Abstract
Breast, colon, and ovarian cancers are among the most prevalent malignancies worldwide, with distinct clinical features. This study aims to identify key proteins as common biomarkers for breast, colon, and ovarian cancer through protein analysis, molecular mechanisms, and patient sample validation. Data mining from curated databases identified 483 proteins for breast cancer, 521 for colon cancer, and 223 for ovarian cancer. Interaction network analysis revealed shared clusters involved in cancer progression, DNA repair, and cell proliferation. A core set of 27 proteins was found to be common across all three cancer types, participating in key biological processes such as DNA damage response, cell proliferation, and apoptosis. Notably, these proteins are implicated in KEGG pathways linked to multiple cancers. Differential gene expression analysis revealed significant alterations in the expressions of MSH2 and KIT across the three cancers, suggesting their potential as common biomarkers. The high expression of these proteins was associated with better survival outcomes, highlighting their potential as common biomarkers for breast, colon, and ovarian cancers. The in-silico methodology integrated various bioinformatic tools—including cluster identification, gene expression profiling, protein network visualization, and biomarker prediction—enhancing the understanding of shared molecular mechanisms and potential therapeutic targets.
1. Introduction
Cancer is a global health issue. Globally, colon and breast cancers are regarded as among the most significant and lethal, alongside lung and prostate cancers. Breast and colon cancer are the most common cancers in women worldwide, ranking first and second, respectively. Colon cancer is the third most prevalent cancer overall [1]. Women with a family history of breast cancer are at a higher risk of developing ovarian cancer [2]. Other risk factors that can increase the likelihood of developing both breast and ovarian cancer include advanced age, not having children, or having children at an older age. Additionally, mutations in specific genes and the use of hormone replacement therapy are also associated with increased risk [2]. There is evidence of a genetic predisposition to develop cancer in the same location or organ in postmenopausal women with family histories of colon, breast, uterine, and ovarian cancers [3]. In this way, patients with breast cancer have a higher risk of developing colorectal neoplasia [4]. As individuals age, the incidence of breast and ovarian cancer tends to rise [5]. Lynch syndrome, also known as hereditary non-polyposis colorectal cancer, and hereditary breast and ovarian cancer syndrome are the most common genetic causes of cancer within families, due to mutations in DNA mismatch repair genes [6]. Approximately 15–20% of cancer patients have a family-related incidence, yet only about 10% inherit mutations in genes that significantly increase their cancer risk. In some instances, malignant pathogenic variants may occur without a discernible family history of cancer [2,6].
The term “synchronous tumors” refers to the simultaneous appearance of multiple primary malignant tumors in the same patient. These tumors occur in different organs, have distinct pathological characteristics, and do not show metastasis between them. There has been a notable increase in the incidence of synchronous tumors, which were previously considered rare [3,4]. These tumors have been associated with various types of cancer, including breast, melanoma, gastrointestinal, colorectal, gynecological, and ovarian cancers. However, the mechanisms underlying these associations between different cancers have not yet been clearly understood [7]. Research shows that ovarian cancer tends to occur alongside other common cancers in families. As a result, having first-degree relatives (such as a father or mother) who have had breast, ovarian, colorectal, or prostate cancer is associated with a higher risk of developing ovarian cancer in female offspring (daughters) [8]. Family medical history should be reviewed to identify susceptibility to gynecological cancer, especially in premenopausal women, due to previously reported genetic predisposition [9]. For instance, Llinás-Quintero et al. [10] reported the case of a woman with a family history of pancreatic and colon cancer who developed synchronous ovarian cancer (bilateral ovarian adenocarcinoma) and breast cancer (triple-negative medullary breast carcinoma), which was associated with a novel mutation in the BRCA2 gene. Some studies examined the incidence of second malignancies in women diagnosed with ovarian cancer. The findings revealed a statistically significant increase in the risk of developing breast cancer after a diagnosis of ovarian serous adenocarcinoma [8,10]. Furthermore, there was an association between ovarian mucinous adenocarcinoma and subsequent rectal cancer. This increased risk may be related to genetic predispositions and underlying susceptibilities associated with ovarian cancer, which may also increase the likelihood of breast and colorectal cancer, thus complicating subsequent medical treatments [11]. The most common primary sites for ovarian metastases include the colon (15–32%), breast (8–28%), stomach (6–22%), and appendix (2–20%) [12]. The prognosis for patients with colon cancer who have ovarian metastases, especially if the metastases are bilateral, is poor. Optimal surgical cytoreduction—removing as much of the cancerous tumor as possible, along with affected organs and tissues in the pelvis and abdomen—significantly improves the survival rate of these patients compared to suboptimal cytoreduction, where not all tumors are removed [12]. Colorectal cancer typically metastasizes to the liver and lungs. However, it sometimes spreads to the ovary and, rarely, to the breast. There has been one reported case of a patient with colon cancer that initially spread to the ovary and, after receiving palliative chemotherapy, further metastasized to the breast [13].
Metastatic breast tumors that originate from other organs are very rare. However, an increased risk of developing secondary primary cancers in other organs has been shown to occur after treatment for ovarian cancer, which may be influenced by the therapy administered as well as by common risk factors [14]. Patients with ovarian cancer who received radiotherapy or chemotherapy had an increased risk of subsequently developing colon cancer, bladder cancer from radiotherapy alone, leukemia from chemotherapy, and lymphoma from either treatment [14]. Mori et al. [15] described a case involving a patient with a p53 mutation who had ovarian cancer that later metastasized to the breast. Mutations in BRCA1 and BRCA2 genes significantly elevate the hereditary risk of developing breast and ovarian cancer. Additionally, these mutations are linked to an increased risk of colon cancer, indicating a broader susceptibility to various types of cancer in individuals who carry this mutation [16]. Defects in the DNA mismatch repair genes can increase the likelihood of developing colorectal cancer as well as other cancers, including those of the endometrium, ovaries, gastrointestinal tract, and kidneys. Furthermore, mutations in certain genes involved in homologous recombination repair pathways, such as CHEK2, PALB2, ATM, RAD51C, RAD51D, and BRIP1, may also contribute to the development of hereditary cancer syndromes [17].
Ovarian, breast, and colon cancers are highly complex and necessitate advanced technologies for diagnosis and treatment [7]. Proteomic biomarkers, therapeutic targets, identification of key signaling pathways, cancer subtype classification, and liquid biopsies are significant contributions of proteomics that have led to substantial progress in breast, ovarian, and colon cancers. Proteomics, in conjunction with genomics, enables precision medicine and personalized treatment predictions, enhancing clinical outcomes for patients [18]. Thus, the identification of biomarkers is essential for diagnosing, predicting, and assessing the prognosis of complex diseases like cancer. There is substantial research potential to better understand different types of tumors by cataloging key mutations and the molecular networks that underlie this complex disease [19]. Nowadays, numerous molecular markers are widely used in cancer evaluation, prediction, monitoring and diagnosis. The levels of these markers are dynamic and fluctuate according to tumor progression and disease stage [5]. However, given the complexity of cancer, its diverse subtypes with multiple genetic backgrounds, and the need for a personalized approach, expanding knowledge of the underlying molecular mechanisms and advancing the search for new biomarkers is essential.
The aim of this study is to identify key proteins that can serve as reliable common biomarkers for breast, colon, and ovarian cancer through a comprehensive analysis of common proteins, molecular mechanisms, and pathways, as well as validating findings using patient samples via an in-silico analysis approach. The ultimate purpose of this research is to identify biomarkers that could be useful for enhancing early detection, tailoring treatments, predicting therapy responses, and/or identifying risk groups, through further studies; this would represent a significant advance in precision medicine and, consequently, in the clinical management of patients.
2. Materials and Methods
2.1. Study Strategy and Analysis
The search for common molecular mechanisms and biomarkers for breast, colon, and ovarian cancer has followed a three-stage analysis strategy [20], as described in Figure 1. First, a data mining process was conducted, followed by a Gene Ontology (GO) enrichment analysis, and a Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis. The third stage involved cluster identification and a protein–protein interaction analysis. Finally, an in-silico validation using patient data was performed.
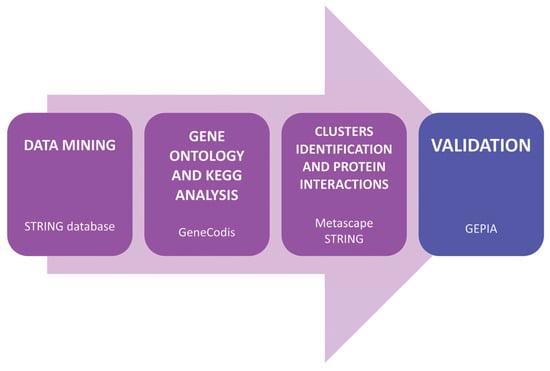
Figure 1.
Diagram of the in-silico analysis process and databases used.
2.2. Data Mining and Protein Screening
The data mining was performed in the Search Tool for the Retrieval of Interacting Genes (STRING) database (https://string-db.org/) (version 12.0) (Accessed on 30 January 2025) [21]. All human proteins involved in breast, colon, and ovarian cancer, obtained from curated databases, were downloaded. To ensure the accuracy and relevance of the analyzed data, a rigorous criterion for the inclusion and exclusion of proteins was applied. Only proteins described in curated databases within STRING were considered, ensuring their support through experimental evidence or systematic reviews. Proteins reported exclusively in scientific literature without validation in curated databases were excluded, avoiding potential biases caused by unverified data. Additionally, only proteins corresponding to the search terms specified in Table 1 were selected, ensuring their direct relevance to the biological processes of breast, colon, and ovarian cancer. This approach enabled the identification of a highly reliable and functionally relevant set of proteins for studying shared mechanisms among these cancer types.

Table 1.
Compendium of search terms used in data mining of breast, colon, and ovarian cancer-related proteins in Homo sapiens.
STRING was selected for its reliability and widespread use in molecular biology research. It provides access to multiple curated databases containing protein interactions supported by both experimental data and in-silico predictions, ensuring a robust foundation for network analysis.
For statistical analysis, STRING automatically applies False Discovery Rate (FDR) correction using the Benjamini–Hochberg procedure. This method controls the expected proportion of false positives among the rejected null hypotheses, ensuring a balance between sensitivity and specificity in biomarker selection. Compared to more conservative approaches like Bonferroni correction, FDR provides greater statistical power while maintaining control over Type I errors. This ensures that the reported associations reflect meaningful biological relevance in the context of cancer research.
While other databases such as CPTAC, TCGA, or HPA provide valuable information on protein expression and clinical data, the objective of our study was not to evaluate expression differences at the tissue level or within clinical cohorts, but rather to identify proteins with strong evidence of interactions across multiple cancer types while considering biological information.
Finally, a protein list for each tumor type was obtained, after removing duplicate proteins. The three protein lists were then compared to identify the common proteins across the different tumor types. This comparison was performed using a Venn diagram (https://bioinfogp.cnb.csic.es/tools/venny/) (Venny 2.1.0) (Accessed on 7 February 2025) [22]. Subsequent analyses were carried out with the proteins common to breast, colon, and ovarian cancer.
2.3. Gene Ontology and KEGG Pathways Analysis
The common proteins found for breast, colon, and ovarian cancer were used to perform a GO functional enrichment analysis and subsequently a functional study of KEGG pathways. Both analyses were carried out using the GeneCodis4 application (https://genecodis.genyo.es/) (Accessed 10 February 2025) [23,24].
The hypergeometric test was employed as a standard method for enrichment analysis to determine whether specific gene sets are represented more frequently in biological categories than expected by chance. This approach ensures a rigorous evaluation of the functional relevance of identified biomarkers. The statistical threshold was set at p < 0.05; however, the results obtained show highly significant associations, with p-values as low as 5 × 10−8 or even lower. These values reinforce the reliability of the identified associations, minimizing the risk of false positives and ensuring high specificity in biomarker selection. Given the large datasets analyzed, applying a highly restrictive statistical filter enhances confidence in the findings, aligning with best practices in biomarker discovery.
The online application SRplot (https://www.bioinformatics.com.cn/en) (version © 2025) (Accessed 12 February 2025) was used to generate the GO and KEGG graphs [25].
2.4. Clusters Identification and Protein-Protein Interaction Analysis
Protein interaction networks and cluster identification were performed using Metascape (http://metascape.org) (version 3.5) (Accessed 18 February 2025) [26]. This application uses the Molecular Complex Detection (MCODE) algorithm to identify neighborhoods where proteins are densely connected. Subsequently, the interaction between the proteins identified in each MCODE cluster was analyzed using the STRING database (version 12.0).
Metascape integrates various functional enrichment resources within a unified analytical framework, facilitating the interpretation of biological processes in an efficient and comprehensive manner. Additionally, Metascape employs the MCODE algorithm to identify core genes and proteins based on biologically meaningful interactions, allowing for the detection of highly interconnected molecular clusters. The combination of these tools enables a systematic approach to biomarker identification.
2.5. Validation
The validation of the proteins found in each MCODE cluster was performed on the Gene Expression Profiling Interactive Analysis (GEPIA2) website (http://gepia2.cancer-pku.cn/#index) (Accessed 25 February 2025) [27]. This application uses RNA-seq data and survival data from over 9000 tumors and more than 8000 patient samples from TCGA and the GTEx project. Differential gene expression analysis was performed to identify genes consistently altered across breast, colon, and ovarian cancer.
To identify potential biomarkers relevant across multiple cancer types, a combined survival analysis was performed, integrating patient data from breast, colon, and ovarian cancer. Despite their biological differences, this approach aimed to highlight molecular mechanisms shared across tumor types. Statistical controls and normalization techniques were applied to minimize confounding effects, ensuring the reliable identification of genes with cross-cancer relevance.
The latest access of all the databases mentioned in the materials and methods section was undertaken in February 2025.
3. Results
3.1. Data Mining and Protein Screening
The data mining performed using the terms presented in Table 1 returned a dataset of annotations related to each of the terms used in STRING. Only results from curated databases were considered. Supplementary Tables S1–S3 show the results obtained for each of the terms used in breast, colon, and ovarian cancer, respectively. A total of 922 proteins were downloaded based on the search terms used for breast cancer. They were grouped by category, including diseases, tissues, and metabolic pathways. The information covered adenocarcinoma (32 proteins), breast cancer in general (636 proteins), carcinoma (112 proteins), neoplasm (72 proteins), sarcoma (4 proteins), lymphoma (4 proteins), and other subtypes (62 proteins). Supplementary Table S2 presents search terms related to different types of colorectal cancer and its variants. They are grouped by category, including diseases, tissues, and metabolic pathways, with a total of 1194 proteins identified for all terms. The information covers adenocarcinoma (280 proteins), carcinoma (62 proteins), neoplasm (218 proteins), colon cancer (98 proteins), colonic cancer cell (323 proteins), colorectal cancer (184 proteins), and other categories and subtypes (29 proteins). The terms related to different types of ovarian cancer and its variants were grouped by category in the same way, with a total of 558 proteins. The results show 112 proteins for carcinoma, 103 proteins for neoplasm, 24 proteins for adenocarcinoma, 268 proteins for ovarian cancer in general and 51 proteins associated with other types and subtypes (Supplementary Table S3).
Duplicate proteins were then removed. Finally, the results show 483 unique proteins for breast cancer, 521 for colon cancer, and 223 for ovarian cancer (Supplementary Table S4).
3.2. Interaction Networks
The analysis of protein interaction networks revealed multiple clusters, with Figure 2 highlighting the twenty most enriched ontology clusters for each tumor type.
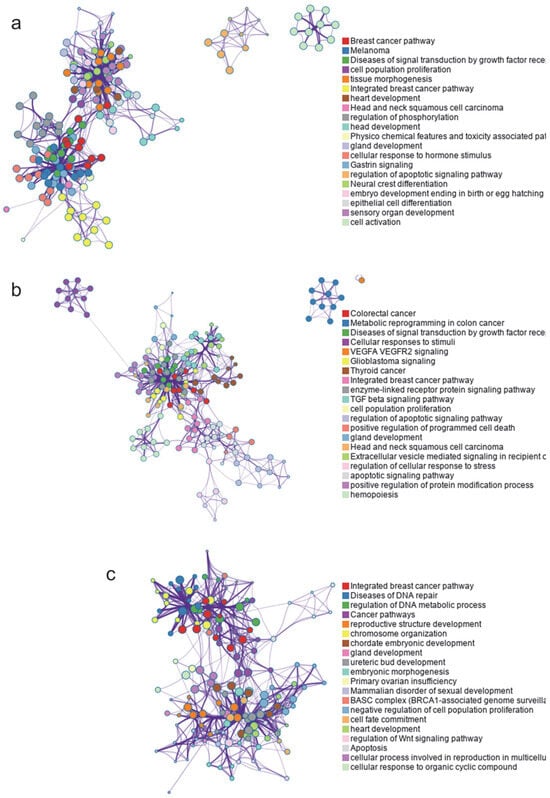
Figure 2.
Protein interaction networks and identified clusters for all proteins involved in each type of cancer. (a) Breast cancer, 483 proteins. (b) Colon cancer, 521 proteins. (c) Ovarian cancer, 223 proteins.
A subset of representative terms from the full cluster was selected and organized into a network layout. Each term is represented as a circular node, with its size proportional to the number of input genes associated with that term. Node colors indicate cluster identity, grouping terms of the same cluster. Terms with a similarity score greater than 0.3 are connected by edges, the thickness of which reflects the similarity score.
The interaction network of proteins related to breast cancer identified the most enriched ontological clusters as those associated with cancer-specific pathways, melanoma, signal transduction diseases via growth factor receptors, and cell proliferation, among others (Figure 2a). For colorectal cancer, the most enriched clusters included metabolic reprogramming, signal transduction diseases, and cellular response to stimuli, among others (Figure 2b). Finally, the enriched clusters for ovarian cancer showed a connection to breast cancer pathways, DNA repair diseases, and the regulation of metabolic processes, as well as the development of reproductive structures and chromosomal organization, etc. (Figure 2c).
As shown in Figure 2, some of the identified clusters are common in breast, colon, and/or ovarian cancer, suggesting the presence of shared molecular mechanisms involved in these cancers. This highlights the need to identify common proteins among these tumor types in order to study the connections between them.
3.3. Identification of Common Proteins
The identification of common proteins among breast, colon, and ovarian cancer was performed using a Venn diagram to compare the protein lists presented in Supplementary Table S4.
The results show 24 common proteins exclusively shared between breast cancer and ovarian cancer, 90 between breast cancer and colon cancer, and 37 common proteins between colon cancer and ovarian cancer. It is essential to emphasize that the diagram identified 27 proteins commonly shared among all three types of cancer (Figure 3). This finding suggests a significant relationship not only between breast and ovarian cancer, as described in the literature, but also with colon cancer.
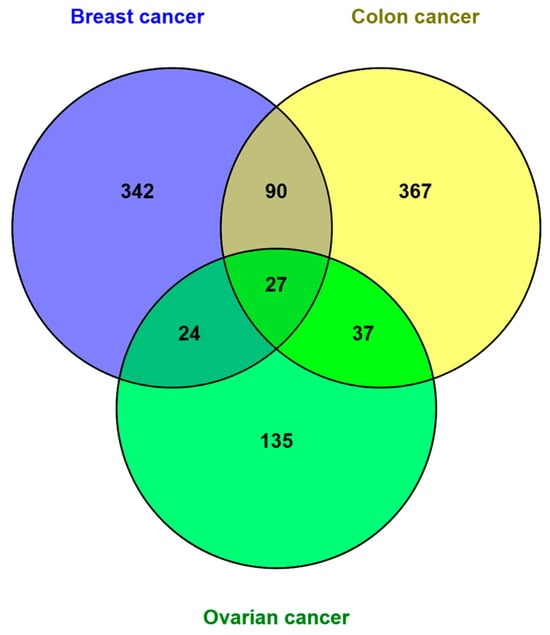
Figure 3.
Comparison of the proteins identified in breast (483), colon (521), and ovarian (223) cancer through the Venn diagram. The diagram shows that the three types of tumors share 27 common proteins.
Table 2 shows the 27 common proteins identified for breast, colon, and ovarian cancer, with a brief description of their function.
Table 2.
List of common proteins identified in breast, colon, and ovarian cancer with description obtained from the STRING database (version 12.0).
3.4. Gene Ontology and KEGG Pathways Analysis
The GO enrichment study showed that the 27 proteins shared between breast, colon, and ovarian cancer participate in 731 biological processes out of a total of 1027 (p < 0.05, hypergeometric test) (Supplementary Table S5). In Figure 4a, the 20 biological processes with the highest statistical significance (p < 9 × 10−5), largest number of implicated genes, and enrichment are shown. The processes with the highest statistical significance included response to gamma radiation (p = 8.66 × 10−10), DNA damage response (p = 8.66 × 10−10), cell population proliferation (p = 2.14 × 10−8), and apoptotic signaling in response to DNA damage (p = 2.27 × 10−8). The most enriched processes were the somatic recombination of immunoglobulin gene segments and DNA damage response mediated by p53 signal transduction. The processes with the highest number of implicated genes were apoptosis and its regulation, as well as the response to DNA damage (Supplementary Table S5).
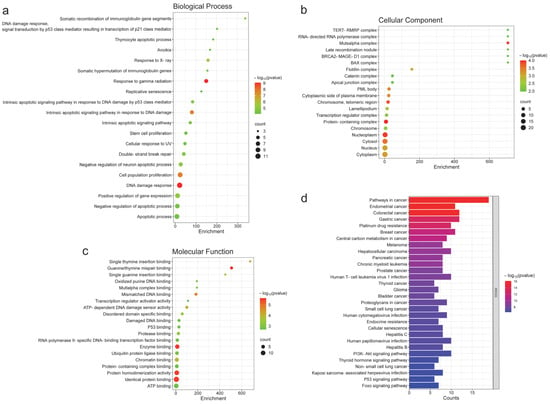
Figure 4.
Gene Ontology and KEGG pathways analysis of the 27 shared proteins across breast, colon, and ovarian cancer. (a) Top 20 biological processes (p < 9 × 10−5). The highest statistical significance included ranged from p = 8.66 × 10−10 to p = 2.27 × 10−8. (b) Top 20 cellular components (p < 0.05). The highest statistical significance included ranged from p = 9.96 × 10−5 to p = 1.19 × 10−4. (c) Top 20 molecular functions (p < 0.005). The highest statistical significance showed a p-value of 3.05 × 10−6. (d) Top 30 KEGG pathways (p < 5 × 10−7). The KEGG pathways with the highest statistical significance ranged from 7.42 × 10−17 to 2.6 × 10−7. This figure only represents the annotations with the highest enrichment, number of genes, and statistical probability (hypergeometric test).
The enrichment analysis of cellular components showed the involvement of these proteins in 80 components out of a total of 159 (p < 0.05, hypergeometric test) (Supplementary Table S6). The most enriched cellular component, which also had the highest probability, was the MutSalpha complex (p = 1.19 × 10−4). However, other less enriched cellular components showed high probability, with more implicated proteins. Notably, these include the telomeric region of chromosomes (p = 1.53 × 10−4), the protein-containing complex (p = 9.96 × 10−5), and the nucleoplasm (p = 1.19 × 10−4) (Supplementary Table S6). Figure 4b shows the 20 most significant components based on their statistical probability (p < 0.01).
The study of molecular functions revealed that the 27 common proteins participate in 120 functions out of a total of 208 (p < 0.05, hypergeometric test) (Supplementary Table S7). Figure 4c presents the 20 most significant functions (p < 0.005). In this regard, guanine/thymine mispair binding stands out as the most probable (p = 2.37 × 10−6) and enriched function. Other functions highlighted due to their high probability and greater number of implicated proteins were enzyme binding (p = 3.05 × 10−6), protein homodimerization activity (p = 3.05 × 10−6), and identical protein binding (p = 3.05 × 10−6) (Supplementary Table S7) (Figure 4c).
The KEGG pathway analysis showed the involvement of these 27 proteins in 107 statistically significant pathways out of a total of 172 (p < 0.05, hypergeometric test) (Supplementary Table S8). The 30 most significant pathways (p < 3.5 × 10−7) with the highest number of implicated proteins are shown in Figure 4d. The KEGG pathways that stand out due to their high probability and number of implicated proteins are related to various types of cancer (p-value ranging from 2.6 × 10−7 to 7.42 × 10−17)—pathways in cancer, endometrial cancer, colorectal cancer, gastric cancer, platinum drug resistance, breast cancer, carbon metabolism in cancer, etc. It is important to highlight the relationship between these common proteins in breast, colon, and ovarian cancer and pathways involved in infections caused by various viruses (p-value ranging from 2.6 × 10−7 to 2.74 × 10−9)—human T-cell leukemia virus 1, human cytomegalovirus, hepatitis C, human papillomavirus, hepatitis B, and Kaposi sarcoma-associated herpesvirus (Supplementary Table S8).
The proteins involved in each KEGG pathway were examined after the analysis. It was found that the number of common proteins implicated was reduced to 24.
3.5. Clusters Identification
A subsequent Metascape analysis of these 24 proteins was conducted to identify clusters. The results reveal that these proteins are involved in multiple clusters, with a significant presence across the three tumor types studied (p-values ranging from 1 × 10−2 to 1 × 10−20) (Figure 5a).
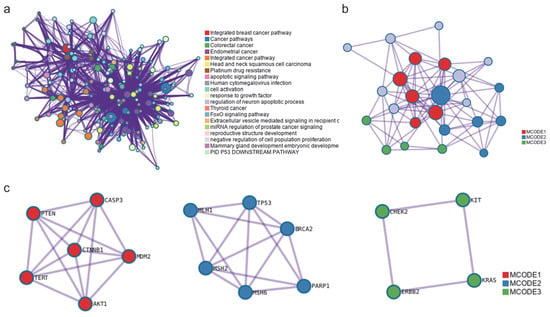
Figure 5.
Ontology enrichment analysis performed on the 24 most common genes identified after the KEGG analysis. (a) The cluster network is displayed with nodes representing individual terms, where node size corresponds to the number of input proteins associated with each term, and the color indicates cluster identity. Terms with a similarity score greater than 0.3 are connected by edges, with edge thickness reflecting the degree of similarity. p-values ranged from 1 × 10−2 to 1 × 10−20. (b) Protein–protein interaction components identified by MCODE algorithm. This network displays in red, blue, and green the proteins that form part of MCODE1, MCODE2, and MCODE3, as identified, along with their connections. The proteins marked in grey, although connected within the network, do not belong to the subset of proteins identified by the MCODE algorithm. (c) MCODE clusters identified. The p-values for MCODE1 ranged from 2 × 10−11 to 1.58 × 10−12, those for MCODE2 ranged from 5.01 × 10−12 to 7.94 × 10−13, and those for MCODE3 ranged from 5.01 × 10−7 to 5.01 × 10−8 (hypergeometric test).
All protein–protein interactions (PPI) among input genes were extracted from the PPI data source and formed a PPI network. GO enrichment analysis was applied to the network to extract “biological meanings”. The MCODE algorithm was then applied to this network to identify neighborhoods where proteins are densely connected. The analysis identified three interrelated MCODE clusters within the network (Figure 5b). Figure 5c shows the independently identified MCODE clusters along with the interactions between the proteins that comprise them. The p-values for MCODE1 ranged from 2 × 10−11 to 1.58 × 10−12, those for MCODE2 ranged from 5.01 × 10−12 to 7.94 × 10−13, and those for MCODE3 ranged from 5.01 × 10−7 to 5.01 × 10−8 (hypergeometric test).
Table 3 lists the proteins included in each of the identified MCODE clusters (Figure 5c), along with the statistical probability range according to the hypergeometric test. From the initial 24 proteins obtained through the KEGG pathway analysis, the algorithm used in Metascape allowed for the identification of the most important core proteins, reducing the number to the 16 listed in Table 3. Six of these correspond to MCODE 1, another six to MCODE 2, and four to MCODE 3.
Table 3.
Proteins included in each MCODE cluster.
3.6. Protein-Protein Interaction Analysis
After identifying the core proteins of the MCODE clusters, a PPI analysis was conducted. Figure 6 illustrates the network of physical interactions among all the proteins. The dashed red lines separate the three MCODE clusters. The group of proteins on the left corresponds to MCODE1, the central group to MCODE2, and the group on the far right to MCODE3. It can be observed that the proteins TERT, CTNNB1, AKT1, MDM2, TP53, MLH1, MSH2, MSH6, CHEK2, and ERBB2 form a subnetwork with stronger interaction lines. The GO enrichment analysis performed on these clusters identified 10 biological processes with significant gene involvement and statistically significant values (False Discovery Rate (FDR) ranging from 3 × 10−6 to 1 × 10−11). Notably, these proteins are involved in radiation response and intrinsic apoptotic signaling in response to DNA damage (Figure 6).
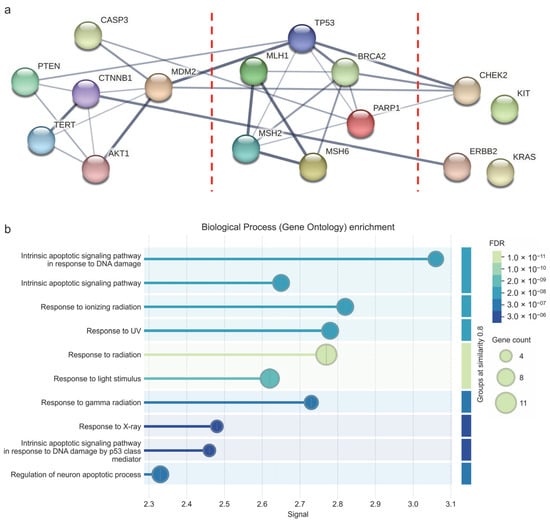
Figure 6.
Protein–protein interaction analysis of MCODE clusters. (a) Physical interaction network. The red dotted lines separate the three MCODE clusters. The leftmost protein cluster corresponds to MCODE1, the middle cluster to MCODE2, and the rightmost cluster to MCODE3. The edges indicate that the proteins are part of a physical complex. Line thickness indicates the strength of data support. The minimum required interaction score was adjusted to medium confidence: 0.400. (b) Gene Ontology enrichment showing the biological processes in which the proteins of the MCODE clusters intervene. FDR values ranged from 3 × 10−6 to 1 × 10−11. FDR: False Discovery Rate. Images obtained from STRING.
Continuing with the PPI analysis in STRING, the involvement of the MCODE cluster proteins in multiple KEGG pathways was evaluated. The study revealed that these proteins participate in pathways related to multiple cancer types, such as colorectal, endometrial, prostate, breast, gastric, glioma, pancreatic, and melanoma, among others (FDR values ranging from 0.0067 to 1.92 × 10−13) (Figure 7a). They are also involved in cancer-related mechanisms, such as platinum drug resistance, carbon metabolism, and apoptosis (FDR values ranging from 1.18 × 10−5 to 3.25 × 10−18) (Figure 7b), as well as cancer signaling pathways, including thyroid hormone signaling, p53 signaling, PI3K-Akt signaling, and MAPK signaling (FDR values ranging from 1.23 × 10−7 to 0.0071) (Figure 7c).
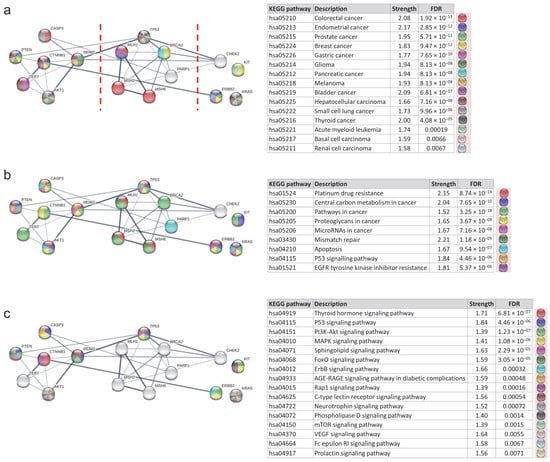
Figure 7.
Protein–protein interaction network of MCODE clusters—analysis of KEGG pathways. (a) KEGG pathways related to different types of cancer. FDR values ranged from 0.0067 to 1.92 × 10−13. The red dotted lines separate the three MCODE clusters. The leftmost protein cluster corresponds to MCODE1, the middle cluster to MCODE2, and the rightmost cluster to MCODE3. (b) KEGG pathways related to mechanisms in cancer. FDR values ranged from 1.18 × 10−5 to 3.25 × 10−18. (c) KEGG pathways related to signaling in cancer. FDR values ranged from 0.0071 to 1.23 × 10−7. FDR: False Discovery Rate.
It is important to highlight the involvement of certain proteins (PTEN, MDM2, TP53, AKT1, and KRAS) in pathways related to cellular senescence (FDR = 1.68 × 10−6) and longevity regulation (FDR = 0.0063) (Supplementary Figure S1a). The analysis also revealed that the MCODE cluster proteins involved in tumor processes were strongly connected to infectious processes. Specifically, a significant role was noted for the proteins of the MCODE1 cluster (CASP3, CTNNB1, MDM2, and AKT1) in infections caused by Salmonella, Shigella, and Mycobacterium tuberculosis (FDR ranging from 0.0036 to 0.0292) (Supplementary Figure S1b), with a particular association of CASP3, PTEN, CTNNB1, MDM2, TERT, AKT1, TP53, and KRAS with multiple viral infections, such as human papillomavirus, cytomegalovirus, hepatitis C, Kaposi sarcoma-associated herpesvirus, Epstein–Barr virus, and human immunodeficiency virus 1 (FDR ranging from 2.54 × 10−9 to 0.0244) (Supplementary Figure S1c).
The REACTOME analysis revealed that the proteins in MCODE2 (TP53, MLH1, BRCA2, MSH2, MSH6, PARP1) were primarily involved in pathways related to DNA repair, such as mismatch repair diseases, DNA repair disorders, and defective mismatch repair associated with various proteins, as well as the regulation of DNA repair genes’ transcription via TP53 (FDR values ranging from 0.0037 to 1.20 × 10−6) (Figure 8a). However, it was primarily the proteins from MCODE1 (CASP3, CTNNB1, AKT1) that were implicated in apoptosis, pyroptosis, and the apoptotic cleavage of cell adhesion proteins (FDR values ranging from 0.00093 to 0.009) (Figure 8b).
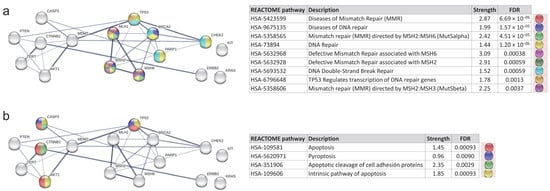
Figure 8.
Protein–protein interaction network of MCODE clusters—analysis of REACTOME pathways. (a) REACTOME pathways related to DNA repair. FDR values ranged from 0.0037 to 1.20 × 10−6. (b) REACTOME pathways related to apoptosis and pyroptosis. FDR values ranged from 0.00093 to 0.009.
3.7. Validation
The PPI analysis revealed the involvement of core proteins in multiple KEGG and REACTOME pathways associated with breast, colon, and ovarian cancer. These results suggest the potential for identifying these proteins as common markers across these types of cancer. However, to clarify this possibility and identify common biomarkers, a preliminary validation using cancer patient samples was performed using the GEPIA2 database. The expression levels (RNA-seq) of 16 proteins from the MCODE1, MCODE2, and MCODE3 clusters in patients with breast cancer, colon cancer, rectum cancer, and ovarian cancer, compared to normal tissue expression levels, were analyzed (Figure 9 and Supplementary Figure S2).
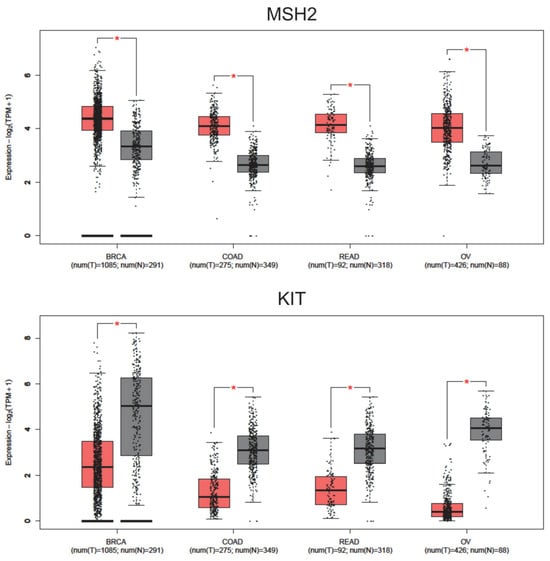
Figure 9.
Expression levels (RNA-seq) of MSH2 and KIT proteins in tumor patient samples with breast invasive carcinoma (BRCA), colon adenocarcinoma (COAD), rectum adenocarcinoma (READ), and ovarian serous cystadenocarcinoma (OV). Num (T): Number of tumor patients (in red). Num (N): Number of normal patients (in grey). * p-value cutoff: 0.01.
Some of the proteins studied showed a statistically significant increase in expression in specific cancer types (p-value cutoff: 0.01), such as CASP3, CTNNB1, BRCA2, and CHEK2 in colon and rectum cancer; MSH6 in breast cancer; PARP1 in breast and ovarian cancer; TP53 in colon, rectum, and ovarian cancer; and ERBB2 in breast and rectal cancer (Supplementary Figure S2). However, it is crucial to highlight that the protein MSH2 exhibited a statistically significant increase in expression (p-value cutoff: 0.01) in samples from patients with all four cancer types (breast, colon, rectal, and ovarian). Additionally, the protein KIT showed a significant alteration in expression levels across all four cancer types, but with a decrease in expression compared to normal tissue (p-value cutoff: 0.01) (Figure 9). The analysis revealed that MSH2 and KIT exhibited statistically significant differential expression across breast, colon, and ovarian cancer. In contrast, other identified proteins displayed variable expression patterns, with some showing differential expression in certain cancers but not in others. The consistent regulation of MSH2 and KIT suggests their potential role as transversal biomarkers.
The study of overall survival in patients with breast, colon, rectum, and ovarian cancer was conducted by considering all of them together. Survival was evaluated based on the gene expression levels (RNA-seq) of the MSH2 and KIT genes, which are potential common biomarkers (Figure 10). The sample of patients (n = 929) with high MSH2 gene expression showed better survival compared to those with low gene expression (n = 929), whose prognosis was poorer (p = 1.7 × 10−5) (Figure 10a). Patients (n = 924) with high KIT gene expression also exhibited a better prognosis compared to those with low expression of the gene (n = 929) (p = 0) (Figure 10b).
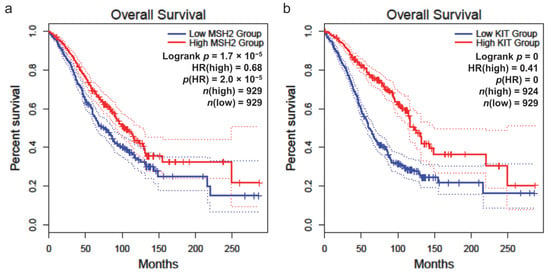
Figure 10.
Overall survival of patients based on gene expression level (RNA-seq). (a) MSH2 gene. (b) KIT gene. The cutoff for high and low expression was set at 50%. The hazard ratio (HR) was calculated using the Cox proportional hazards (Cox PH) model. The confidence interval (CI) was set at 95%, represented by dotted lines. The graphs include all patients (n) with breast, colon, rectum, and ovarian cancer combined. The red curves represent patients with high gene expression, while the blue curves represent those with low expression.
The survival analyses in this study were conducted using the GEPIA2 platform, which employs the Log-rank test (Mantel–Cox test) for hypothesis testing, and automatically calculates hazard ratios (HR) along with 95% confidence intervals (CI). The cutoff for high and low gene expression levels was set at 50%. The survival curves generated by GEPIA2 include dotted lines representing the 95% CI, ensuring the robustness of the statistical evaluation. Given that GEPIA automatically performs these calculations based on curated datasets, we directly report the HR and CI values provided by the platform without additional model adjustments, as shown in Figure 10.
Survival analysis revealed a potential link between MSH2 and KIT expression levels and patient outcomes. Although the observed association was not strongly pronounced, the identified trends suggest that these genes may play a role in prognosis. Further studies with larger datasets will be necessary to confirm these findings and explore their clinical relevance in greater detail.
4. Discussion
With the improvement in life expectancy for patients who survive cancer, the risk of developing a second primary cancer has also increased. Notably, individuals with a history of colorectal cancer have a higher susceptibility to gynecological cancers, such as ovarian and uterine cancer, as well as breast cancer, compared to the general population [28]. Similarly, the risk of colorectal cancer is higher in breast cancer patients [3,4]. Between 5% and 10% of ovarian, breast, and colorectal cancers are believed to have genetic causes. Therefore, to effectively prevent and manage these types of cancer, it is important to periodically implement surveillance protocols for early detection, with particular emphasis on identifying hereditary forms of cancer in individuals with a family history of these diseases [29].
In a 30-year study, the results of diagnostic tests for 15 genes associated with high-risk breast and gastrointestinal tract cancer were analyzed, including the following: BRCA1, BRCA2, PALB2, PTEN, TP53, APC, BMPR1A, CDH1, MLH1, MSH2, MSH6, PMS2, SMAD4, STK11, and MUTYH. This was implemented as a strategy for the prevention and early detection of cancer in patients with a family history of cancer [30]. Studies have highlighted that germline variants in BRCA1 and BRCA2 increase the predisposition to hereditary breast and ovarian cancer, particularly due to the high incidence of genomic aberrations that lead to a worse prognosis. It has been described that some proteins involved in genomic integrity, primarily in the accurate repair of DNA, play a crucial role [31]. This underscores the need to study the biological processes and molecular functions of proteins involved in the mechanisms of carcinogenesis and, subsequently, in tumor biology.
The results obtained in our study provide a comprehensive insight into the common molecular mechanisms in breast, colon, and ovarian cancers. This in-silico study identified common proteins among breast, colon, and ovarian cancers, providing a robust basis for multi-cancer therapeutic and diagnostic applications. The hypothesis of this study centers on identifying common molecular mechanisms in three prevalent cancer types—breast, colon, and ovarian—and exploring the possibility of developing shared biomarkers. In our study, 27 common proteins were identified among the three cancer types, reinforcing the idea that some molecular alterations are universal across various cancers. These proteins are involved in essential biological processes such as DNA damage response, apoptosis, cell proliferation, and cell cycle regulation, which are shared features in these three cancer types. The objectives outlined at the beginning of the study included identifying commonly altered genes and proteins across these cancers, and exploring their potential as biomarkers.
While TP53, BRCA2, and MSH2 are well-known cancer-related genes, this study offers a novel perspective by systematically analyzing their common molecular mechanisms across breast, colon, and ovarian cancers. Unlike previous studies that have focused on individual tumor types, our integrative approach identifies cross-cancer patterns and highlights potential common biomarkers that could aid in diagnostic and therapeutic strategies. By employing in-silico methodologies and curated datasets, our findings contribute to a broader understanding of the molecular similarities among these cancers.
The study of common molecular mechanisms in cancer has been addressed in previous research, suggesting the existence of shared pathways and proteins across different types of cancer. According to several prior studies, signaling pathways involved in cell cycle control, apoptosis, and DNA repair, such as the p53 pathway, are commonly altered in multiple cancer types [32]. The results of our study largely align with these observations, as PPI network analyses and KEGG pathway studies showed a prominent involvement of key proteins such as TP53, MSH2, and BRCA2, which are involved in DNA damage response and cell cycle regulation. Moreover, previous studies have highlighted the importance of shared biomarkers between these cancers, such as HER2 in breast cancer, which has also been implicated in colon cancer [33].
GO and KEGG analyses have shown that these processes are involved in a variety of cancer-related pathways, suggesting that alterations in these pathways could play a crucial role in the oncogenesis of multiple cancer types. In particular, proteins involved in DNA repair, such as MSH2 and BRCA2, could represent key therapeutic targets for the treatment of various cancers, not only in breast or colon cancer but also in ovarian cancer. The relationship between these proteins and pathways associated with infections, particularly viral ones, is especially noteworthy.
The identification of MSH2 and KIT proteins as shared biomarkers suggests their potential utility in both diagnosis and prognosis across multiple cancer types, thereby enabling the development of broader targeted treatments. The results of this study open the door for further research in patient samples to confirm their applicability in clinical settings. The identification of proteins involved in multiple signaling pathways, such as the cell cycle and apoptosis, also opens up the possibility of using targeted therapies that could simultaneously affect several cancer types, optimizing resources and therapeutic approaches. The opposing expression patterns of MSH2 and KIT across breast, colon, and ovarian cancer may reflect distinct tumor regulatory mechanisms. MSH2, involved in DNA mismatch repair, plays a critical role in maintaining genomic stability, whereas KIT, a receptor tyrosine kinase, is involved in proliferative signaling. These differences suggest potential implications in treatment responses, particularly regarding therapies targeting genomic integrity and proliferative pathways.
To ensure the reliability of the findings, data were exclusively obtained from curated databases. Additionally, an in-silico validation was performed using real patient data through the GEPIA platform, providing further evidence of the relevance of the identified biomarkers. While the importance of experimental validation is recognized, the results presented offer a solid foundation for future studies that could confirm their clinical applicability. Furthermore, this approach stands out for its simultaneous integration of data from breast, colon, and ovarian cancer, enabling a comprehensive comparison of shared molecular mechanisms and presenting a novel perspective on the search for common biomarkers.
Although the results obtained in this study provide validation based on patient samples, it is essential to conduct additional experimental studies with clinical samples from other cohorts of patients with the three cancer types to validate the proteins identified as common biomarkers. Expression analysis in human tumors and preclinical models could provide a deeper understanding of their clinical relevance. The identification of key proteins involved in the oncogenesis of multiple cancers suggests that targeted therapies could be a viable option. Designing inhibitors for these common proteins could offer a way to treat several cancer types simultaneously. Identifying interactions between common proteins may shed light on the underlying signaling network in these cancer types. Some of these proteins have been previously described in the literature as key players in cancer biology, further validating the approach of this research [34,35,36,37,38]. These shared proteins participate in a variety of key biological processes, including those related to DNA damage response, cell proliferation, apoptosis, and DNA repair. Among the most significant biological processes, the response to gamma radiation and the regulation of apoptosis in response to genetic damage stand out. These processes are fundamental to carcinogenesis and could be implicated in resistance to treatments, such as radiotherapy and chemotherapy. Through the PPI analysis and the application of the MCODE algorithm, three key clusters were identified, grouping densely interconnected proteins. These clusters showed a strong relation to several metabolic and signaling pathways, including those involved in the regulation of apoptosis, cellular metabolism, and cellular senescence. These observations suggest that cellular control processes like apoptosis and DNA repair could be crucial points in the progression of the three cancer types studied.
The validation of gene expression using the GEPIA2 database showed that several of the proteins studied exhibited significant differential expression in tumor tissues compared to normal tissues. Although the differential expression levels of MSH2 and KIT may be considered modest, their consistent presence across all three cancer types and involvement in key biological processes, such as DNA repair and cell proliferation, support their functional relevance. This consistency, unlike the variability observed in other proteins, strengthens the rationale for further validation studies to explore their potential as shared biomarkers in cancer research. More importantly, survival analysis revealed that the higher expression of MSH2 and KIT was associated with a better prognosis in patients, suggesting that these genes could serve as key indicators of disease progression and treatment response. By analyzing survival data collectively, this study identifies trends that may be relevant across multiple cancer types, highlighting potential biomarkers with cross-cancer applicability. Despite biological differences, this approach allows for the detection of shared molecular mechanisms that could inform future research into diagnosis and treatment strategies.
The identification of shared proteins also enabled the evaluation of their involvement in various signaling pathways, including those related to cancer and responses to viral infections. These pathways include PI3K-Akt, MAPK, P53, and pathways related to drug resistance, such as platinum drug resistance. In this regard, previous reports from our laboratory have documented the relationship between multiple dysregulated proteins and resistance to cisplatin [39], as well as the expression of multiple resistance mechanisms to radiation in tumor cells [40]. Although some studies have suggested potential interactions between these phenomena, these associations remain speculative and require further mechanistic validation. Friedenson [41] describes that the transformation caused by the Epstein–Barr virus in mammary cells induces the genetic disruption of essential functions for protection against carcinogenesis, suggesting a possible link to tumor progression, though additional experimental studies are necessary to confirm these mechanisms. While the identified viral pathways and radiation resistance mechanisms provide interesting insights, it is important to interpret these findings with caution. The current evidence is based on bioinformatics analyses and previously reported associations, but experimental validation is required to confirm direct mechanistic links. Future studies should include functional assays to determine the extent to which viral-related signaling pathways influence resistance mechanisms in breast, colon, and ovarian cancer. Until such validation is available, these observations should be considered as preliminary hypotheses for further exploration.
The fact that breast, colon, and ovarian cancers share a significant set of proteins involved in similar biological processes opens the door to identifying common biomarkers. While clinical sample validation is crucial, the findings suggest that MSH2 and KIT could be used as biomarkers for a wide range of cancers, not only for diagnosis but also for prognosis and therapy customization. This approach could not only improve diagnostic accuracy, but also enable the implementation of more specific therapies for these cancer types. The approach of identifying shared proteins between different cancer types holds enormous potential in the field of personalized medicine. By identifying key proteins involved in various stages of carcinogenesis, it is possible to develop more precise and targeted therapies that consider both the shared mechanisms among cancer types and the unique characteristics of each tumor. This approach could also facilitate the treatment of cancers that are currently difficult to treat due to their molecular heterogeneity.
Our study does not aim to identify biomarkers that are superior to those described in previous research focused on a single tumor type, but rather to provide a rigorous analysis that reveals the robust markers present in multiple cancer types. The identification of shared proteins among breast, colon, and ovarian cancer not only highlights common molecular mechanisms, but also presents new perspectives on the biological interconnection between these tumors. Specifically, the differential expression of MSH2 and KIT across the three cancer types suggests their potential role in patient stratification and the design of therapeutic strategies. Furthermore, the presence of common biomarkers in multiple tumors could be key in assessing a patient’s risk of developing another cancer with similar biological characteristics.
While these findings are promising, additional validation in preclinical and clinical models is essential to confirm their applicability. Additionally, expanding the analysis to other cancer types would be valuable in confirming whether the proteins identified in this study play a common role in a broader range of pathologies. Longitudinal studies are also recommended to assess how alterations in the expressions of these proteins affect disease progression and treatment response. Combining these strategies will provide a deeper understanding of the molecular foundations of cancer and contribute to the advancement of more effective and personalized treatments.
The advantages of this work lie in its comprehensive approach using advanced bioinformatics tools, enabling the identification of common proteins among three different cancer types efficiently. The use of artificial intelligence applied to the search for molecular markers through the analysis of proteomic data represents a very promising advancement [19]. Proteomic research currently requires advanced technologies due to its complexity [18]. The protein–protein interaction network analysis, the GO/KEGG enrichment analyses, and the identification of MCODE clusters provided an in-depth understanding of shared molecular mechanisms, representing an advantage over more traditional studies that may focus on a single cancer type.
The methodology employed in this study integrates multiple bioinformatics tools to ensure a comprehensive and reliable analysis. By combining STRING, GeneCodis4, GEPIA2, and Metascape, the approach allows for robust data filtering and interpretation, minimizing the inclusion of irrelevant information. While these tools rely on publicly available databases, their curation, consistent updates and validation processes maintain high reliability in biomedical research. Furthermore, the sequential nature of the analysis strengthens the identification of meaningful biomarkers, providing a solid foundation for future experimental validation. This study prioritized validated data by using STRING’s “curated” subset, minimizing noise and ensuring accuracy. GeneCodis was chosen for its multi-category analysis capability, allowing a broader evaluation of common cancer biomarkers. These methodological choices strengthen the reliability of the findings and their future experimental validation.
The filtering process applied in this study has enabled the identification of biomarkers with the highest robustness and biological relevance in breast, colon, and ovarian cancer. The selection of 27 proteins and the validation of MSH2 and KIT as differential markers reinforce the existence of shared molecular mechanisms among these tumor types. The presence of an excessive number of biomarkers would have indicated a deficient filtering process, compromising the specificity of the analysis. On the contrary, the strict application of methodological and bioinformatics criteria ensures that the selected markers are representative of key processes, providing a solid foundation for future research and clinical validations.
The statistical approach employed in this study, including the hypergeometric test and the application of a highly restrictive p-value threshold, reinforces the robustness of the identified associations. By minimizing the risk of false positives and ensuring high specificity in biomarker selection, these methodological choices strengthen confidence in the reported findings. Such stringent criteria align with best practices in biomarker discovery, and have been applied in previous studies to enhance result reliability and interpretability.
However, one of the main limitations of this study is that the results are based on bioinformatics analyses, and the experimental validation in clinical samples performed is limited to patients from a single project (TCGA). Although the association between MSH2 and KIT expression and patient survival was modest, the observed trends suggest a possible prognostic value. These results highlight the need for further research with larger datasets and experimental validation to confirm their potential as reliable biomarkers. Nonetheless, this study provides an initial framework for considering MSH2 and KIT as potential biomarkers across breast, colon, and ovarian cancer. Therefore, it is essential to confirm the relevance of the identified biomarkers in other patient cohorts. Additionally, patient variability and tumor heterogeneity could affect the applicability of these biomarkers in all cases. Although RNA-seq expression analysis through GEPIA2 provides valuable insights into gene expression differences between tumor and normal tissues, it does not establish protein-level relevance, which is essential for understanding functional implications. As gene expression does not always correlate directly with protein abundance or activity, future studies should incorporate proteomic analyses, such as mass spectrometry, to confirm protein expression patterns in clinical samples. Additionally, immunohistochemical validation in independent patient cohorts could provide further evidence of the biological significance of MSH2 and KIT as potential biomarkers. These complementary approaches will strengthen the translational impact of the findings and improve their applicability in personalized oncology.
The association of high MSH2 and KIT expression with better survival outcomes in our analysis contrasts with previous reports wherein MSH2 loss leads to mismatch-repair deficiency, and KIT overexpression is implicated in oncogenesis in certain gastrointestinal tumors. This apparent paradox underscores the context-dependent role of these proteins across different cancer types. While MSH2 is essential for DNA repair, its differential expression may reflect compensatory mechanisms in tumors attempting to maintain genomic integrity. Similarly, KIT signaling can contribute to oncogenesis in specific tissues, but its regulation varies depending on genetic alterations, tumor microenvironment, and cellular context. The organ-specific variability of KIT expression highlights the need for a nuanced approach to its modulation in cancer therapy. Rather than assuming a simple activation or inhibition strategy, future studies should explore the functional implications of KIT signaling in different tumor types. The targeted modulation of KIT, considering its oncogenic or tumor-suppressive roles in specific cancers, could provide a more refined therapeutic approach. In cases where KIT overexpression correlates with oncogenesis, inhibition remains a clinically relevant strategy for controlling tumor progression, as demonstrated by current tyrosine kinase inhibitors such as Imatinib, Sunitinib, Dasatinib, and Sorafenib. Conversely, in cancers where KIT is underexpressed, its potential functional restoration should be carefully evaluated through proteomic and immunohistochemical studies to determine its relevance in disease progression and response to therapy. These discrepancies underscore the complexity of tumor biology and reinforce the need for experimental validation to clarify the functional significance of MSH2 and KIT expression in breast, colon, and ovarian cancer. Future studies integrating proteomic analyses, mechanistic experiments, and clinical cohort validation will be essential to defining their precise roles in personalized oncology.
Although breast and ovarian cancers predominantly affect women, the inclusion of colon cancer in this analysis allows for the exploration of shared molecular mechanisms without gender being a determining factor. Our approach does not aim to compare sex-based differences, but rather to identify common proteins that could have implications across multiple cancer types. Additionally, while these tumors are not traditionally considered synchronous, the identification of shared biomarkers suggests the presence of common biological mechanisms that may influence the development of tumors with similar characteristics, providing new perspectives in cancer research.
5. Conclusions
The analysis conducted on common proteins involved in breast, colon, and ovarian cancers has revealed significant findings that contribute to the understanding of the molecular similarities and shared biological mechanisms between these pathologies.
Data mining identified 483 proteins for breast cancer, 521 for colon cancer, and 223 for ovarian cancer. Interaction network analysis revealed shared clusters, including pathways for cancer progression, DNA repair, and cell proliferation.
The analysis found 27 proteins to be common across the three cancer types. These proteins participate in crucial biological processes like DNA damage response, cell proliferation, and apoptosis. Notably, they are involved in KEGG pathways linked to multiple cancers and infections.
The study identified MSH2 and KIT as proteins that showed significant changes in expression across breast, colon, and ovarian cancer. The high expression of these proteins correlated with better survival outcomes, suggesting their potential as common biomarkers for all three cancer types. The opposing expression patterns of MSH2 and KIT across breast, colon, and ovarian cancer highlight potential implications for diagnosis and therapeutic strategies. Their distinct regulatory roles in genomic stability and proliferative signaling suggest that assessing their combined expression could provide valuable insights for patient stratification and treatment selection. Future studies are needed to explore their clinical applicability and validate their relevance as biomarkers in personalized oncology.
While this study is based on a rigorous in-silico approach, we acknowledge that experimental validation is essential to confirm the clinical relevance of the identified biomarkers. Future research should include validation in independent patient cohorts and experimental models to strengthen the applicability of these findings. Expanding the dataset and performing longitudinal studies could provide deeper insights into the role of these biomarkers in disease progression and treatment response. The methodological framework presented here lays the foundation for further studies that will validate and refine these results in a clinical setting.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/app15137018/s1, Table S1: Complete list of curated ontologies from the STRING Database for 15 breast cancer-related search terms; Table S2: Complete list of curated ontologies from the STRING Database for 24 colon cancer-related search terms; Table S3: Complete list of curated ontologies from the STRING Database for 15 ovarian cancer-related search terms; Table S4: List of unique proteins found for breast cancer (483), colon cancer (521), and ovarian cancer (223); Table S5: List of biological processes identified through the functional enrichment analysis of Gene Ontology for proteins shared among breast, colon, and ovarian cancer; Table S6: List of cellular components identified through the functional enrichment analysis of Gene Ontology for proteins shared among breast, colon, and ovarian cancer; Table S7: List of molecular functions identified through the functional enrichment analysis of Gene Ontology for proteins shared among breast, colon, and ovarian cancer; Table S8: List of KEGG pathways identified through the functional analysis of proteins shared among breast, colon, and ovarian cancer; Figure S1: Protein-protein interaction network of MCODE clusters: analysis of KEGG pathways; Figure S2: Expression levels (RNAseq) of additional proteins from the MCODE1, MCODE2, and MCODE3 clusters in tumor patient samples with breast invasive carcinoma (BRCA), colon adenocarcinoma (COAD), rectum adenocarcinoma (READ), and ovarian serous cystadenocarcinoma (OV).
Author Contributions
Conceptualization, V.M.G.-C.; methodology, V.M.G.-C. and M.J.R.-G.; validation, V.M.G.-C. and M.J.R.-G.; formal analysis, V.M.G.-C., A.G.-V., A.M.B.-M., S.M.-S., F.S.-P. and M.J.R.-G.; investigation, V.M.G.-C., A.G.-V., A.M.B.-M., S.M.-S., F.S.-P. and M.J.R.-G.; resources, V.M.G.-C. and M.J.R.-G.; data curation, V.M.G.-C., A.G.-V., A.M.B.-M., S.M.-S., F.S.-P. and M.J.R.-G.; writing—original draft preparation, V.M.G.-C. and M.J.R.-G.; writing—review and editing, V.M.G.-C. and M.J.R.-G.; visualization, V.M.G.-C. and M.J.R.-G.; supervision, M.J.R.-G.; project administration, M.J.R.-G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Acknowledgments
Thanks to Ana I. López-Sesé (I.H.S.M. “La Mayora”, Consejo Superior de Investigaciones Científicas–Universidad de Málaga) for her critical review of the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| FDR | False Discovery Rate |
| GEPIA2 | Gene Expression Profiling Interactive Analysis |
| GO | Gene Ontology |
| GTEx | Genotype-Tissue Expression Project |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| MCODE | Molecular Complex Detection |
| PPI | Protein-Protein Interactions |
| STRING | Search Tool for the Retrieval of Interacting Genes |
| TCGA | The Cancer Genome Atlas |
References
- Labianca, R.; Beretta, G.D.; Kildani, B.; Milesi, L.; Merlin, F.; Mosconi, S.; Pessi, M.A.; Prochilo, T.; Quadri, A.; Gatta, G.; et al. Colon cancer. Crit. Rev. Oncol. 2010, 74, 106–133. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.; Sellers, T.; Rich, S.; Potter, J.; McGovern, P.; Kushi, L. Familial clustering of colon, breast, uterine, and ovarian cancers as assessed by family history. Genet. Epidemiol. 1993, 10, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Segelman, J.; Nordgren, A.; Lindström, L.; Frisell, J.; Martling, A. Increased risk of colorectal cancer in patients diagnosed with breast cancer in women. Cancer Epidemiol. 2016, 41, 57–62. [Google Scholar] [CrossRef]
- Abu-Sbeih, H.; Ali, F.S.; Ge, P.S.; Barcenas, C.H.; Lum, P.; Qiao, W.; Bresalier, R.S.; Bhutani, M.S.; Raju, G.S.; Wang, Y. Patients with breast cancer may be at higher risk of colorectal neoplasia. Ann. Gastroenterol. 2019, 32, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, A.; Lendorf, M.E.; Couchman, J.R.; Multhaupt, H.A. Breast and ovarian cancers: A survey and possible roles for the cell surface heparan sulfate proteoglycans. J. Histochem. Cytochem. 2012, 60, 9–21. [Google Scholar] [CrossRef]
- Farrell, C.; Lyman, M.; Freitag, K.; Fahey, C.; Piver, M.S.; Rodabaugh, K.J. The role of hereditary nonpolyposis colorectal cancer in the management of familial ovarian cancer. Genet. Med. 2006, 8, 653–657. [Google Scholar] [CrossRef]
- Iorga, C.; Iorga, C.R.; Grigorescu, A.; Bengulescu, I.; Constantin, T.; Strambu, V. Synchronous breast and colorectal malignant tumors-A systematic review. Life 2024, 14, 1008. [Google Scholar] [CrossRef]
- Tung, K.H.; Goodman, M.T.; Wu, A.H.; McDuffie, K.; Wilkens, L.R.; Nomura, A.M.; Kolonel, L.N. Aggregation of ovarian cancer with breast, ovarian, colorectal, and prostate cancer in first-degree relatives. Am. J. Epidemiol. 2004, 159, 750–758. [Google Scholar] [CrossRef]
- Auranen, S.A.; Grénman, S.K.; Mäkinen, J.I.; Salmi, T.A. Primary breast and colon cancer associated with endometrial or ovarian cancer. Ann. Chir. Gynaecol. Suppl. 1994, 208, 5–9. [Google Scholar]
- Llinás-Quintero, N.; Cabrera-Florez, E.; Mendoza-Fandiño, G.; Matute-Turizo, G.; Vasquez-Trespalacios, E.M.; Gallón-Villegas, L.J. Synchronous ovarian and breast cancers with a novel variant in BRCA2 gene: A case report. Case Rep. Oncol. Med. 2019, 2019, 6958952. [Google Scholar] [CrossRef]
- Travis, L.; Curtis, R.; Boice, J.; Platz, C.; Hankey, B.; Fraumeni, J. Second malignant neoplasms among long-term survivors of ovarian cancer. Cancer Res. 1996, 56, 1564–1570. [Google Scholar] [PubMed]
- Lagkadas, A.; Mantas, D.; Pittokopitou, S.; Papadimitriou, A.; Daglas, K.; Chionis, A.; Giannakopoulos, K. Ovarian metastasis from colorectal carcinoma: A case report and review of the literature. Int. J. Gynecol. Cancer 2019, 29, A484–A485. [Google Scholar] [CrossRef]
- Cheng, H.; Chang, C.; Wang, P.; Hung, P.; Chang, K. Breast metastasis and ovary metastasis of primary colon cancer. J. Cancer Res. Pract. 2015, 2, 330–334. [Google Scholar] [CrossRef]
- Reimer, R.; Hoover, R.; Fraumeni, J.; Young, R. Second primary neoplasms following ovarian cancer. J. Natl. Cancer Inst. 1978, 61, 1195–1197. [Google Scholar] [CrossRef]
- Mori, R.; Futamura, M.; Morimitsu, K.; Saigo, C.; Miyazaki, T.; Yoshida, K. The diagnosis of a metastatic breast tumor from ovarian cancer by the succession of a p53 mutation: A case report. World J. Surg. Oncol. 2017, 15, 117. [Google Scholar] [CrossRef]
- Ford, D.; Easton, D.F.; Bishop, D.T.; Narod, S.A.; Goldgar, D.E. Risks of cancer in BRCA1-mutation carriers. Lancet 1994, 343, 692–695. [Google Scholar] [CrossRef]
- Infante, M.; Arranz-Ledo, M.; Lastra, E.; Olaverri, A.; Ferreira, R.; Orozco, M.; Hernández, L.; Martínez, N.; Durán, M. Profiling of the genetic features of patients with breast, ovarian, colorectal and extracolonic cancers: Association to CHEK2 and PALB2 germline mutations. Clin. Chim. Acta 2024, 552, 117695. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y. The application and outlook of proteomics in Ovarian Cancer, Breast Cancer, and Colon Cancer. BIO Web Conf. 2024, 111, 03004. [Google Scholar] [CrossRef]
- Pawar, S.; Liew, T.O.; Stanam, A.; Lahiri, C. Common cancer biomarkers of breast and ovarian types identified through artificial intelligence. Chem. Biol. Drug Des. 2020, 96, 995–1004. [Google Scholar] [CrossRef]
- González-Vidal, A.; Mercado-Sáenz, S.; Burgos-Molina, A.M.; Alamilla-Presuel, J.C.; Alcaraz, M.; Sendra-Portero, F.; Ruiz-Gómez, M.J. Molecular Mechanisms of Resistance to Ionizing Radiation in S. cerevisiae and Its Relationship with Aging, Oxidative Stress, and Antioxidant Activity. Antioxidants 2023, 12, 1690. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Annika, G.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Oliveros, J.C.; Venny. An Interactive Tool for Comparing Lists with Venn’s Diagrams. 2007–2015. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 7 February 2025).
- Kanehisa, M.; Sato, Y. KEGG Mapper for inferring cellular functions from protein sequences. Protein Sci. 2020, 29, 28–35. [Google Scholar] [CrossRef]
- Garcia-Moreno, A.; López-Domínguez, R.; Villatoro-García, J.A.; Ramirez-Mena, A.; Aparicio-Puerta, E.; Hackenberg, M.; Pascual-Montano, A.; Carmona-Saez, P. Functional Enrichment Analysis of Regulatory Elements. Biomedicines 2022, 10, 590. [Google Scholar] [CrossRef]
- Tang, D.; Chen, M.; Huang, X.; Zhang, G.; Zeng, L.; Zhang, G.; Wu, S.; Wang, Y. SRplot: A free online platform for data visualization and graphing. PLoS ONE 2023, 18, e0294236. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef]
- Shin, D.W.; Choi, Y.J.; Kim, H.S.; Han, K.D.; Yoon, H.; Park, Y.S.; Kim, N.; Lee, D.H. Secondary breast, ovarian, and uterine cancers after colorectal cancer: A nationwide population-based cohort study in Korea. Dis. Colon Rectum 2018, 61, 1250–1257. [Google Scholar] [CrossRef]
- Vasen, H.F. Inherited forms of colorectal, breast, and ovarian cancer: Guidelines for surveillance. Surg. Oncol. Clin. N. Am. 1994, 3, 501–521. [Google Scholar] [CrossRef]
- Woodward, E.R.; Green, K.; Burghel, G.J.; Bulman, M.; Clancy, T.; Lalloo, F.; Schlecht, H.; Wallace, A.J.; Evans, D.G. 30 year experience of index case identification and outcomes of cascade testing in high-risk breast and colorectal cancer predisposition genes. Eur. J. Hum. Genet. 2022, 30, 413–419. [Google Scholar] [CrossRef]
- Barili, V.; Ambrosini, E.; Bortesi, B.; Minari, R.; De Sensi, E.; Cannizzaro, I.R.; Taiani, A.; Michiara, M.; Sikokis, A.; Boggiani, D.; et al. Genetic Basis of Breast and Ovarian Cancer: Approaches and Lessons Learnt from Three Decades of Inherited Predisposition Testing. Genes 2024, 15, 219. [Google Scholar] [CrossRef]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting p53 pathways: Mechanisms, structures and advances in therapy. Signal Transduct. Target. Ther. 2023, 8, 92. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Chapman, A.; Pickard, L.A.; Porplycia, D.; McConkey, H.; Jarosz, P.; Sinfield, J.; Lauzon-Young, C.; Cecchini, M.J.; Howlett, C.; et al. Evaluation of HER2 immunohistochemistry expression in non-standard solid tumors from a Single-Institution Prospective Cohort. Explor. Target. Anti-Tumor Ther. 2024, 5, 1100–1109. [Google Scholar] [CrossRef]
- Kanapathy Pillai, S.K.; Tay, A.; Nair, S.; Leong, C.O. Triple-negative breast cancer is associated with EGFR, CK5/6 and c-KIT expression in Malaysian women. BMC Clin. Pathol. 2012, 12, 18. [Google Scholar] [CrossRef] [PubMed]
- Shnaider, P.V.; Petrushanko, I.Y.; Aleshikova, O.I.; Babaeva, N.A.; Ashrafyan, L.A.; Borovkova, E.I.; Dobrokhotova, J.E.; Borovkov, I.M.; Shender, V.O.; Khomyakova, E. Expression level of CD117 (KIT) on ovarian cancer extracellular vesicles correlates with tumor aggressiveness. Front. Cell Dev. Biol. 2023, 11, 1057484. [Google Scholar] [CrossRef] [PubMed]
- Cheo, S.W.; Zhao, J.J.; Ong, P.Y.; Ow, S.G.W.; Ow, C.J.L.; Chan, G.H.J.; Walsh, R.J.; Lim, J.S.J.; Lim, S.E.; Lim, Y.W.; et al. Pathogenic germline variants in cancer predisposition genes in patients with multiple primary cancers in an Asian population and the role of extended panel genetic testing. ESMO Open 2025, 10, 104495. [Google Scholar] [CrossRef]
- Lepage, M.; Uhrhammer, N.; Molnar, I.; Privat, M.; Ponelle-Chachuat, F.; Gay-Bellile, M.; Bidet, Y.; Cavaillé, M. Prevalence of constitutional pathogenic variant in a cohort of 348 patients with multiple primary cancer addressed in oncogenetic consultation. Mol. Genet. Genom. Med. 2025, 13, e70086. [Google Scholar] [CrossRef]
- Nakano, Y.; Nishikawa, G.; Degawa, K.; Moriyoshi, K.; Kuriyama, K.; Watanabe, Y.; Miyamoto, S. A case of multiple advanced colon cancers with spontaneous regression of only one lesion after biopsy: A case report and literature review. Clin. J. Gastroenterol. 2025, 18, 393–398. [Google Scholar] [CrossRef]
- Burgos-Molina, A.M.; Mercado-Sáenz, S.; Cárdenas, C.; López-Díaz, B.; Sendra-Portero, F.; Ruiz-Gómez, M.J. Identification of new proteins related with cisplatin resistance in Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 2021, 105, 1965–1977. [Google Scholar] [CrossRef]
- Alamilla-Presuel, J.C.; Burgos-Molina, A.M.; González-Vidal, A.; Sendra-Portero, F.; Ruiz-Gómez, M.J. Factors and molecular mechanisms of radiation resistance in cancer cells. Int. J. Radiat. Biol. 2022, 98, 1301–1315. [Google Scholar] [CrossRef]
- Friedenson, B. Identifying safeguards disabled by Epstein-Barr virus infections in genomes from patients with breast cancer: Chromosomal bioinformatics analysis. JMIRx Med 2025, 6, e50712. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).