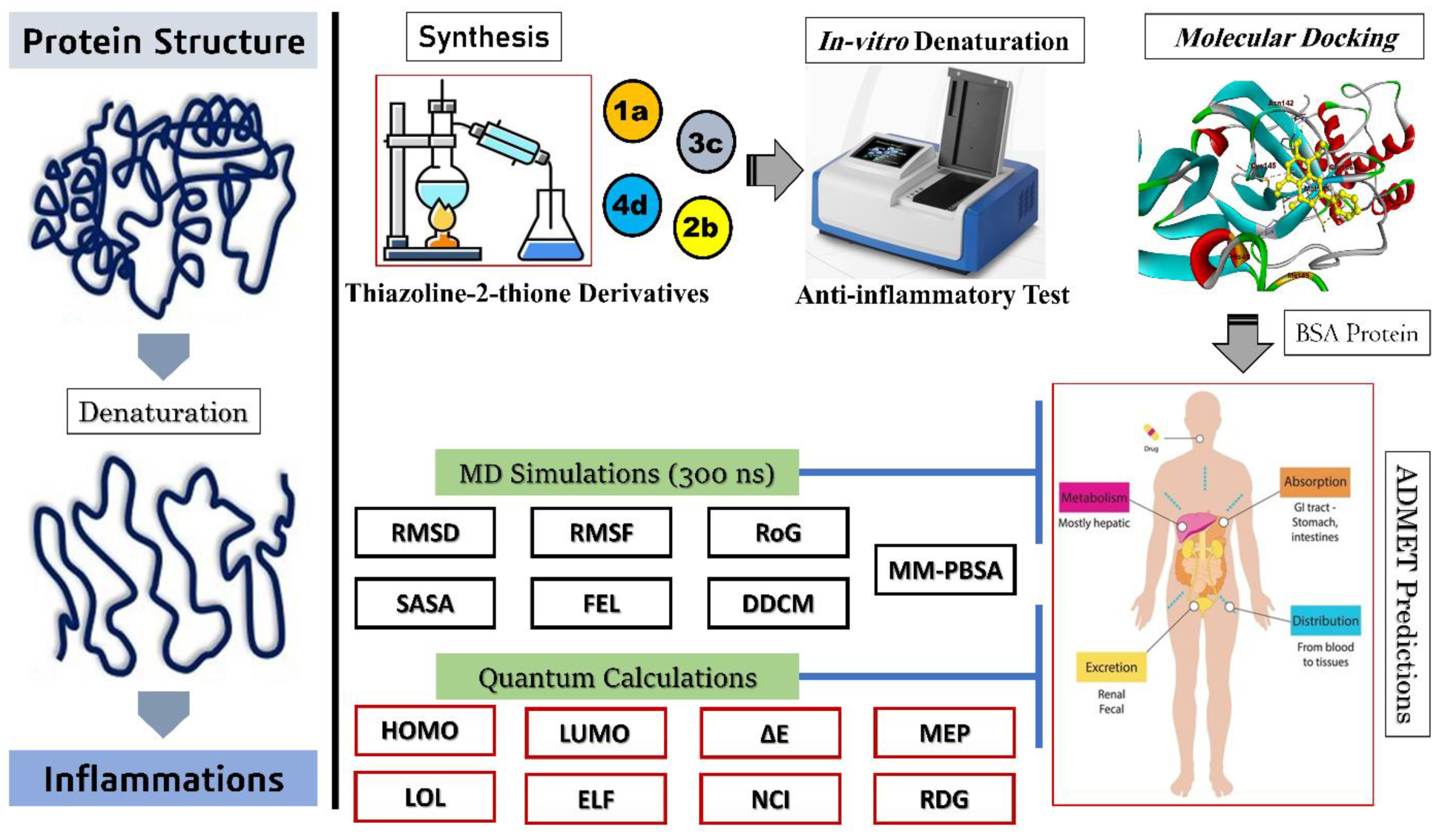
Synthesis, In Vitro Anti-Inflammatory Activity, Molecular Docking, Molecular Dynamics and DFT Calculations of Thiazoline-2-Thione Derivatives

 ,
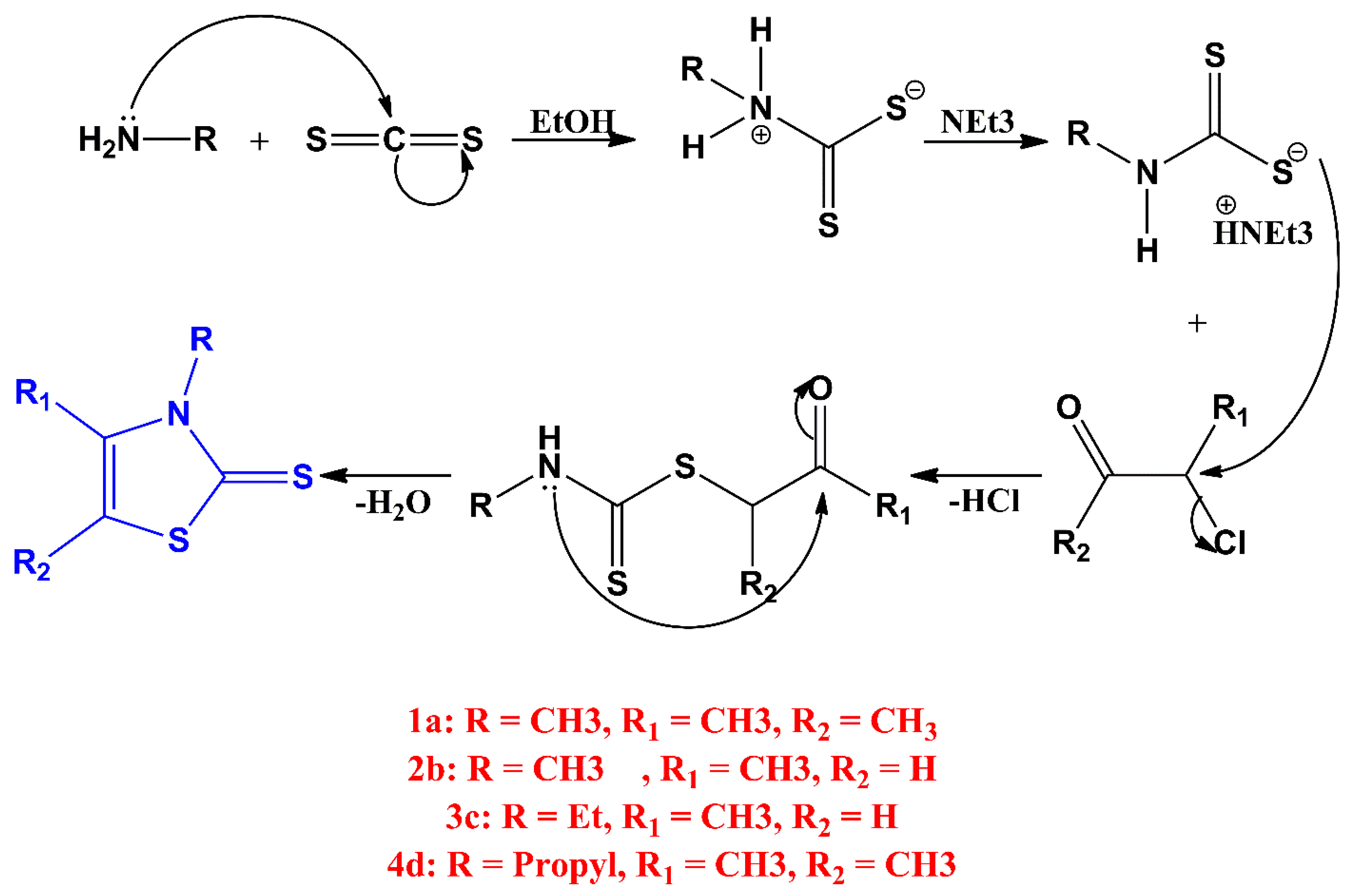
,  , , , ,
, , , ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Procedures
2.1.1. Synthesis
2.1.2. In Vitro Inhibition of Albumin Denaturation
2.1.3. Binding Free Energy and Binding Constant
2.2. Theoretical Procedures
2.2.1. Protein and Ligand Preparation
2.2.2. Preparation of Grid and Docking Modeling
2.2.3. ADME-Tox Features
2.2.4. MD Simulation and Stability
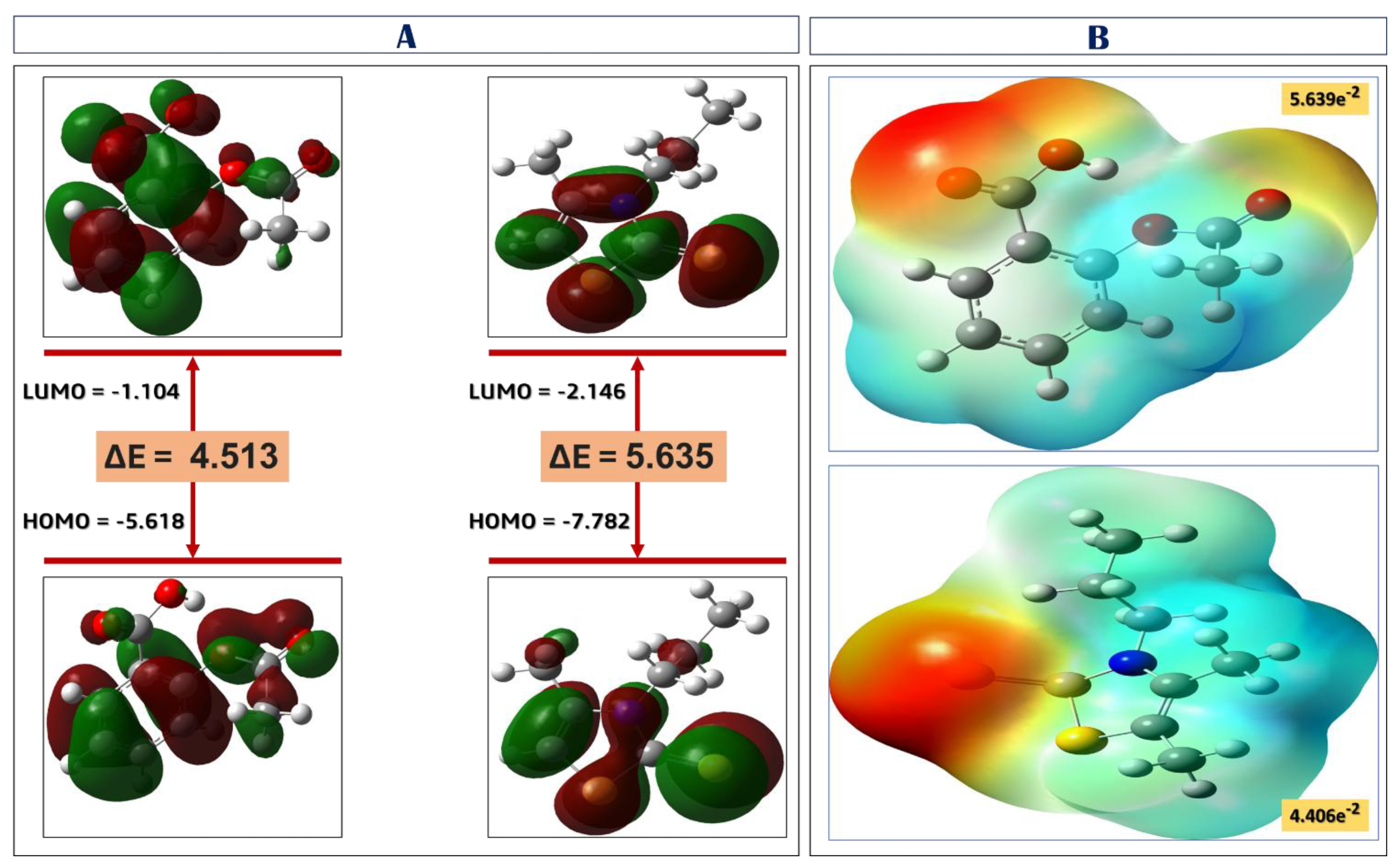
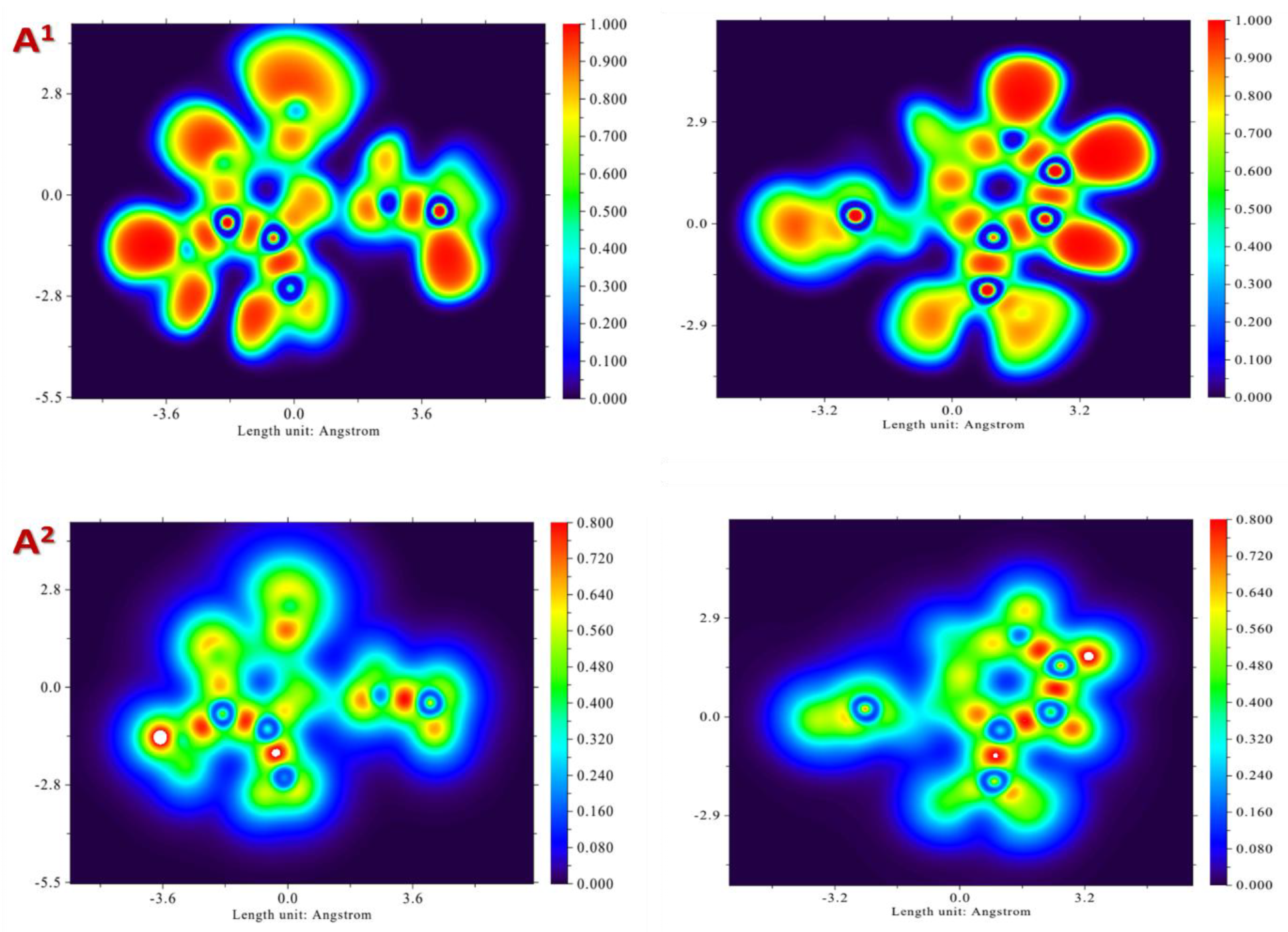
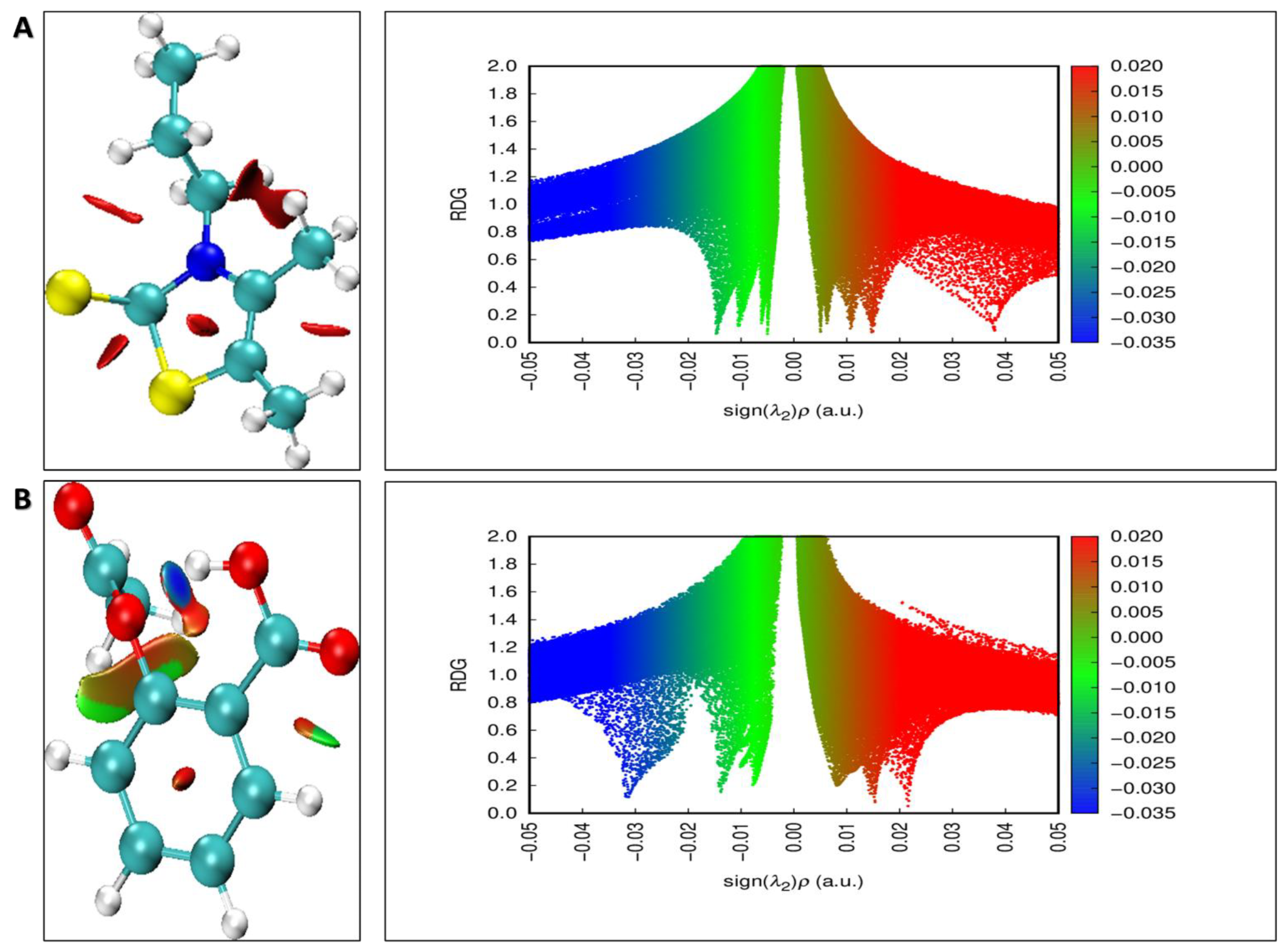
2.2.5. Density Functional Theory (DFT) Calculations
3. Results
3.1. Anti-Inflammatory Activity and the Experimental Validation
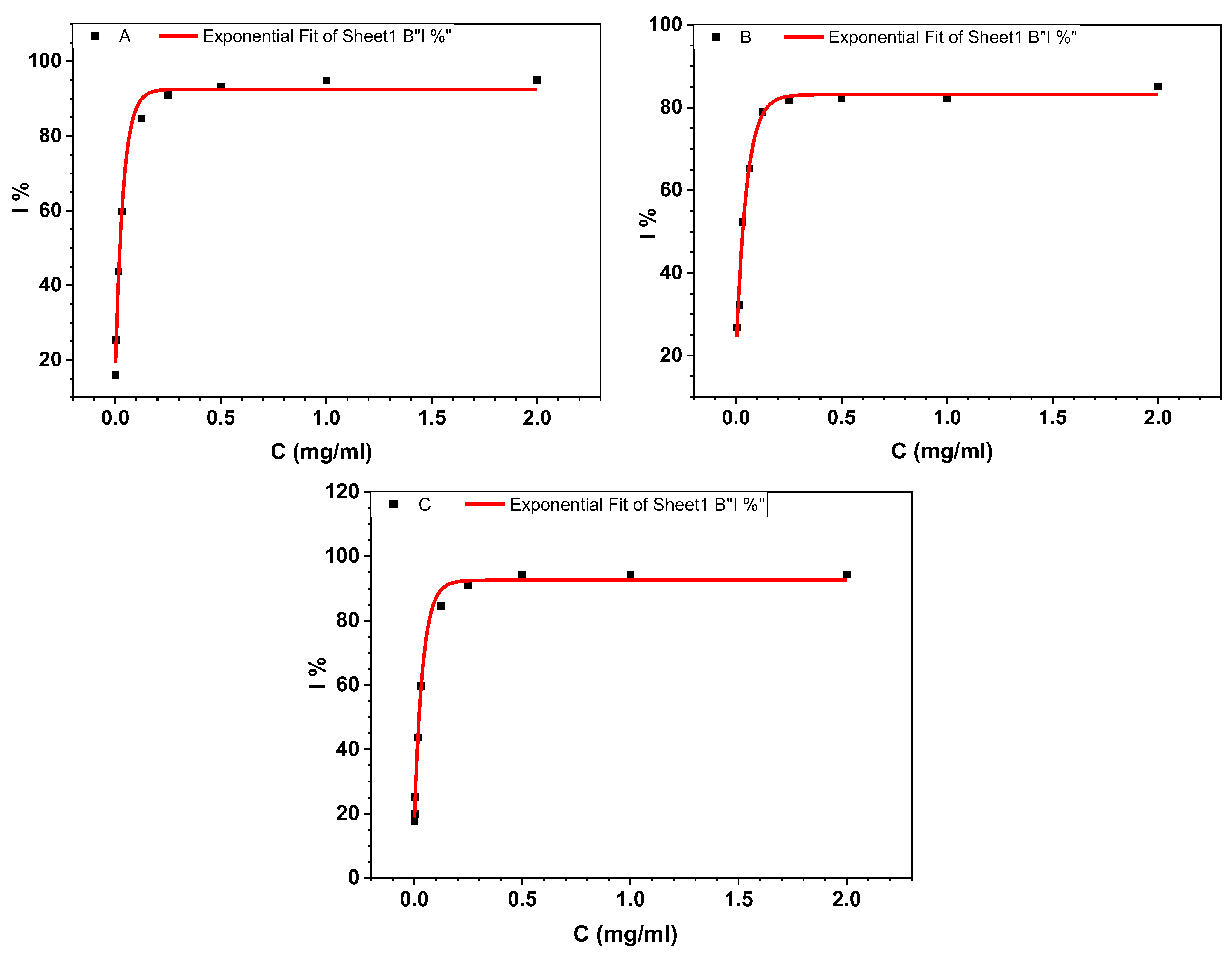
3.1.1. BSA Denaturation Assay
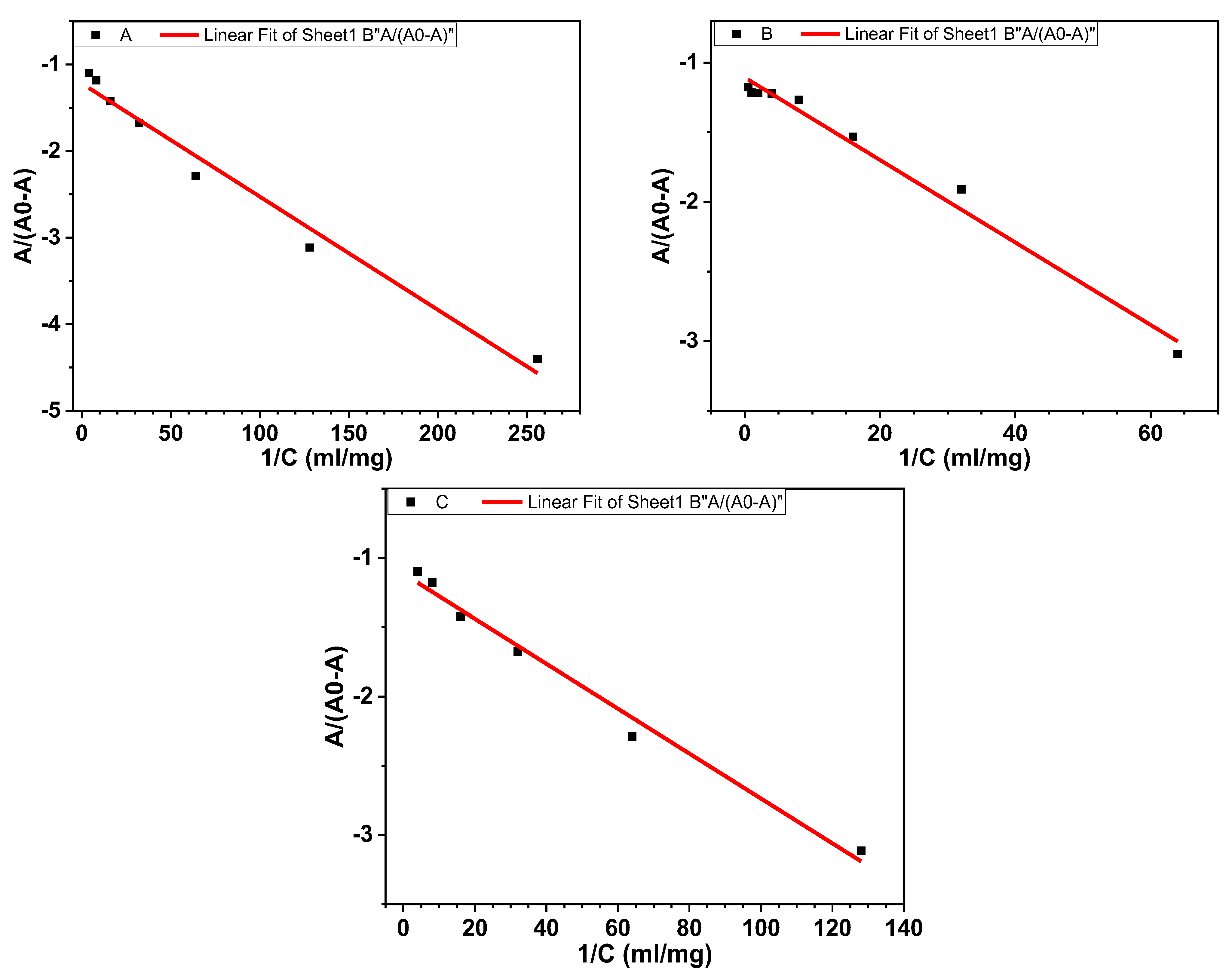
3.1.2. Binding Free Energy and Binding Constant
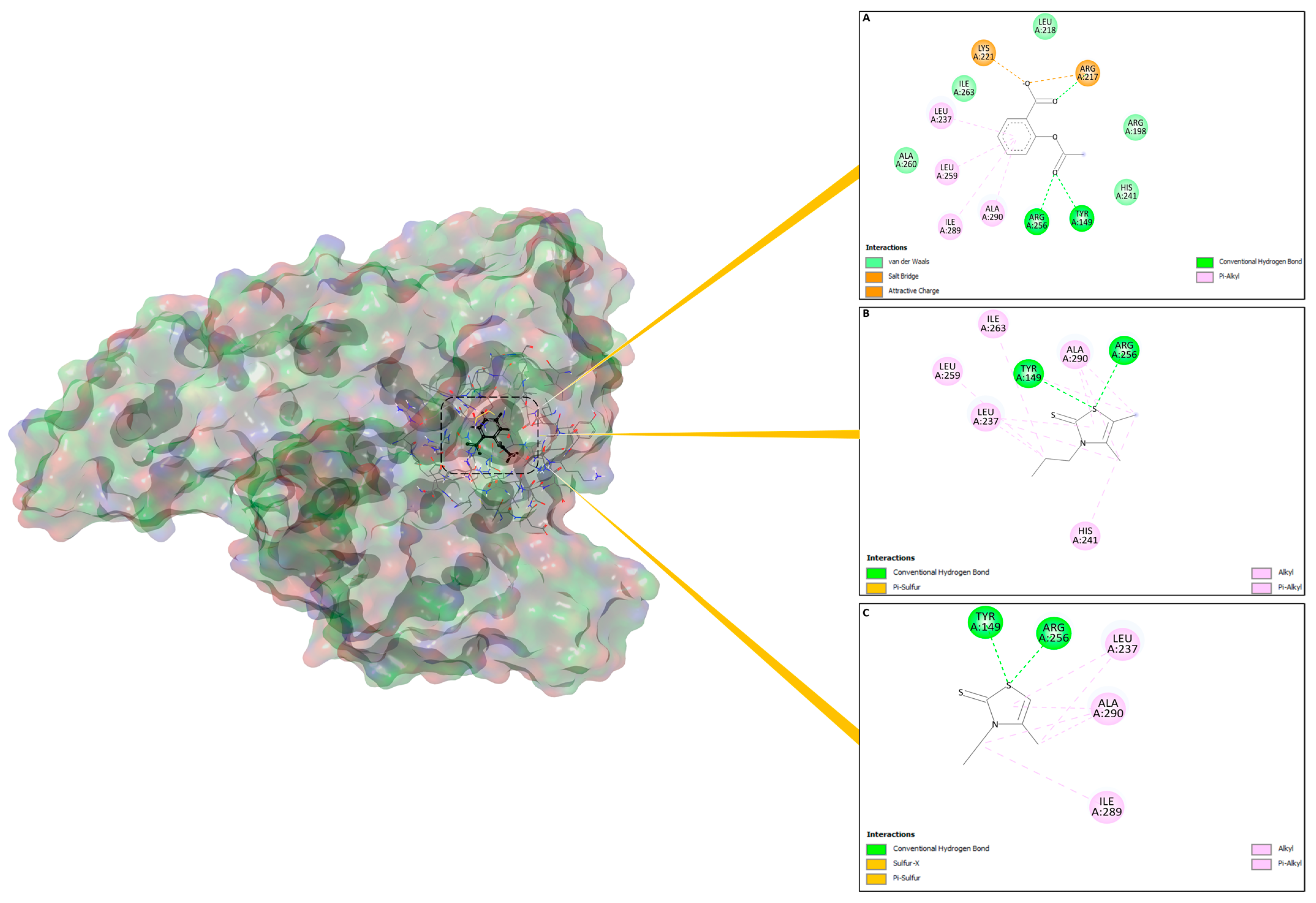
3.2. Molecular Docking Studies
3.3. ADME-Tox and Bioavailability Prediction
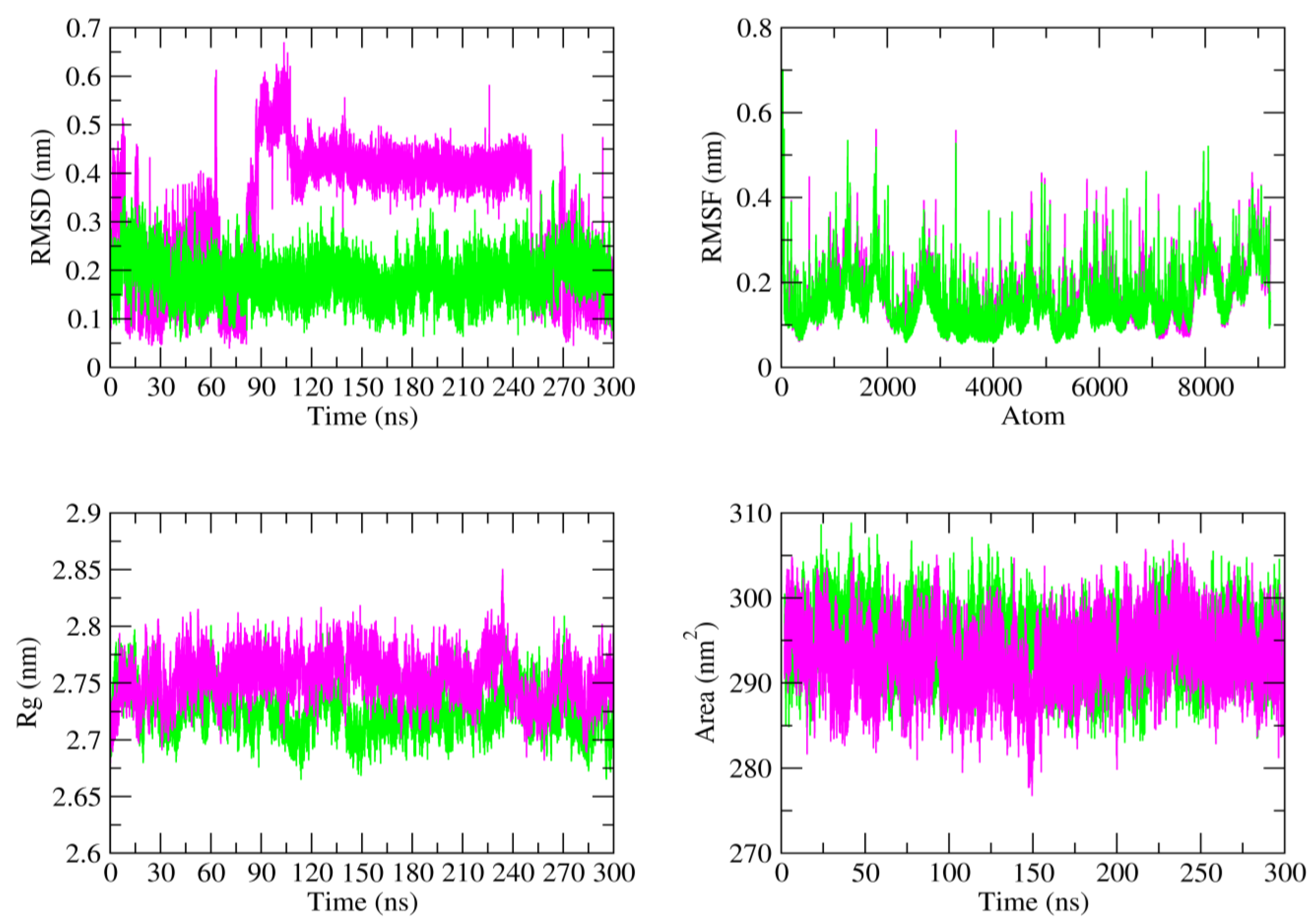
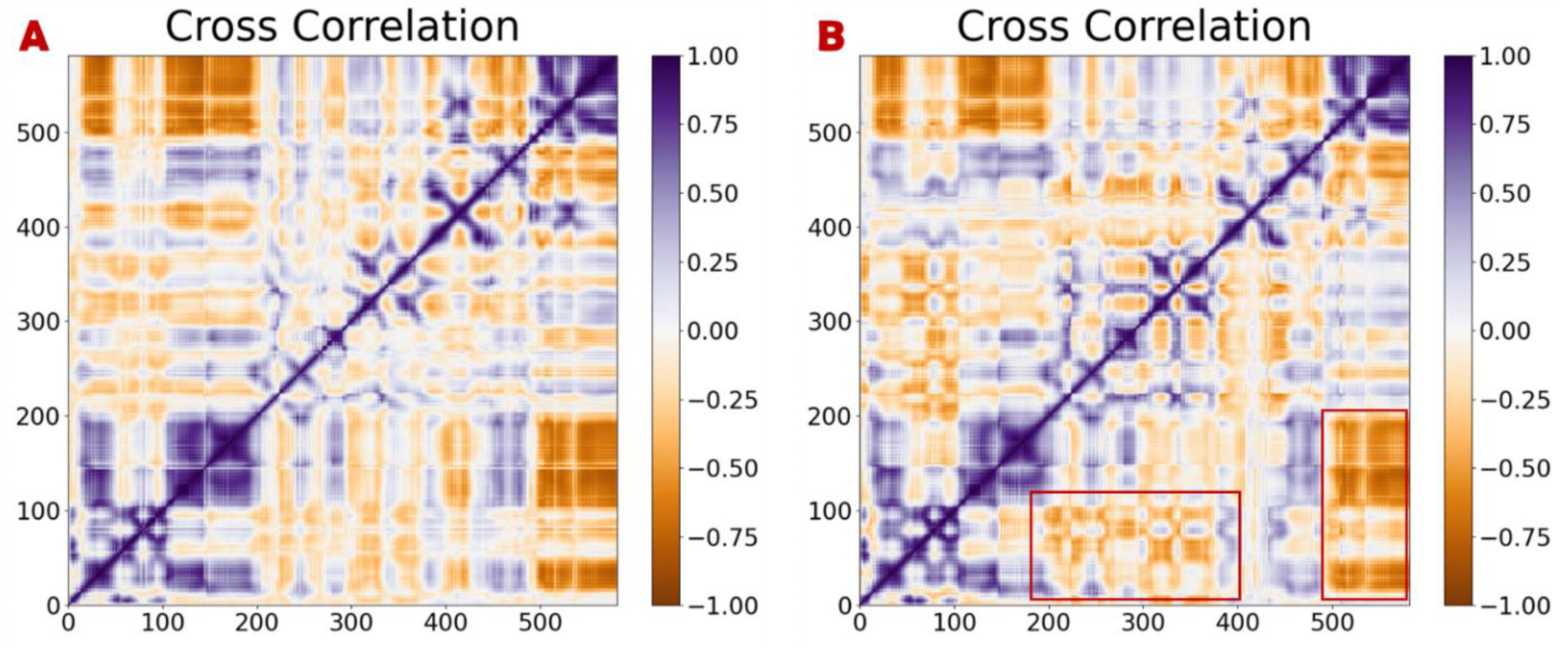
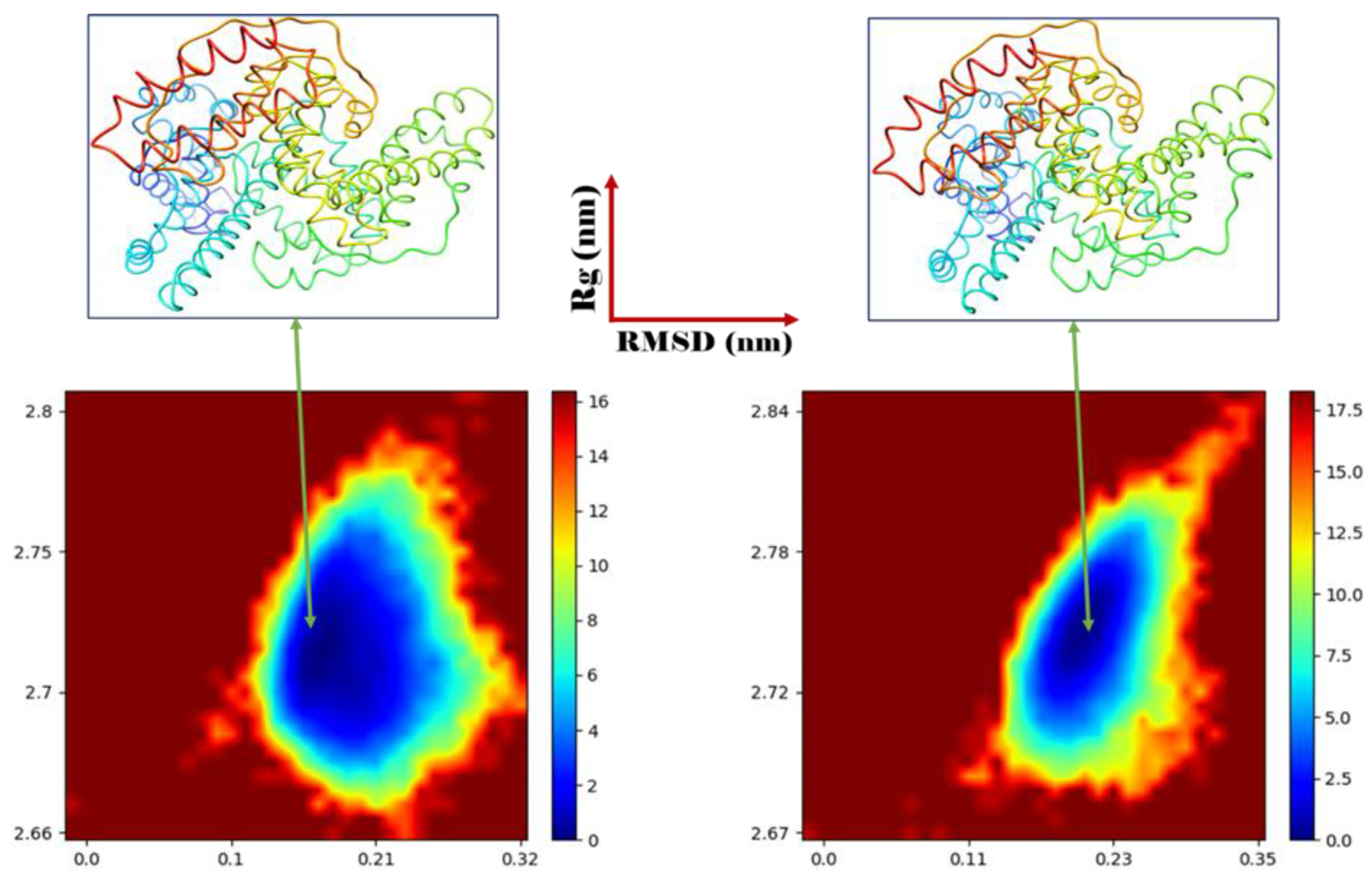
3.4. MD Simulation and Stability
3.5. Density Functional Theory (DFT) Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Broughton, H.B.; Watson, I.A. Selection of Heterocycles for Drug Design. J. Mol. Graph. Model. 2004, 23, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Arora, A.; Sapra, S.; Kumar, R.; Singh, B.K.; Singh, S.K. Recent Advances in the Synthesis and Utility of Thiazoline and Its Derivatives. RSC Adv. 2024, 14, 902–953. [Google Scholar] [CrossRef]
- Chugh, V.; Pandey, G.; Rautela, R.; Mohan, C. Heterocyclic Compounds Containing Thiazole Ring as Important Material in Medicinal Chemistry. Mater. Today Proc. 2022, 69, 478–481. [Google Scholar] [CrossRef]
- Amin, M.Z.; Rity, T.I.; Uddin, M.R.; Rahman, M.d.M.; Uddin, M.J. A Comparative Assessment of Anti-Inflammatory, Anti-Oxidant and Anti-Bacterial Activities of Hybrid and Indigenous Varieties of Pumpkin (Cucurbita Maxima Linn.) Seed Oil. Biocatal. Agric. Biotechnol. 2020, 28, 101767. [Google Scholar] [CrossRef]
- Lekouaghet, A.; Boutefnouchet, A.; Bensuici, C.; Gali, L.; Ghenaiet, K.; Tichati, L. In Vitro Evaluation of Antioxidant and Anti-Inflammatory Activities of the Hydroalcoholic Extract and Its Fractions from Leuzea Conifera L. Roots. S. Afr. J. Bot. 2020, 132, 103–107. [Google Scholar] [CrossRef]
- Singh, H.; Kumar, S.; Arya, A. Evaluation of Antibacterial, Antioxidant, and Anti-Inflammatory Properties of GC/MS Analysis of Extracts of Ajuga. Integrifolia Buch.-Ham. Leaves. Sci. Rep. 2024, 14, 16754. [Google Scholar] [CrossRef]
- Bakhouche, I.; Aliat, T.; Boubellouta, T.; Gali, L.; Şen, A.; Bellik, Y. Phenolic Contents and in Vitro Antioxidant, Anti-Tyrosinase, and Anti-Inflammatory Effects of Leaves and Roots Extracts of the Halophyte Limonium Delicatulum. S. Afr. J. Bot. 2021, 139, 42–49. [Google Scholar] [CrossRef]
- Kaur, R.; Desai, D.; Amin, S.; Raza, K.; Bhalla, A.; Yadav, P.; Kaushal, N. Selenocoxib-3, a Novel Anti-Inflammatory Therapeutic Effectively Resolves Colitis. Mol. Cell Biochem. 2023, 478, 621–636. [Google Scholar] [CrossRef]
- Aladejana, E.B. Biological Properties of Polyherbal Formulations: A Review of Their Antimicrobial, Anti-Inflammatory, Antioxidant, and Toxicological Activities. Pharmacogn. J. 2023, 15, 933–963. [Google Scholar] [CrossRef]
- Bindu, S.; Mazumder, S.; Bandyopadhyay, U. Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) and Organ Damage: A Current Perspective. Biochem. Pharmacol. 2020, 180, 114147. [Google Scholar] [CrossRef]
- Panchal, N.K.; Prince Sabina, E. Non-Steroidal Anti-Inflammatory Drugs (NSAIDs): A Current Insight into Its Molecular Mechanism Eliciting Organ Toxicities. Food Chem. Toxicol. 2023, 172, 113598. [Google Scholar] [CrossRef] [PubMed]
- Muhammed, M.T.; Aki-Yalcin, E. Molecular Docking: Principles, Advances, and Its Applications in Drug Discovery. Lett. Drug Des. Discov. 2024, 21, 480–495. [Google Scholar] [CrossRef]
- Prieto-Martínez, F.D.; López-López, E.; Eurídice Juárez-Mercado, K.; Medina-Franco, J.L. Computational Drug Design Methods—Current and Future Perspectives. In Silico Drug Design; Elsevier: Amsterdam, The Netherlands, 2019; pp. 19–44. [Google Scholar]
- Deore, A.B.; Dhumane, J.R.; Wagh, R.; Sonawane, R. The Stages of Drug Discovery and Development Process. Asian J. Pharm. Res. Dev. 2019, 7, 62–67. [Google Scholar] [CrossRef]
- Ferreira, L.; Dos Santos, R.; Oliva, G.; Andricopulo, A. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Alqahtani, S. In Silico ADME-Tox Modeling: Progress and Prospects. Expert. Opin. Drug Metab. Toxicol. 2017, 13, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Van Gunsteren, W.F.; Berendsen, H.J.C. Computer Simulation of Molecular Dynamics: Methodology, Applications, and Perspectives in Chemistry. Angew. Chem. Int. Ed. Engl. 1990, 29, 992–1023. [Google Scholar] [CrossRef]
- Kuhlman, B.; Bradley, P. Advances in Protein Structure Prediction and Design. Nat. Rev. Mol. Cell Biol. 2019, 20, 681–697. [Google Scholar] [CrossRef]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Tegegn, D.F.; Belachew, H.Z.; Salau, A.O. DFT/TDDFT Calculations of Geometry Optimization, Electronic Structure and Spectral Properties of Clevudine and Telbivudine for Treatment of Chronic Hepatitis B. Sci. Rep. 2024, 14, 8146. [Google Scholar] [CrossRef]
- Siddique, F.; Anwaar, A.; Bashir, M.; Nadeem, S.; Rawat, R.; Eyupoglu, V.; Afzal, S.; Bibi, M.; Bin Jardan, Y.A.; Bourhia, M. Revisiting Methotrexate and Phototrexate Zinc15 Library-Based Derivatives Using Deep Learning in-Silico Drug Design Approach. Front. Chem. 2024, 12, 1380266. [Google Scholar] [CrossRef]
- Chou, L.-K.; Quijada, M.A.; Clevenger, M.B.; de Oliveira, G.F.; Abboud, K.A.; Tanner, D.B.; Talham, D.R. Dication Salts of the Organic Donor Bis(Ethylenedithio)Tetrathiafulvalene. Chem. Mater. 1995, 7, 530–534. [Google Scholar] [CrossRef]
- Bellec, N.; Lorcy, D.; Boubekeur, K.; Carlier, R.; Tallec, A.; Los, S.z.; Pukacki, W.; Trybula, M.; Piekara-Sady, L.; Robert, A. Dicationic State of Dithiadiazafulvalene within a TCNQ Charge-Transfer Complex: Generation and Characterization. Chem. Mater. 1999, 11, 3147–3153. [Google Scholar] [CrossRef]
- Anokwah, D.; Kwatia, E.A.; Amponsah, I.K.; Jibira, Y.; Harley, B.K.; Ameyaw, E.O.; Obese, E.; Biney, R.P.; Mensah, A.Y. Evaluation of the Anti-Inflammatory and Antioxidant Potential of the Stem Bark Extract and Some Constituents of Aidia Genipiflora (DC.) Dandy (Rubiaceae). Heliyon 2022, 8, e10082. [Google Scholar] [CrossRef]
- Benesi, H.A.; Hildebrand, J.H. A Spectrophotometric Investigation of the Interaction of Iodine with Aromatic Hydrocarbons. J. Am. Chem. Soc. 1949, 71, 2703–2707. [Google Scholar] [CrossRef]
- Ahmad, S.; Raza, K. Identification of 5-Nitroindazole as a Multitargeted Inhibitor for CDK and Transferase Kinase in Lung Cancer: A Multisampling Algorithm-Based Structural Study. Mol. Divers. 2024, 28, 1189–1202. [Google Scholar] [CrossRef]
- Chitre, T.S.; Hirode, P.V.; Lokwani, D.K.; Bhatambrekar, A.L.; Hajare, S.G.; Thorat, S.B.; Priya, D.; Pradhan, K.B.; Asgaonkar, K.D.; Jain, S.P. In-Silico Studies of 2-Aminothiazole Derivatives as Anticancer Agents by QSAR, Molecular Docking, MD Simulation and MM-GBSA Approaches. J. Biomol. Struct. Dyn. 2024, 42, 11396–11414. [Google Scholar] [CrossRef] [PubMed]
- Musthafa, M.; Aneesrahman, K.N.; Perumalsamy, B.; Ramasamy, T.; Ganguly, R.; Sreekanth, A. Synthesis, Crystal Structure, DFT Study, in Vitro and in Silico Molecular Docking of Novel Bis (Aroyl Selenourea) Derivatives. J. Mol. Struct. 2019, 1180, 585–594. [Google Scholar] [CrossRef]
- Hossain, M.d.J.; Sultan, M.d.Z.; Rashid, M.A.; Kuddus, M.d.R. Interactions of Linagliptin, Rabeprazole Sodium, and Their Formed Complex with Bovine Serum Albumin: Computational Docking and Fluorescence Spectroscopic Methods. Anal. Sci. Adv. 2021, 2, 480–494. [Google Scholar] [CrossRef] [PubMed]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Kumar, S.; Manoharan, A.; J, J.; Abdelgawad, M.A.; Mahdi, W.A.; Alshehri, S.; Ghoneim, M.M.; Pappachen, L.K.; Zachariah, S.M.; Aneesh, T.P.; et al. Exploiting Butyrylcholinesterase Inhibitors through a Combined 3-D Pharmacophore Modeling, QSAR, Molecular Docking, and Molecular Dynamics Investigation. RSC Adv. 2023, 13, 9513–9529. [Google Scholar] [CrossRef]
- Amani, A.M.; Gholamzadeh, S.; Zarenezhad, M.; Malekpour, A.; Javidnezhad, E. DFT and Molecular Docking Studies on Biological Active Acridines: The Interaction with Bovine Serum Albumin. J. Adv. Med. Sci. Appl. Technol. 2015, 1, 78. [Google Scholar] [CrossRef]
- B, M.; Manandhar, S.; Hari, G.; Priya, K.; Kumar B, H.; Pai, K.S.R. Virtual Structure-Based Docking, WaterMap, and Molecular Dynamics Guided Identification of the Potential Natural Compounds as Inhibitors of Protein-Tyrosine Phosphatase 1B. J. Mol. Struct. 2021, 1226, 129396. [Google Scholar] [CrossRef]
- Ni, Y.; Zhu, R.; Kokot, S. Competitive Binding of Small Molecules with Biopolymers: A Fluorescence Spectroscopy and Chemometrics Study of the Interaction of Aspirin and Ibuprofen with BSA. Analyst 2011, 136, 4794. [Google Scholar] [CrossRef]
- Ayodele, P.F.; Bamigbade, A.; Bamigbade, O.O.; Adeniyi, I.A.; Tachin, E.S.; Seweje, A.J.; Farohunbi, S.T. Illustrated Procedure to Perform Molecular Docking Using PyRx and Biovia Discovery Studio Visualizer: A Case Study of 10kt With Atropine. Progress. Drug Discov. Biomed. Sci. 2023, 6. [Google Scholar] [CrossRef]
- Xu, L.; Zhao, L.; Che, J.; Zhang, Q.; Cao, R.; Li, X. Identification of Novel Influenza Polymerase PB2 Inhibitors Using Virtual Screening Approach and Molecular Dynamics Simulation Analysis of Active Compounds. Bioorg Med. Chem. 2021, 52, 116515. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A Webserver for the Prediction of Toxicity of Chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Biophys. J. 2017, 112, 175a–176a. [Google Scholar] [CrossRef]
- Bugnon, M.; Goullieux, M.; Röhrig, U.F.; Perez, M.A.S.; Daina, A.; Michielin, O.; Zoete, V. SwissParam 2023: A Modern Web-Based Tool for Efficient Small Molecule Parametrization. J. Chem. Inf. Model. 2023, 63, 6469–6475. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Bourougaa, L.; Ouassaf, M.; Alhatlani, B.Y. Clinical Informatics and Molecular Hybridization of Established Clinical DPP-4 Inhibitors to Generate next-Level Diabetes Type 2 Drugs. Chem. Pap. 2024, 78, 8485–8503. [Google Scholar] [CrossRef]
- Elangovan, N.; Sowrirajan, S.; Arumugam, N.; Rajeswari, B.; Mathew, S.; Priya, C.G.; Venkatraman, B.R.; Mahalingam, S.M. Theoretical Investigation on Solvents Effect in Molecular Structure (TD-DFT, MEP, HOMO-LUMO), Topological Analysis and Molecular Docking Studies of N-(5-((4-Ethylpiperazin-1-Yl)Methyl)Pyridin-2-Yl)-5-Fluoro-4-(4-Fluoro-1-Isopropyl-2-Methyl-1H-Benzo[d] Imidazol-6-Yl) Pyrimidin-2-Amine. Polycycl. Aromat. Compd. 2024, 44, 4467–4490. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Castaneda, S.M.B.; Alvarenga, E.S.; Demuner, A.J.; Guimaraes, L.M. Vibrational Spectra and Theoretical Calculations of a Natural Pentacyclic Triterpene Alcool Isolated from Mucuna Pruriens. Struct. Chem. 2020, 31, 599–607. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Ouassaf, M.; Bourougaa, L.; Bahaz, F.; Alhatlani, B.Y. Exploring the Antiviral Potential of Artemisia Annua Through JAK-STAT Pathway Targeting: A Network Pharmacology Approach. Pharmaceuticals 2024, 17, 1539. [Google Scholar] [CrossRef]
- Lotfi, B.; Mebarka, O.; Khan, S.U.; Htar, T.T. Pharmacophore-Based Virtual Screening, Molecular Docking and Molecular Dynamics Studies for the Discovery of Novel Neuraminidase Inhibitors. J. Biomol. Struct. Dyn. 2024, 42, 5308–5320. [Google Scholar] [CrossRef] [PubMed]
- Lotfi, B.; Mebarka, O.; Alhatlani, B.Y.; Abdallah, E.M.; Kawsar, S.M.A. Pharmacoinformatics and Breed-Based De Novo Hybridization Studies to Develop New Neuraminidase Inhibitors as Potential Anti-Influenza Agents. Molecules 2023, 28, 6678. [Google Scholar] [CrossRef]
- Choudhary, V.; Bhatt, A.; Dash, D.; Sharma, N. DFT Calculations on Molecular Structures, HOMO–LUMO Study, Reactivity Descriptors and Spectral Analyses of Newly Synthesized Diorganotin(IV) 2-chloridophenylacetohydroxamate Complexes. J. Comput. Chem. 2019, 40, 2354–2363. [Google Scholar] [CrossRef]
- Feixas, F.; Matito, E.; Duran, M.; Solà, M.; Silvi, B. Electron Localization Function at the Correlated Level: A Natural Orbital Formulation. J. Chem. Theory Comput. 2010, 6, 2736–2742. [Google Scholar] [CrossRef]
- Poater, J.; Duran, M.; Solà, M.; Silvi, B. Theoretical Evaluation of Electron Delocalization in Aromatic Molecules by Means of Atoms in Molecules (AIM) and Electron Localization Function (ELF) Topological Approaches. Chem. Rev. 2005, 105, 3911–3947. [Google Scholar] [CrossRef] [PubMed]
- Janani, S.; Rajagopal, H.; Muthu, S.; Aayisha, S.; Raja, M. Molecular Structure, Spectroscopic (FT-IR, FT-Raman, NMR), HOMO-LUMO, Chemical Reactivity, AIM, ELF, LOL and Molecular Docking Studies on 1-Benzyl-4-(N-Boc-Amino)Piperidine. J. Mol. Struct. 2021, 1230, 129657. [Google Scholar] [CrossRef]
- Reeda, V.J.; Sakthivel, S.; Divya, P.; Javed, S.; Jothy, V.B. Conformational Stability, Quantum Computational (DFT), Vibrational, Electronic and Non-Covalent Interactions (QTAIM, RDG and IGM) of Antibacterial Compound N-(1-Naphthyl)Ethylenediamine Dihydrochloride. J. Mol. Struct. 2024, 1298, 137043. [Google Scholar] [CrossRef]
- Suja, R.; Rathika, A.; Reeda, V.S.J.; Kumar, A.A.; Divya, P. Synthesis, Spectroscopic Analysis (FT-IR, FT-Raman, UV, NMR), Non-Covalent Interactions (RDG, IGM) and Dynamic Simulation on Bis(8-hydroxy Quinoline) Salicylate Salicylic Acid. J. Mol. Struct. 2024, 1310, 138231. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Equations | R2 | IC50 (µg/mL) |
|---|---|---|---|
| 4d | 0.993 | 21.9 | |
| 3c | 0.990 | 31.7 | |
| Aspirin | 0.996 | 22 |
| Compounds | Equations | R2 | K (M−1) | ΔG (Kcal.mol−1) |
|---|---|---|---|---|
| 4d | 0.979 | 16.30 × 103 | −5.75 | |
| 3c | 0.984 | 5.95 × 103 | −5.16 | |
| Aspirin | 0.987 | 12.40 × 103 | −5.59 |
| Molecules | ∆G (XP) | H-Bond | Distance (Å) | Pi–sulfur | Distance (Å) | Hydrophobic | Distance (Å) |
|---|---|---|---|---|---|---|---|
| 4d | −5.274 | Tyr149, Arg256 | [2.0–2.1] | Tyr149, | [2.9–3.1] | Tyr149, Leu237, His241, Ile263, Ile289, Ala290, | [3.7–4.8] |
| 3c | −4.731 | Tyr149 Arg256 | 2.2 | Tyr149 | 2.1 | Tyr149, His241, Leu237, Leu259, Ile263, Ile289, Ala290 | [4.1–5.0] |
| Aspirinref | −4.641 | Tyr149, Arg256 | [2.2–2.5] | - | - | Leu237 and Ala290 | [4.3–5.2] |
| 4d | Aspirin | |
|---|---|---|
| MW (g/mol) | 187.33 | 180.16 |
| Consensus Log Po/w | 2.36 | 1.28 |
| Log S | −2.77 | −1.85 |
| Bioavailability | 0.55 | 0.85 |
| GI absorption | High | High |
| Cytochrome P450 inhibition | No | No |
| Lipinski | Yes | Yes |
| Synthetic accessibility | 2.47 | 1.52 |
| Hepatotoxicity | Neurotoxicity | Cardiotoxicity | Carcinogenicity | Cytotoxicity | LD50 (mg/kg) | Class | |
|---|---|---|---|---|---|---|---|
| 4d | Inactive | Inactive | Inactive | Inactive | Inactive | 960 | 4 |
| 4d_BSA Complex | Aspirin_BSA Complex | |
|---|---|---|
| RMSD (nm) | 0.180 ± 0.01 | 0.324 ± 0.02 |
| RMSF (nm) | 0.161 ± 0.01 | 0.169 ± 0.01 |
| Rg (nm) | 2.757 ± 0.03 | 2.725 ± 0.02 |
| SASA (nm2) | 294.606 ± 1.25 | 292.994 ± 1.10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farial, B.; Lotfi, B.; Takoua, B.; Sameh, H.; Zinelaabidine, C.; Mandumpal, J.; Abdelfattah, M.A.O.; Maroua, F.; Abdelkrim, G. Synthesis, In Vitro Anti-Inflammatory Activity, Molecular Docking, Molecular Dynamics and DFT Calculations of Thiazoline-2-Thione Derivatives. Appl. Sci. 2025, 15, 6095. https://doi.org/10.3390/app15116095
Farial B, Lotfi B, Takoua B, Sameh H, Zinelaabidine C, Mandumpal J, Abdelfattah MAO, Maroua F, Abdelkrim G. Synthesis, In Vitro Anti-Inflammatory Activity, Molecular Docking, Molecular Dynamics and DFT Calculations of Thiazoline-2-Thione Derivatives. Applied Sciences. 2025; 15(11):6095. https://doi.org/10.3390/app15116095
Chicago/Turabian StyleFarial, Bahaz, Bourougaa Lotfi, Belghit Takoua, Hadjar Sameh, Cheraiet Zinelaabidine, Jestin Mandumpal, Mohamed A. O. Abdelfattah, Fattouche Maroua, and Gouasmia Abdelkrim. 2025. "Synthesis, In Vitro Anti-Inflammatory Activity, Molecular Docking, Molecular Dynamics and DFT Calculations of Thiazoline-2-Thione Derivatives" Applied Sciences 15, no. 11: 6095. https://doi.org/10.3390/app15116095
APA StyleFarial, B., Lotfi, B., Takoua, B., Sameh, H., Zinelaabidine, C., Mandumpal, J., Abdelfattah, M. A. O., Maroua, F., & Abdelkrim, G. (2025). Synthesis, In Vitro Anti-Inflammatory Activity, Molecular Docking, Molecular Dynamics and DFT Calculations of Thiazoline-2-Thione Derivatives. Applied Sciences, 15(11), 6095. https://doi.org/10.3390/app15116095