
The Study of the Synthesis of SiC by the Carbonization of Si(111) Substrates: The Role of Native Silicon Oxide

 ,
,  , , , , , , , , , ,
, , , , , , , , , , 
Abstract
Featured Application
Abstract
1. Introduction
2. Materials and Methods
3. Results
- -
- Due to the removal of material from the thin films by a directed plasma flow, a significant predominance of Si atoms sputtered from the SiC as a part from the thin film is expected compared to those of carbon.
- -
- The elemental analysis in the GDOES method refers to the atoms sputtered from the sample surface, while in the depth-dependent concentration profiles, the solid phase (unsputtered) of the samples is analyzed, and in this sense, the two methods, when analyzing identical samples, give a picture as close as possible to the real one.
- (i)
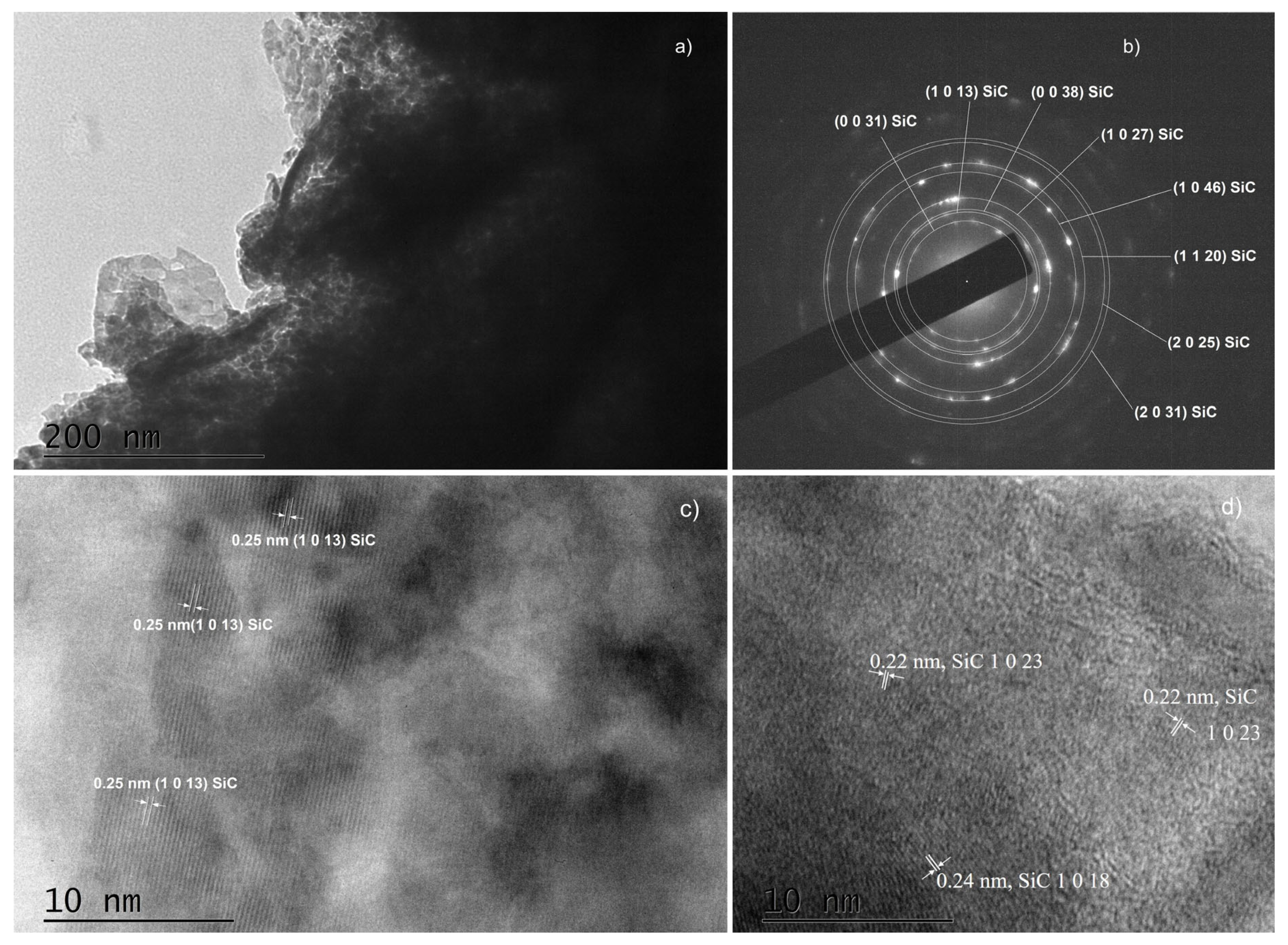
- The crystallite size in the thin films synthesized for 60 and 90 min is much larger than in the other samples.
- (ii)
- In the thin films synthesized for 90 min, there are a lot of areas in which a two-dimensional network of crystal planes is clearly distinguished, which indicates an enhanced crystalline ordering (Figure 9c and Figure S7b).
4. Discussion
- -
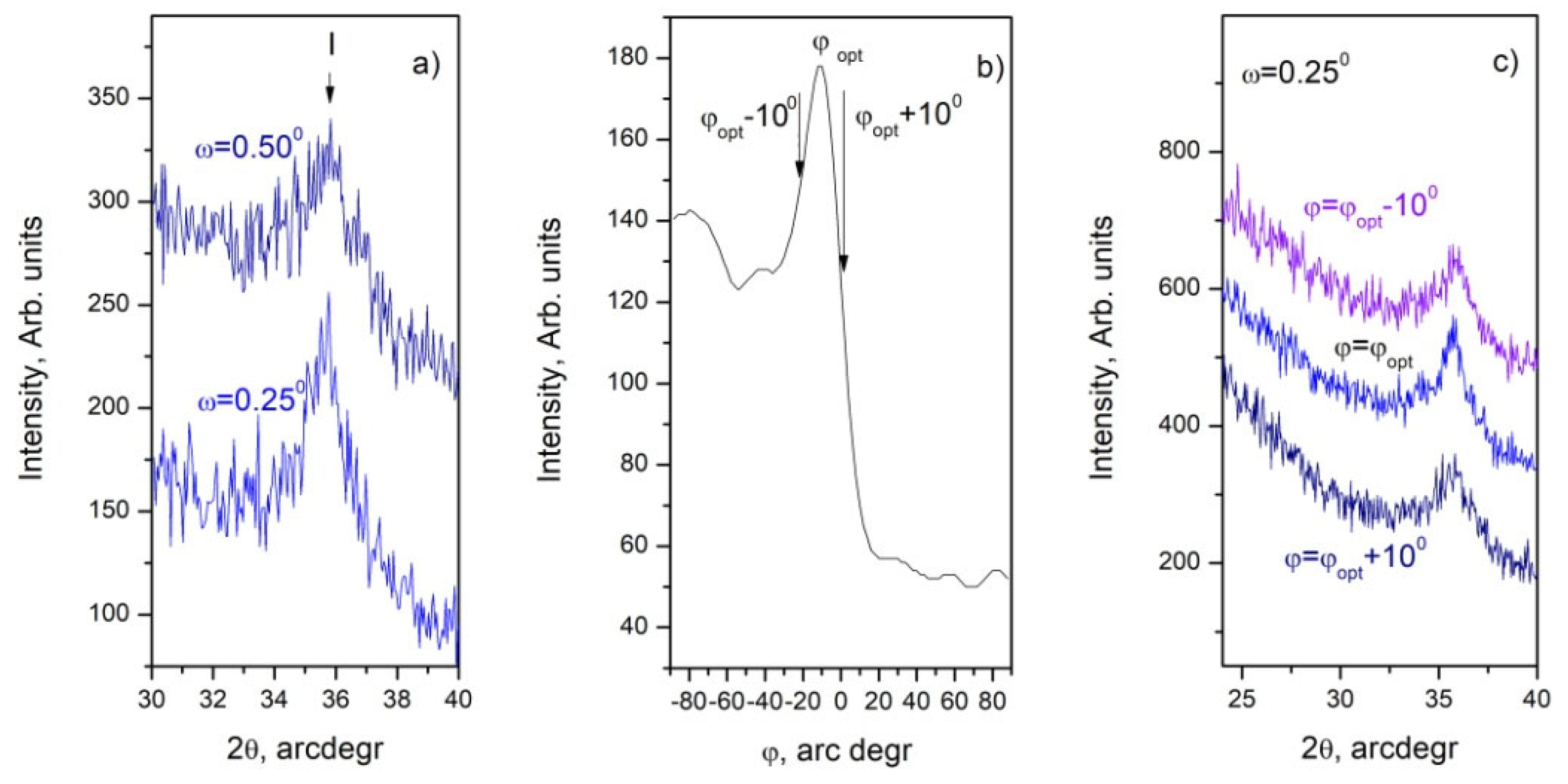
- Regardless of the substrate preparation and the synthesis time, a fraction of SiC is present in the thin films. It is most likely formed in its vast part at the Si substrate/thin film interface from gas phase carbon and Si atoms from the substrate. SiC as a separate phase probably forms a continuous film only in samples cleaned according to the protocols mentioned in the experimental section. Continuous films with a well-ordered crystal structure are formed only when the synthesis time equals or exceeds 60 min. The XRD peaks from the XRD patterns and the SAED and HRTEM images taken from these experiments clearly show a well-ordered crystalline phase, and in both cases, the identified crystalline phase is of the polytype 36H–SiC.
- -
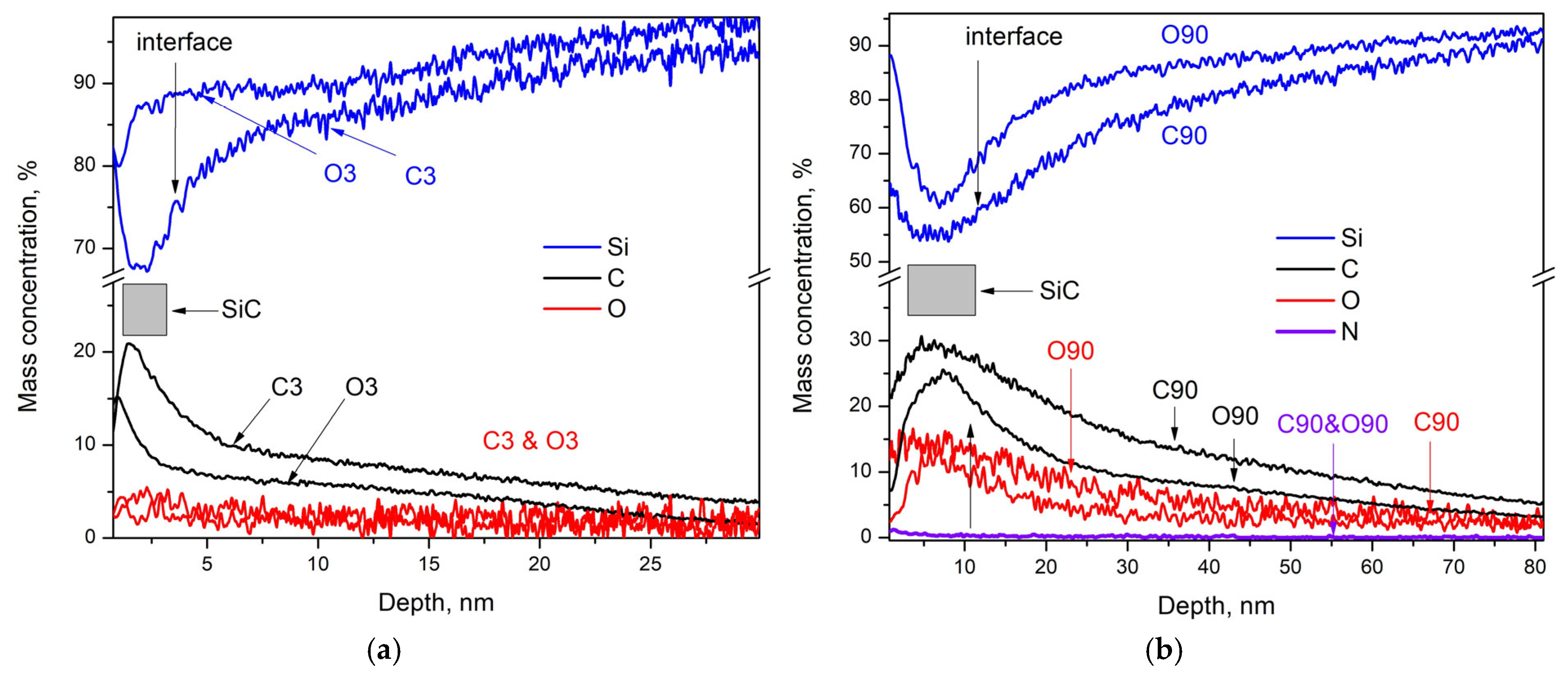
- The processes of permanent reconstruction of the Si(111) slab at high temperatures determine the deep penetration of C and O atoms into the substrate and the blurring of the thin film/Si-substrate interface. This gives rise to certain difficulties in measuring the thickness of these thin films. The detailed distribution of C- and O-containing species (see Figure 4a–f) unequivocally confirms that oxygen and carbon diffuse into the Si substrate, while the SiC phase predominantly remains within the thin film. This process results in the formation of a region extending from the interface into the substrate depth, where Si is doped with C and O. We assume that this region begins at the interface (or at the point where the carbon concentration decreases by 2–4% from its maximum, while the silicon concentration increases correspondingly), and it extends until the C and O concentrations stabilize at their constant residual values. Based on this, the thickness of these regions ranges from approximately 50–60 nm (for C3 samples) to over 100–110 nm (for C90 samples) (see Figure 5a,b and Figure S5d) in the samples with properly cleaned native oxides and is larger in the uncleaned samples (see Figure S5c). This also explains why no clear results are obtained when measuring the thickness of thin films synthesized in long processes (20 min or more) (see the beginning of the Section 3) using SEM, AFM, and optical profilometry. In these methods, thickness determination relies on mechanical surface scratching; however, the high concentration of C and O near the substrate surface significantly alters the silicon properties and affects thin film adhesion, preventing the scratches from accurately defining the thin film/substrate interface. Similarly, ellipsometric measurements yield slightly increased thickness values for thin films—even those with small thicknesses (up to 15 nm)—because for reasons similar to those mentioned above, the upper part of the substrate exhibits characteristics resembling those of the thin film. Consequently, the ellipsometrically determined thicknesses of samples O3, C3, O5, and C5 were 12, 12, 13.5, and 15 nm, respectively, whereas GDOS measurements determined them to be 4, 5, 7, and 8 nm. This suggests that in ellipsometric measurements, part of the heavily doped region of the Si substrate is perceived as part of the thin film.
- -
- As mentioned above, our experiments showed that the thin films consist of crystalline/nanocrystalline/amorphous SiC doped with some O, which is on a C and O co- doped Si- substrate. From general considerations, we can suppose that the complex composition of the thin films and highly enriched-with-C-and-O silicon area will more easily compensate for the difference in the lattice parameters of the SiC and Si substrates as well as their coefficients of thermal expansion than a monophase thin film of nanocrystalline/crystalline SiC. From this, we can conclude that the applied method is promising for depositing intermediate thin films of SiC on Si(111) using quartz equipment, i.e., compatible with the Si technology.
- -
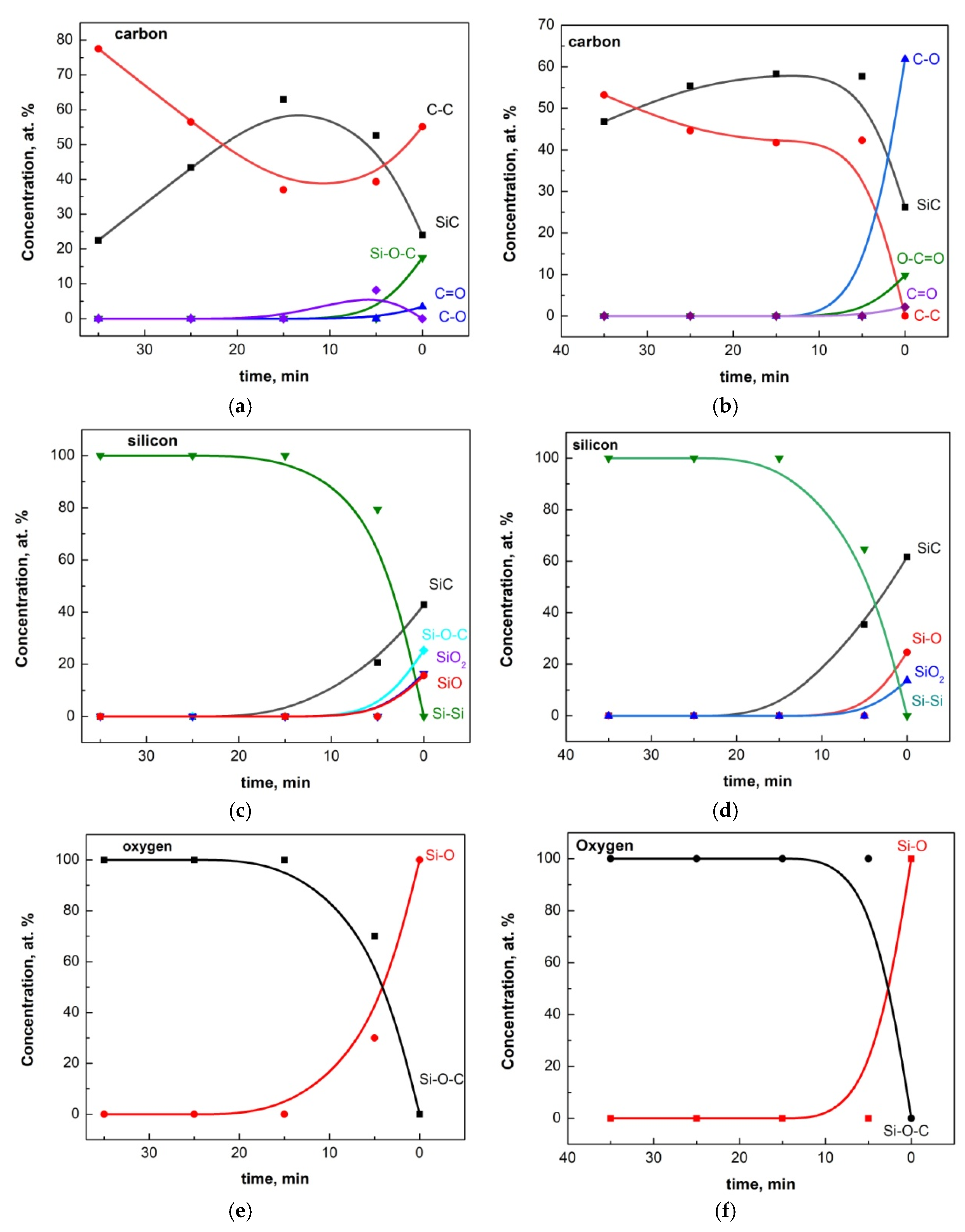
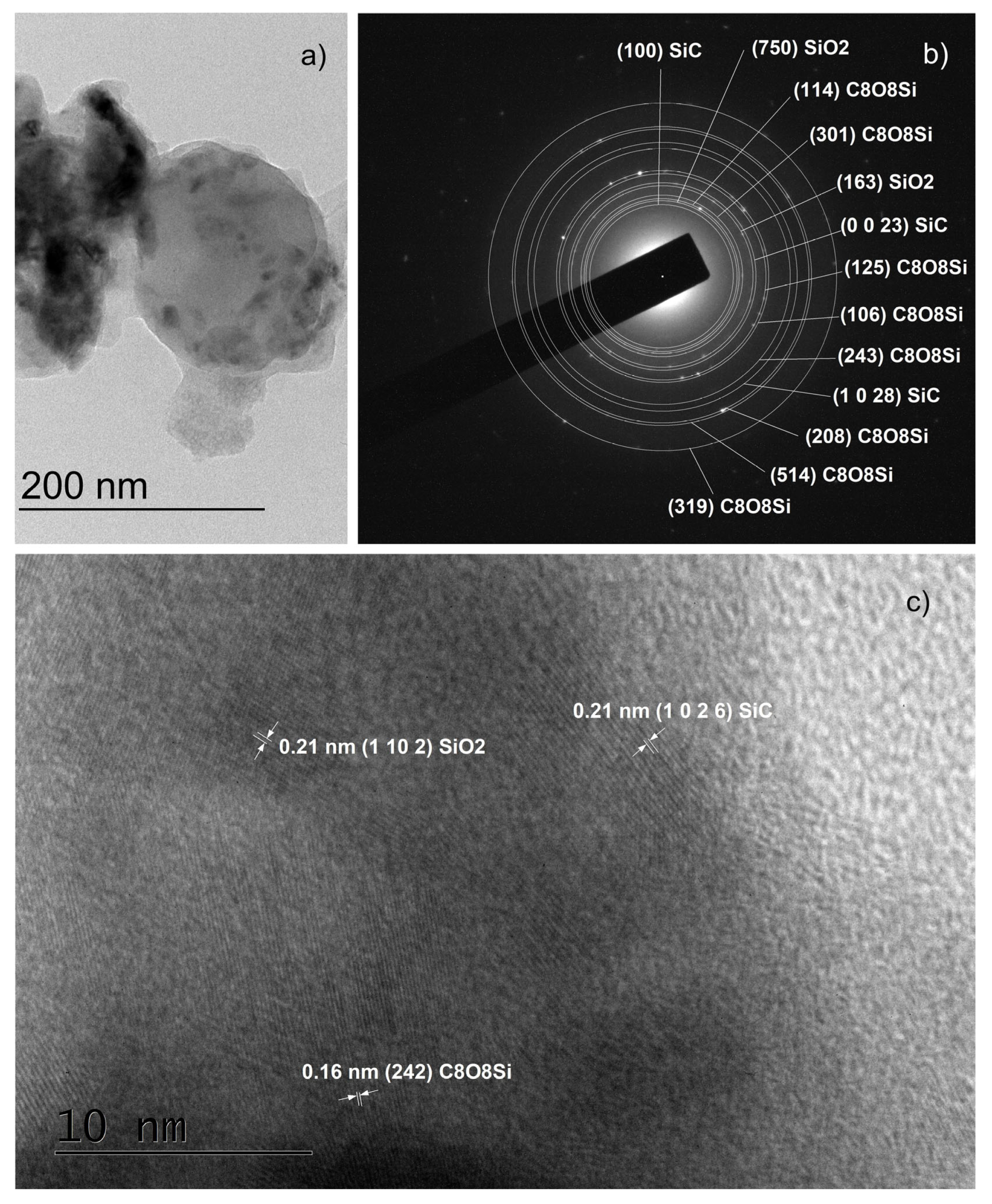
- Carbon and oxygen were observed in the thin films both in the form of impurities, C, O, C–O, C=O, and Si–O–C (see the XPS results), as well as crystalline SiO2 (in the SAED and HRTEM images) and as the complex compound Si(OCOCH3)4 (in the SAED and HRTEM images). The latter compound has a low melting point of 160 °C and most likely is formed in those areas where its components were present in the necessary proportions during the cooling of the thin films. In this sense, its synthesis can be considered accidental rather than typical, and it was found only in two of the samples mentioned above. Moreover, we observed crystalline SiO2 and Si(OCOCH3)4 only in samples with improperly removed native SiO2.
- -
- The XPS results (see Figure 3c and Figure S3a–d) clearly indicate the presence of SiO2 in all studied samples with thin films deposited on substrates where the native oxide was not properly removed. Some of these findings are further supported by other local analysis methods (see the CAED insets and the XRD images referenced above). Although we lack X-ray data confirming crystalline or amorphous silicon oxide, powder XRD is not expected to detect such small quantities of a given phase. Therefore, we can reasonably assume that in all samples with natural oxide—following established protocols—SiO2 is present within the thin films in the form of Si–O, SiO2, and Si–O–C particles. We attribute their presence to the diffusion of residual oxygen, left after etching at 1135–1140 °C in a hydrogen (H2) flow, into the substrate during subsequent heating above 900–1000 °C. The deposition of carbon atoms and their further migration likely amplify this process.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| PVD | Physical Vapor Deposition |
| CVD | Chemical Vapor Deposition |
| UHSS | Ultra-High Strength Steel |
| XPS | X-ray Photoelectron spectroscopy |
| GDOES | Glow Discharge Optical Emission Spectroscopy |
| XRD | X-ray Diffraction |
| GIXRD | Grazing Incidence X-ray diffraction |
| TEM | Transmission Electron Microscopy |
| HRTEM | High-Resolution Transmission Electron Microscopy |
| SAED | Selected Area Electron Diffraction |
References
- Acheson, E.G. Process of Making Graphite. U.S. Patent # US 711031, 14 October 1902. [Google Scholar]
- Xu, X.; Hu, X.; Chen, X. SiC Single Crystal Growth and Substrate Processing. In Light-Emitting Diodes; Solid State Lighting Technology and Application Series; Li, J., Zhang, G.Q., Eds.; Springer: Cham, Switzerland, 2019; Volume 4. [Google Scholar]
- Rebecca, C. Silicon Carbide Microelectromechanical Systems for Harsh Environments; Imperial College Press: London, UK, 2006; p. 3. ISBN 1-86094-624-0. [Google Scholar]
- Käckell, B.; Wenzien, F.B. Electronic properties of cubic and hexagonal SiC polytypes from ab initio calculations. Phys. Rev. B 1994, 50, 10761. [Google Scholar]
- Nishino, S.; Powell, J.A.; Will, H.A. Production of large-area single-crystal wafers of cubic SiC for semiconductor devices. Appl. Phys. Lett. 1983, 42, 460. [Google Scholar]
- Severino, A.; D’Arrigo, G.; Bongiorno, C.; Scalese, S.; La Via, F.; Foti, G. Thin crystalline 3C-SiC layer growth through carbonization of differently oriented Si substrates. J. Appl. Phys. 2007, 102, 023518. [Google Scholar] [CrossRef]
- Kukushkin, S.A.; Osipov, A.V. New method for growing silicon carbide on silicon by solid-phase epitaxy: Model and experiment. Phys. Solid State 2008, 50, 1238. [Google Scholar] [CrossRef]
- Deura, M.; Fukuyama, H. Formation of SiC layer by carbonization of Si surface using CO gas. J. Cryst. Growth 2016, 434, 77. [Google Scholar] [CrossRef]
- Kukushkin, S.A.; Osipov, A.V. Quantum mechanical theory of epitaxial transformation of silicon to silicon carbide. J. Phys. D Appl. Phys. 2017, 50, 464006. [Google Scholar] [CrossRef]
- Kukushkin, S.A.; Osipov, A.V. Nanoscale Single-Crystal Silicon Carbide on Silicon and Unique Properties of This Material. Inorg. Mater. 2021, 57, 1319. [Google Scholar] [CrossRef]
- Kukushkin, S.A.; Markov, L.K.; Pavlyuchenko, A.S.; Smirnova, I.P.; Osipov, A.V.; Grashchenko, A.S.; Nikolaev, A.E.; Sakharov, A.V.; Tsatsulnikov, A.F.; Sviatets, G.V. SiC/Si Hybrid Substrate Synthesized by the Method of Coordinated Substitution of Atoms: A New Type of Substrate for LEDs. Coatings 2023, 13, 1142. [Google Scholar] [CrossRef]
- Molina, S.I.; Morales, F.M.; Araújo, D. SiC thin films obtained by Si carbonization. Mater. Sci. Eng. B 2001, 80, 342–344. [Google Scholar] [CrossRef]
- Severino, A.; Bongiorno, C.; Leone, S.; Mauceri, M.; Pistone, G.; Condorelli, G.; Abbondanza, G.; Portuese, F.; Foti, G.; La Via, F. Carbonization Study of Different Silicon Orientations. Mater. Sci. Forum 2007, 556–557, 171–174. [Google Scholar] [CrossRef]
- Sato, H.; Goto, T.; Okuno, A.; Yoshikawa, A. Preparation of SiC coatings on graphite substrates via CVD using polysilaethylene. J. Cryst. Growth 2025, 649, 127931. [Google Scholar] [CrossRef]
- Steiner, J.; Schultheiß, J.; Wang, S.; Wellmann, P.J. Fabrication of SiC-on-Insulator (SiCOI) Layers by Chemical Vapor Deposition of 3C-SiC on Si-in-Insulator Substrates at Low Deposition Temperatures of 1120_C. Crystals 2023, 13, 1590. [Google Scholar] [CrossRef]
- Kablov, E.N.; Kuznetcov, N.T.; Sarkisov, P.D.; Graschenkov, D.V.; Sevast’yanov, V.G.; Orlova, L.A.; Simonenko, E.P. Method for Protecting Carbon-Containing Materials with Silicon Carbide. Patent RU 2350580, 27 March 2009. [Google Scholar]
- Shikunov, S.; Kaledin, A.; Shikunova, I.; Straumal, B.; Kurlov, V. Novel Method for Deposition of Gas-Tight SiC Coatings. Coatings 2023, 13, 354. [Google Scholar] [CrossRef]
- Chen, B.Y.; Chi, C.C.; Hsu, W.K.; Ouyang, H. Synthesis of SiC/SiO2 core–shell nanowires with good optical properties on Ni/SiO2/Si substrate via ferrocene pyrolysis at low temperature. Sci. Rep. 2021, 11, 233. [Google Scholar] [CrossRef]
- Koo, J.; Chinkyo Kim, C. A new critical growth parameter of H2/CH4 gas flow ratio and mechanistic model for SiC nanowire synthesis via Si substrate carbonization. Sci. Rep. 2024, 14, 29629. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, X.; Xu, N.; Han, C.; Wu, N.; Wang, B.; Wang, Y. Progress of One-Dimensional SiC Nanomaterials: Design, Fabrication and Sensing Applications. Nanomaterials 2024, 14, 187. [Google Scholar] [CrossRef]
- Yang, X.; Liu, R.; Liu, B.; Liu, M. Synthesis of Ultra-Thin Two-Dimensional SiC Using the CVD Method. Energies 2022, 15, 6351. [Google Scholar] [CrossRef]
- Kalchevski, D.A.; Trifonov, D.V.; Kolev, S.K.; Aleksandrov, H.A.; Milenov, T.I. Ab initio study of the mechanism of carbonization of {111} Si-substrate at high temperature. Mater. Chem. Phys. 2024, 317, 129180. [Google Scholar]
- The University of Texas at El Paso, Nanofabrication Facility. Available online: https://www.utep.edu/engineering/nanomil/processes/cleaning.html (accessed on 25 April 2025).
- The Integrated Nanosystems Research Facility at The University of California, Irvine. Available online: https://www.inrf.uci.edu/wordpress/wp-content/uploads/sop-wet-silicon-solvent-clean.pdf (accessed on 25 April 2025).
- Krishna, P.; Verma, A.R. A novel determination of the structure of an anomalous polytype of silicon carbide. Acta Crystallogr. 1964, 17, 51–57. [Google Scholar] [CrossRef]
- Singh, G.; Verma, A.R. The structure of a new silicon carbide polytype 96R. Acta Crystallogr. 1964, 17, 49–51. [Google Scholar] [CrossRef]
- Ramsdell, L.S.; Mitchell, R.S. Studies on Silicon Carbide. Am. Mineral. 1947, 32, 64–82. [Google Scholar]
- Braekken, H. Zur Kristallstruktur des kubischen Karborunds. Z. Krist. 1930, 75, 572–573. [Google Scholar]
- Scherrer, P. Bestimmung der Grösse und der inneren Struktur von Kolloidteilchen mittels Röntgenstrahlen. Nachr. Ges. Wiss. Göttingen 1918, 26, 98–100. [Google Scholar]
- National Institute of Standards and Technology. NIST X-Ray Photoelectron Spectroscopy Database; National Institute of Standards and Technology: Gaitherburg, MD, USA, 1997.
- Önneby, C.; Pantano, C.G. Silicon oxycarbide formation on SiC surfaces and at the SiC/SiO2 interface. J. Vac. Sci. Technol. A 1997, 15, 1597. [Google Scholar] [CrossRef]
- University of Twente, Mesa+ Institute, The Netherlands. Available online: https://www.utwente.nl/en/mesaplus/nanolab/analysis/nanolab-mc-pictures/xps-application-depth-profiling.pdf (accessed on 25 April 2025).
- Foster, M.D.; Treacy, M.M.J.; Higgins, J.B.; Rivin, I.; Balkovsky, E.; Randall, K.H. A systematic topological search for the framework of ZSM-10. J. Appl. Crystallogr. 2005, 38, 1028–1030. [Google Scholar] [CrossRef]
- Kamenar, B.; Bruvo, M. Lattice parameters and space groups of the acetates of IVb group elements. Acta Crystallogr. B 1972, 28, 321. [Google Scholar] [CrossRef]
- Nakashima, S.; Harima, H. Raman Investigation of SiC Polytypes. Phys. Status Solidi A 1997, 162, 39. [Google Scholar] [CrossRef]
- Ferrari, A.; Basko, D. Raman spectroscopy as a versatile tool for studying the properties of graphene. Nat. Nanotechnol. 2013, 8, 235. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Robertson, J. Raman spectroscopy of amorphous, nanostructured, diamond-like carbon, and nanodiamond. Philos. Trans. R. Soc. Lond. A 2004, 362, 2477. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | Plasma Etching | Deposition Time (min) | CH4 Gas Flow (sccm) |
|---|---|---|---|
| C3 | + | 1 × 3 | 10 |
| O3 | + | 1 × 3 | 10 |
| C5 | + | 1 × 5 | 10 |
| O5 | + | 1 × 5 | 10 |
| C20 | + | 1 × 20 | 10 |
| O20 | + | 1 × 20 | 10 |
| C60 | + | 1 × 60 | 10 |
| O60 | + | 1 × 60 | 10 |
| C90 | + | 1 × 90 | 10 |
| O90 | + | 1 × 90 | 10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milenov, T.; Mehandzhiev, V.; Rafailov, P.; Avramova, I.; Zahariev, I.; Avdeev, G.; Karashanova, D.; Georgieva, B.; Terziyska, P.; Kirilov, K.; et al. The Study of the Synthesis of SiC by the Carbonization of Si(111) Substrates: The Role of Native Silicon Oxide. Appl. Sci. 2025, 15, 7078. https://doi.org/10.3390/app15137078
Milenov T, Mehandzhiev V, Rafailov P, Avramova I, Zahariev I, Avdeev G, Karashanova D, Georgieva B, Terziyska P, Kirilov K, et al. The Study of the Synthesis of SiC by the Carbonization of Si(111) Substrates: The Role of Native Silicon Oxide. Applied Sciences. 2025; 15(13):7078. https://doi.org/10.3390/app15137078
Chicago/Turabian StyleMilenov, Teodor, Vladimir Mehandzhiev, Peter Rafailov, Ivalina Avramova, Ivan Zahariev, Georgi Avdeev, Daniela Karashanova, Biliana Georgieva, Penka Terziyska, Kiril Kirilov, and et al. 2025. "The Study of the Synthesis of SiC by the Carbonization of Si(111) Substrates: The Role of Native Silicon Oxide" Applied Sciences 15, no. 13: 7078. https://doi.org/10.3390/app15137078
APA StyleMilenov, T., Mehandzhiev, V., Rafailov, P., Avramova, I., Zahariev, I., Avdeev, G., Karashanova, D., Georgieva, B., Terziyska, P., Kirilov, K., Blagoev, B., Kolev, S., Dimov, D., Kalchevski, D., Karaivanova, D., & Popov, V. (2025). The Study of the Synthesis of SiC by the Carbonization of Si(111) Substrates: The Role of Native Silicon Oxide. Applied Sciences, 15(13), 7078. https://doi.org/10.3390/app15137078