidation (ISCO). Chem. Eng. J. 2019, 369, 344–352. [Google Scholar] [CrossRef]
- Dowideit, P.; von Sonntag, C. Reaction of Ozone with Ethene and Its Methyl- and Chlorine-Substituted Derivatives in Aqueous Solution. Environ. Sci. Technol. 1998, 32, 1112–1119. [Google Scholar] [CrossRef]
- Smith, M.M.; Silva, J.A.K.; Munakata-Marr, J.; McCray, J.E. Compatibility of Polymers and Chemical Oxidants for Enhanced Groundwater Remediation. Environ. Sci. Technol. 2008, 42, 9296–9301. [Google Scholar] [CrossRef]
- Mata-Perez, F.; Perez-Benito, J.F. Identification of the product from the reduction of permanganate ion by trimethylamine in aqueous phosphate buffers. Can. J. Chem. 1985, 63, 988–992. [Google Scholar] [CrossRef]
- Jiang, J.; Pang, S.-Y.; Ma, J. Oxidation of Triclosan by Permanganate (Mn(VII)): Importance of Ligands and In Situ Formed Manganese Oxides. Environ. Sci. Technol. 2009, 43, 8326–8331. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lu, J.; Wu, L.; Wang, T.; Liu, Z.; Wang, C.; Yan, K. Developing a new controlled-release KMnO4 for groundwater DNAPL remediation. Environ. Technol. Innov. 2021, 24, 102064. [Google Scholar] [CrossRef]
- Siegrist, R.L.; Crimi, M.; Thomson, N.R.; Clayton, W.S.; Marley, M.C. IN SITU Chemical Oxidation. In Chlorinated Solvent Source Zone Remediation; Kueper, B.H., Stroo, H.F., Vogel, C.M., Ward, C.H., Eds.; Springer: New York, NY, USA, 2014; pp. 253–305. [Google Scholar] [CrossRef]
- Struse Amanda, M.; Siegrist Robert, L.; Dawson Helen, E.; Urynowicz Michael, A. Diffusive Transport of Permanganate during In Situ Oxidation. J. Environ. Eng. 2002, 128, 327–334. [Google Scholar] [CrossRef]
- Thullner, M.; Centler, F.; Richnow, H.-H.; Fischer, A. Quantification of organic pollutant degradation in contaminated aquifers using compound specific stable isotope analysis—Review of recent developments. Org. Geochem. 2012, 42, 1440–1460. [Google Scholar] [CrossRef]
- Mahmoodlu, M.G.; Hassanizadeh, S.M.; Hartog, N. Evaluation of the kinetic oxidation of aqueous volatile organic compounds by permanganate. Sci. Total Environ. 2014, 485–486, 755–763. [Google Scholar] [CrossRef]
- Harte, P.T.; Smith, T.E.; Williams, J.H.; Degnan, J.R. Time series geophysical monitoring of permanganate injections and in situ chemical oxidation of PCE, OU1 area, Savage Superfund Site, Milford, NH, USA. J. Contam. Hydrol. 2012, 132, 58–74. [Google Scholar] [CrossRef]
- Xu, X.; Chen, J.; Wang, S.; Ge, J.; Qu, R.; Feng, M.; Sharma, V.K.; Wang, Z. Degradation kinetics and transformation products of chlorophene by aqueous permanganate. Water Res. 2018, 138, 293–300. [Google Scholar] [CrossRef]
- Chen, J.; Qu, R.; Pan, X.; Wang, Z. Oxidative degradation of triclosan by potassium permanganate: Kinetics, degradation products, reaction mechanism, and toxicity evaluation. Water Res. 2016, 103, 215–223. [Google Scholar] [CrossRef]
- Jiang, J.; Gao, Y.; Pang, S.-Y.; Wang, Q.; Huangfu, X.; Liu, Y.; Ma, J. Oxidation of Bromophenols and Formation of Brominated Polymeric Products of Concern during Water Treatment with Potassium Permanganate. Environ. Sci. Technol. 2014, 48, 10850–10858. [Google Scholar] [CrossRef]
- Lee, E.S.; Seol, Y.; Fang, Y.C.; Schwartz, F.W. Destruction Efficiencies and Dynamics of Reaction Fronts Associated with the Permanganate Oxidation of Trichloroethylene. Environ. Sci. Technol. 2003, 37, 2540–2546. [Google Scholar] [CrossRef] [PubMed]
- Baciocchi, R.; D’Aprile, L.; Innocenti, I.; Massetti, F.; Verginelli, I. Development of technical guidelines for the application of in-situ chemical oxidation to groundwater remediation. J. Clean. Prod. 2014, 77, 47–55. [Google Scholar] [CrossRef]
- Christenson, M.; Kambhu, A.; Reece, J.; Comfort, S.; Brunner, L. A five-year performance review of field-scale, slow-release permanganate candles with recommendations for second-generation improvements. Chemosphere 2016, 150, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Christenson, M.D.; Kambhu, A.; Comfort, S.D. Using slow-release permanganate candles to remove TCE from a low permeable aquifer at a former landfill. Chemosphere 2012, 89, 680–687. [Google Scholar] [CrossRef]
- Woo, N.C.; Hyun, S.G.; Park, W.W.; Lee, E.S.; Schwartz, F.W. Characteristics of permanganate oxidation of TCE at low reagent concentrations. Environ. Technol. 2009, 30, 1337–1342. [Google Scholar] [CrossRef] [PubMed]
- Poulson, S.R.; Naraoka, H. Carbon Isotope Fractionation during Permanganate Oxidation of Chlorinated Ethylenes (cDCE, TCE, PCE). Environ. Sci. Technol. 2002, 36, 3270–3274. [Google Scholar] [CrossRef]
- Yan, Y.E.; Schwartz, F.W. Kinetics and Mechanisms for TCE Oxidation by Permanganate. Environ. Sci. Technol. 2000, 34, 2535–2541. [Google Scholar] [CrossRef]
- Hunkeler, D.; Aravena, R.; Parker, B.L.; Cherry, J.A.; Diao, X. Monitoring Oxidation of Chlorinated Ethenes by Permanganate in Groundwater Using Stable Isotopes: Laboratory and Field Studies. Environ. Sci. Technol. 2003, 37, 798–804. [Google Scholar] [CrossRef]
- Yan, Y.E.; Schwartz, F.W. Oxidative degradation and kinetics of chlorinated ethylenes by potassium permanganate. J. Contam. Hydrol. 1999, 37, 343–365. [Google Scholar] [CrossRef]
- Zhang, M.; Dong, J. Phase-transfer catalysis enhanced remediation of trichloroethylene polluted groundwater by potassium permanganate. Environ. Technol. 2020, 41, 3431–3442. [Google Scholar] [CrossRef] [PubMed]
- Meckenstock, R.U.; Morasch, B.; Griebler, C.; Richnow, H.H. Stable isotope fractionation analysis as a tool to monitor biodegradation in contaminated acquifers. J. Contam. Hydrol. 2004, 75, 215–255. [Google Scholar] [CrossRef] [PubMed]
- Hofstetter, T.B.; Reddy, C.M.; Heraty, L.J.; Berg, M.; Sturchio, N.C. Carbon and Chlorine Isotope Effects During Abiotic Reductive Dechlorination of Polychlorinated Ethanes. Environ. Sci. Technol. 2007, 41, 4662–4668. [Google Scholar] [CrossRef] [PubMed]
- Braeckevelt, M.; Fischer, A.; Kästner, M. Field applicability of Compound-Specific Isotope Analysis (CSIA) for characterization and quantification of in situ contaminant degradation in aquifers. Appl. Microbiol. Biotechnol. 2012, 94, 1401–1421. [Google Scholar] [CrossRef]
- Mariotti, A.; Germon, J.C.; Hubert, P.; Kaiser, P.; Letolle, R.; Tardieux, A.; Tardieux, P. Experimental determination of nitrogen kinetic isotope fractionation: Some principles; illustration for the denitrification and nitrification processes. Plant Soil 1981, 62, 413–430. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, A.; Gan, Y.; Li, X. Effects of inorganic anions on carbon isotope fractionation during Fenton-like degradation of trichloroethene. J. Hazard. Mater. 2016, 308, 187–191. [Google Scholar] [CrossRef]
- Wang, Y.; Dong, Y.; Xing, L.; Guan, Y.; Liu, C. Photocatalytic Degradation of Trichloroethylene Under Different Environmental Conditions: Kinetics and Carbon Isotope Effects. Water 2025, 17, 1533. [Google Scholar] [CrossRef]
- Pang, S.-Y.; Duan, J.-B.; Zhou, Y.; Gao, Y.; Jiang, J. Oxidation kinetics of anilines by aqueous permanganate and effects of manganese products: Comparison to phenols. Chemosphere 2019, 235, 104–112. [Google Scholar] [CrossRef]
- Sun, B.; Zhang, J.; Du, J.; Qiao, J.; Guan, X. Reinvestigation of the Role of Humic Acid in the Oxidation of Phenols by Permanganate. Environ. Sci. Technol. 2013, 47, 14332–14340. [Google Scholar] [CrossRef]
- Vane, L.M.; Giroux, E.L. Henry’s Law Constants and Micellar Partitioning of Volatile Organic Compounds in Surfactant Solutions. J. Chem. Eng. Data 2000, 45, 38–47. [Google Scholar] [CrossRef]
- Kashiyama, N.; Boving, T.B. Hindered Gas-Phase Partitioning of Trichloroethylene from Aqueous Cyclodextrin Systems: Implications for Treatment and Analysis. Environ. Sci. Technol. 2004, 38, 4439–4444. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhong, S.; Dai, Y.; Liu, C.-C.; Zhang, H. Effect of MnO2 Phase Structure on the Oxidative Reactivity toward Bisphenol A Degradation. Environ. Sci. Technol. 2018, 52, 11309–11318. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Chen, Y.; Fu, M.-L. Degradation efficiencies and mechanisms of trichloroethylene (TCE) by controlled-release permanganate (CRP) oxidation. Chem. Eng. J. 2012, 192, 276–283. [Google Scholar] [CrossRef]
- Wang, L.; Jiang, J.; Pang, S.-Y.; Gao, Y.; Zhou, Y.; Li, J.; Yang, Y.; Ma, J.; Zhang, T. Further insights into the combination of permanganate and peroxymonosulfate as an advanced oxidation process for destruction of aqueous organic contaminants. Chemosphere 2019, 228, 602–610. [Google Scholar] [CrossRef]
- Cao, W.; Wu, N.; Qu, R.; Sun, C.; Huo, Z.; Ajarem, J.S.; Allam, A.A.; Wang, Z.; Zhu, F. Oxidation of benzophenone-3 in aqueous solution by potassium permanganate: Kinetics, degradation products, reaction pathways, and toxicity assessment. Environ. Sci. Pollut. Res. 2021, 28, 31301–31311. [Google Scholar] [CrossRef]
- Zhou, Y.; Zeng, Z.; Fu, J.; Gao, Y.; Ma, J.; Zhang, Z.; Zu, D.; Han, B.; Lu, X.; Ma, J.; et al. New Insights into the Role of Humic Acid in Permanganate Oxidation of Diclofenac: A Novel Electron Transfer Mechanism. Environ. Sci. Technol. 2024, 58, 4019–4028. [Google Scholar] [CrossRef]
- Oldham, V.E.; Jones, M.R.; Tebo, B.M.; Luther, G.W. Oxidative and reductive processes contributing to manganese cycling at oxic-anoxic interfaces. Mar. Chem. 2017, 195, 122–128. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, Y.; Wang, Y.; Xing, L.; Uddin, G.; Guan, Y.; E, Z.; Liang, J.; Li, P.; Liu, C.; Fan, Q. Stable Carbon Isotope Fractionation of Trichloroethylene Oxidized by Potassium Permanganate Under Different Environmental Conditions. Appl. Sci. 2025, 15, 7142. https://doi.org/10.3390/app15137142
Dong Y, Wang Y, Xing L, Uddin G, Guan Y, E Z, Liang J, Li P, Liu C, Fan Q. Stable Carbon Isotope Fractionation of Trichloroethylene Oxidized by Potassium Permanganate Under Different Environmental Conditions. Applied Sciences. 2025; 15(13):7142. https://doi.org/10.3390/app15137142
Chicago/Turabian StyleDong, Yaqiong, Yufeng Wang, Lantian Xing, Ghufran Uddin, Yuanxiao Guan, Zhengyang E, Jianjun Liang, Ping Li, Changjie Liu, and Qiaohui Fan. 2025. "Stable Carbon Isotope Fractionation of Trichloroethylene Oxidized by Potassium Permanganate Under Different Environmental Conditions" Applied Sciences 15, no. 13: 7142. https://doi.org/10.3390/app15137142
APA StyleDong, Y., Wang, Y., Xing, L., Uddin, G., Guan, Y., E, Z., Liang, J., Li, P., Liu, C., & Fan, Q. (2025). Stable Carbon Isotope Fractionation of Trichloroethylene Oxidized by Potassium Permanganate Under Different Environmental Conditions. Applied Sciences, 15(13), 7142. https://doi.org/10.3390/app15137142