Abstract
Technological advancements have shifted human living and working environments from outdoor to indoor. Although indoor spaces offer protection from unfavorable weather conditions, they also present new health challenges. Stale, humid, and warm indoor air creates an ideal breeding ground for bacteria and fungi, leading to health issues such as asthma and bacterial infections. Although proper ventilation is crucial, a comprehensive inspection of local indoor air quality is necessary to prevent widespread diseases. In vitro experiments involving bacteria and fungi collected from indoor air yield accurate results but are time- and cost-intensive. In silico methods offer faster results and provide valuable insights for guiding further in vitro experiments. In this study, we conduct an in vitro cytotoxicity assay on 32 fungi species and compare its results with a memory-efficient in silico modeling method using parameter-efficient fine-tuning (PEFT) and ProtBERT. This study suggests a potential methodology for predicting the toxicity of indoor airborne fungi when their identities are known.
1. Introduction
Over centuries, the living and working environments of human beings have gradually shifted from outdoor to indoor. At present, the majority of people spend approximately 20 h indoors. Although it is advantageous to stay indoors for protection from rain, heat, or other environmental factors, staying indoors for prolonged periods may result in certain health hazards. Indoor air pollution is the cause of various cardiovascular and respiratory diseases, which accounted for 3.2 million deaths in 2020 [1,2,3,4]. Indoor air pollution is caused by burning combustion devices, new furniture, and tobacco, which release chemical pollutants such as carbon monoxide and sulfur dioxide. There are also biological pollutants, which include allergens, such as animal fur and house dust mites, and microbes, such as viruses, bacteria, and fungi.
Type A influenza is considered seasonal in a majority of the Korean population. The growing number of patients each year has increased the awareness regarding the prevention of bacterial diseases. For viral diseases, the 2015 MERS [5,6,7] outbreak followed by the 2019 COVID-19 pandemic [8,9,10] has prompted research on viral outbreak prevention. In contrast, fungal infections are often neglected, owing to few reported cases. However, this does lower the threat posed by fungal infections to human health. Aspergillosis, caused by the common household mold Aspergillus, may not be an imminent threat to healthy individuals. However, for individuals with a weakened immune system, allergic reactions or lung damage may occur [11,12]. More fatal diseases include Pneumocystis pneumonia, which is caused by Pneumocystis jirovecii [13,14]. Reports from the Center for Disease Control and Prevention (CDC) highlight an increase in reported fungal infections in the US, cautioning of a possible fungal disease outbreak [15].
Protein sequencing plays a crucial role in understanding the structure and biological function of proteins. In particular, identifying the functions of new microbes is crucial as the slightest mutation may cause microbes to act in a hazardous manner. However, traditional protein sequence analysis methods, such as Edman degradation and X-ray crystallography, require a significant amount of time and resources. This results in the challenge of deciding which microbes are worth analyzing, because it is inefficient to invest in research on microbes that are not well known or perceived to be harmless to other organisms. In silico modeling can be used to address this issue, as state-of-the-art computer simulations can provide a rough estimate of the function of a given protein sequence input. These results may not provide an accurate insight into the protein’s function but can guide the in vitro and in vivo researchers to devote their resources to other, more likely proteins.
In recent years, deep learning technology has resulted in many innovations in computer vision and natural language processing. The transformer model established in 2017 [16] provides an attention mechanism for machine text translation. Many deep learning models that adapt their architecture have been proposed. BERT, RoBERTa, and DistilBERT focus on creating contextualized word embeddings through multiple encoder attention blocks from the original transformer model [17,18,19]. On the contrary, the generative pre-trained transformer (GPT) relies significantly on the decider region of the transformer and is typically used for various generation tasks such as question and answering [20,21].
Because of the versatility of many large language models, any data in the form of text contain contextual data that can be used for pre-training. These models include ChemBERTa [22], MolBERT [23], and SolvBERT [24] from the field of molecular representation learning, which uses simplified molecular-input line-entry system (SMILES) data. Protein sequences can also be trained because they share many similarities with human text, such as repetitive regions and contextual data [25]. Using publicly available protein sequence data, several models have been proposed, such as ProtBERT, ProtT5, and Ankh [26,27].
Such large language models are pre-trained on massive amounts of data and, often, for many deep learning applications, fine-tuning the pre-trained weights with a specific dataset is sufficient for yielding satisfactory results. A major problem in fine-tuning is that, in the absence of layer freezing, all the parameters must be trained. This task is not only time-consuming but also increases the hardware barrier for anyone willing to fine-tune the model for their application. Thus, for a cost-effective fine-tuning method, parameter-efficient fine-tuning (PEFT) was introduced. The concept of PEFT is based on the idea that all the parameters of a pre-trained model are frozen. By adding and training a few trainable parameters, results similar to those of a fine-tuned model can be obtained; however, this results in a drastic reduction in the trainable parameters. This was proven in [28], where adaptors included additional trainable layers in the transformer block. Low-rank adaptation (LoRA) further reduces the parameters by breaking down the adaptation mechanism and optimizing the rank-decomposition matrices [29].
In this study, we attempted to verify the reliability of deep learning models as an appropriate in silico method for predicting the toxicity of fungal species by comparing the prediction results to those of the in vitro experiments. To train the in silico model, we focused on two major tasks. Initially, we trained the in silico model on fungal protein data and then improved its time efficiency using PEFT. For the in vitro experiments, we evaluated the cytotoxicity of fungal species with a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay.
2. Materials and Methods
2.1. In Vitro Data Collection and Experiment Setup
2.1.1. Collecting Indoor Airborne Fungi
The indoor air samples were collected between January 2020 and May 2021 on selected days to ensure similar average humidity and temperature. A microbial air sampler (KAS-110, Kemik Co., Seoul, Republic of Korea) was used to collect 100 L of air from the surfaces of various sites within a 1 m radius, where no obstacles were present. For the selective incubation of fungi, potato dextrose agar (PDA), malt extract agar with streptomycin, potato dextrose agar with streptomycin, sabouraud dextrose, and tryptic soy agar media were used at each site. Each medium was incubated at 28 °C for 5 days before analysis.
2.1.2. Indoor Airborne Fungal Gene Sequencing
Distilled water (100 mL) containing a fungal strain was heated at 100 °C for 10 min and centrifuged at 13,000 rpm for 5 min to precipitate the impurities for strain analysis. The nucleotide sequences of ITS1 and ITS2 were determined in both directions using ABI 3730XL DNA Analyzer (Applied Biosystems, Waltham, MA, USA). The sequencing service was provided by Solgent Co., Ltd., Daejeon, Republic of Korea.
2.1.3. Preparation of Fungal Samples for Measuring Their Effect on Cellular Activity
The 32 most frequently identified fungal species were selected. These species were inoculated onto PDA plates and allowed to grow until mycelial formation was observed. Subsequently, 5 mL of phosphate-buffered saline was added to each plate. Using a spreader, the suspended fungal spores were collected in the liquid state. The spores were then filtered using Miracloth to obtain a spore suspension for use in cell experiments. The suspended fungal spores were inactivated by heating at 100 °C for 10 min before the cytotoxicity assay to prevent unwanted fungal contamination in animal cell culture systems and to simulate fungal static metabolism in the air, where fungi encounter harsh conditions, leading to minimal cellular activity.
2.1.4. In Vitro Cytotoxicity Assay
To assess the impact of indoor airborne fungi on cellular activity, we employed the MTT assay, a widely used method to measure cytotoxicity by toxic agents. Human cell lines MRC5 and HeLa were cultured in minimum essential medium Eagle (MEM, WELGENE) media supplemented with 10% FBS and 1% penicillin–streptomycin solution (WELGENE). Cells were seeded into a 96-well plate and cultured with 5% CO2 at 37 °C for 24 h [30]. Heat-inactivated fungal samples were added after serial dilution. After 24 h of incubation, the optical density of the wells was quantified at 580 nm.
2.2. In Silico Data Preparation
Creating a new dataset for protein sequence analysis is painstaking as a significant amount of time and resources are required to cultivate and sequence the target organism. It has attracted considerable attention worldwide as a unified public database for researchers to share their sequencing results. The Universal Protein Resource Knowledgebase (UniProtKB) [31] is a centralized database of protein sequences, each labeled with their unique functional description. The database comprises data gathered from the European Bioinformatics Institute (EMBL-EBI), Swiss Institute of Bioinformatics (SIB), and Protein Information Resource (PIR); the database provides both reviewed (Swiss-Prot) and computer-annotated unreviewed protein sequences (TrEMBL).
In our previous work, we collected bacterial proteins from UniProt by searching all relevant organisms under the bacteria domain. From the data gathered, we labeled the sequences according to their Gene Ontology (GO) matchings [32]. For the fungi data that we use in this paper, we followed the same technique that we have used in our previous paper and collected all relevant organisms under the kingdom fungi. However, unlike our previous work, we did not label fungal protein sequences with their GO. While GO provides great insights into the properties of a certain protein, our goal was to tag a protein sequence with a comprehensive description such as virulence or toxin. We defined certain keywords and opted to identify every protein sequence with such keywords in their description.
The main focus of an in silico model is to determine whether a protein can create toxins or cause virulent activity. Thus, we chose to label protein sequences with “virulent” for virulent activity or virulence factors, “toxic” for toxic activity or toxins, and “normal” for non-virulent activity. The keywords used to find such protein sequences are “virul” for virulent activity and “toxi” for toxic activity. When labeling the protein sequences, we labeled any sequences with the keyword “anti-toxi” as “normal” because they are not related to any toxic activity. Algorithm 1 shows the pseudocode for our protein sequence labeling strategy.
| Algorithm 1 Pseudo Code of In Silico Data Collection |
|
In total, 15,660,390 sequences were collected using this process. Before utilizing the collected data to train the in silico model, we excluded the sequences of 32 species of fungi from the initial training data. The purpose of this study was to use the data of the same 32 species collected during the indoor airborne fungi collection mentioned above in Section 2.1.1 as inference data for a fair comparison of the reliability of the in silico model with the in vitro experiments. As a portion of the collected fungi species are native to the Korean peninsula, half of the species protein data were unknown or unavailable in the UniProtKB database. Therefore, we made predictions for the sequences of the 15 species in which data were available in UniProtKB. Using the trained in silico model, we compared the results of the 15 fungi species data with the in vitro experiments. Table 1 shows the sequences collected for each class of the 15 species. After excluding the above-mentioned data, we collected 15,643,956 of normal sequences, 11,828 of toxin sequences, and 4606 of virulent sequences for each class. However, in this case, the dataset is highly imbalanced. It is well known that imbalanced data can result in the failure of a model to classify protein sequences accurately in the minority class. Hence, to balance the dataset, we randomly selected 4606 sequences from the normal and toxin classes because there were only 4606 sequences in the virulence class. We split the balanced dataset using a common data splitting ratio of 7:2:1 for the training, validation, and test sets. The training set was used to train the model, whereas the validation set was used to evaluate the performance of the trained model at each epoch. Finally, the test set was used to evaluate the general performance of the model. The data used to train the model is made available online. The link to the data is available below in the Supplementary Materials below.

Table 1.
Number of protein sequences collected for the 15 fungal species excluded from the training dataset for comparison with in vitro experiment results.
2.3. In Silico Model Development
In a previous study [30], we used the hugging face [33] implementation of ProtBERT [26]. We again used the hugging face implementation of ProtBERT. ProtBERT, unlike the original BERT model, used 30 encoder layers, whereas the BERT-based model only used 12 encoder layers. With more encoder layers, more in-depth contextual data can be drawn from the inputs. Unlike the BERT model pre-trained on natural language, ProtBERT is pre-trained using protein sequences. In this paper, we chose ProtBERT-BFD, which is pre-trained on the Big Fantastic Database (BFD) data for fine-tuning experiments conducted in [26]. The study reveals that the BFD pre-trained ProtBERT model yields a better understanding of the protein sequence contextual data. To further improve the efficiency of the model, we applied LoRA to the ProtBERT model. Adaptors allow one to train on significantly fewer parameters with minor to no degradation in model performance [28,29]. Much like the original LoRA paper, we applied low-rank decomposition to the query and value attention heads. The model structure of both ProtBERT and ProtBERT with LoRA is shown below in Figure 1.
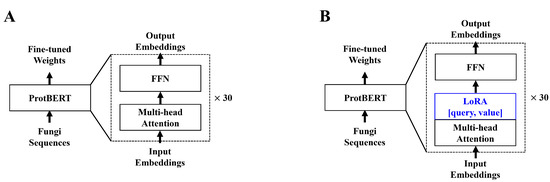
Figure 1.
(A) depicts the original ProtBERT model with an additional classification layer. (B) depicts the modified ProtBERT model with LoRA. Both models take protein sequences with a maximum length of 1024. We apply zero padding to sequences shorter than 1024. For (B), we apply LoRA to the query and value attention heads in the feed-forward layer.
2.4. In Silico Model Training and Evaluation
We trained ProtBERT with and without LoRA to observe any significant increases in the efficiency. Both models were trained for 20 epochs at a learning rate of 1 × using the Adam optimizer. We used the NVIDIA RTX 3090 (Santa Clara, CA, USA) for the hardware, which offers 24 GB of VRAM suitable for training many deep learning models. We used accuracy, F1-score, Matthews correlation coefficient (MCC), and auROC metrics to evaluate both models. In addition, we investigated the number of trainable parameters in both models because they indicate the time required to train the model. For a further visual representation of the outputs, we used the t-distributed stochastic neighbor embeddings (t-SNEs) and reduced the dimension of the last hidden state embeddings of the model to a 2-dimensional space.
3. Results
3.1. In Silico Results
Regarding the model efficiency, training the ProtBERT model with normal fine-tuning required an average of 34 min per epoch, with 420,983,811 trainable parameters used. For ProtBERT with LoRA, an average of 27 min was required, with 983,040 trainable parameters. We present the results for both models with the lowest validation set loss within 20 epochs. Figure 2 shows the t-distributed stochastic neighbor embedding (t-SNE) visualization of the prediction results for both models with the test dataset. Both figures show clear decision boundaries for the classification of classes. For a more objective examination of the performance of the model, we ran the particular trained weights on the test set. Performance measures of both models are presented below in Table 2.
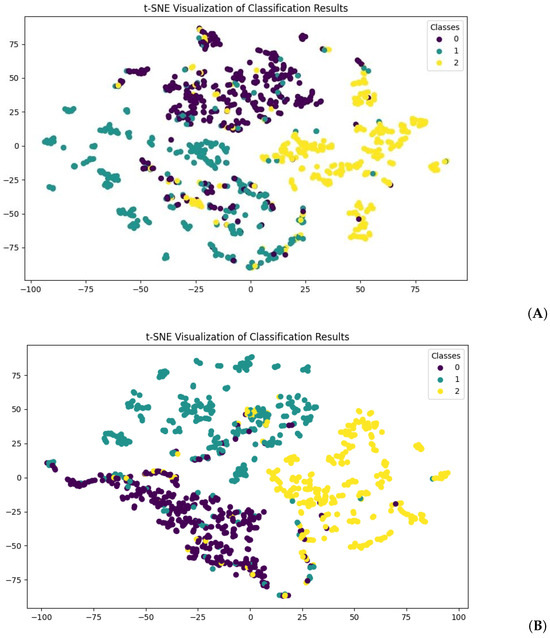
Figure 2.
t-SNE visualization of test set data prediction. (A) shows predictions made from normal fine-tuned ProtBERT, and (B) shows predictions made from ProtBERT with LoRA. For both visualizations, class 0 stands for normal protein sequences (purple), class 1 stands for toxic protein sequences (green), and class 2 stands for virulent protein sequences (yellow).
Table 2.
In silico model performance of ProtBERT with normal fine-tuning and ProtBERT with LoRA.
It is noticeable that the ProtBERT model with LoRA outperformed the normal fine-tuned ProtBERT model by a margin in every performance measure. The test set results showed a 0.0568 increase in accuracy, 0.0555 increase in F1-score, 0.0819 in MCC, and 0.0166 in auROC. Originally, we presumed a significant decrease in training time and a slight decrease in performance for the ProtBERT model with LoRA. However, the experimental results contradicted this, and a significant increase was observed for all performance measures and a slight reduction was observed in the training time. The discussion section below presents more presumptions regarding the in silico model training results.
3.2. In Silico Results on Inference Data
To assess the in silico model’s reliability in predicting hazardous fungal proteins, we used ProtBERT with LoRA because it exhibited better performance in every evaluation metric. We ran the inference data of all 15 fungal species. A sample of the prediction results with the highest probabilities of being in each class is presented in Table 3, Table 4 and Table 5. The three right columns of each table represent the different probabilites of each of the classes. The prediction values are derived from the softmax layer outputs of the model. All prediction results for all the protein sequences of 15 fungal species are available online. The link to access results is noted below in the supplementary materials section.
Table 3.
Inference results for possible toxic proteins.
Table 4.
Inference results for possible virulent proteins.
Table 5.
Inference results for possible normal proteins.
3.3. The Effect of Algorithm-Predicted Fungi on Cellular Activity of Human Cell Lines
To validate the predictive capacity of our algorithm for putative toxicity-related proteins in fungi, we selected 14 fungal species that exhibited significant protein numbers for normal, toxic, and virulent proteins according to our algorithm. For assessing the impact of these fungal species on cell viability in human cell lines MRC5 and Hela, we initially established a threshold for cytotoxic concentrations of fungal samples using serial concentrations of the C. cladosporioides sample. C. cladosporioides is known to cause seasonal allergic reactions but does not cause invasive infections in animals (Figure 3A). We found a discernible decrease in cell viability for both MRC5 and Hela cells at the concentration of 1 × CFU of C. cladosporioides. Utilizing this concentration as the standard experimental condition for observing cell viability, we observed consistent patterns of cell viability changes across both human cell lines in response to respective fungal samples (Figure 3B). This suggests the possibility of common signaling or damaging pathways of both cell types affected by fungal components. A. niger, B. Adusta, C. cladosporioides, and F. equiseti exhibited minimal effects on the viability of both cell lines. In contrast, the remaining 10 species caused a notable reduction, hovering around 50% in cell viability across both cell lines. This result underscores the variability in the cytotoxicity of fungal content, highlighting fungal-species-dependent responses in this in vitro evaluation test for fungi.
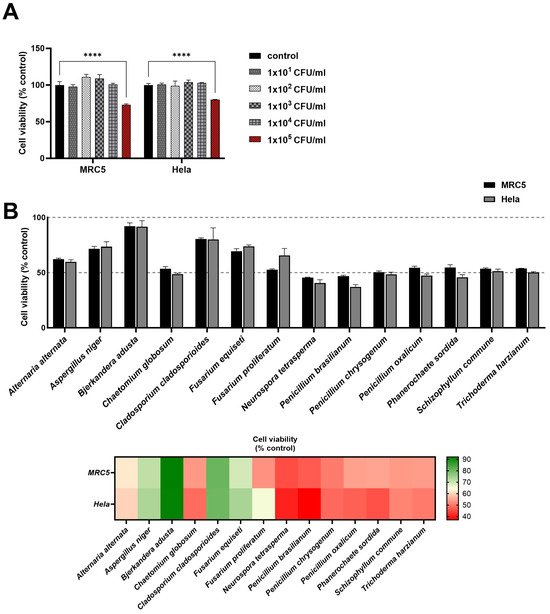
Figure 3.
Assessing fungal toxicity in human cell lines through in vitro analysis. Effects of selected fungal species on the viability of MRC5 and Hela cells. For all assays, heat-inactivated fungal samples were added to cell lines pre-seeded in 96-well plates after 24 h of incubation. (A) The cytotoxicity threshold was determined by conducting with various concentrations of C. cladosporioides for establishing a standard concentration of fungal samples. (B) Cell viability rates of 14 fungi are shown as bars and heat maps. Results are represented as cell viability as a percentage of cells incubated without fungi. Experiments were reiterated twice, evaluating each condition in triplicate. Data are shown as the mean ± SD. **** p < 0.0001, as determined by two-way analysis of variance. CFU; colony forming unit.
4. Discussion
We hypothesized that applying LoRA to ProtBERT would improve the model efficiency at the cost of decreasing the model performance. However, the in silico training results presented in Section 3.1 proved otherwise, as all the performance measures increased as compared with those of normal fine-tuning. We presume that this is because of the difference in the number of trainable parameters and how the LoRA works. Normal fine-tuned ProtBERT has too many parameters to train as compared to that for ProtBERT with LoRA, which has approximately 400 times fewer parameters. With fewer parameters to train, the likelihood of catastrophic forgetting reduces. Additionally, LoRA trains only newly added layers and does not include the original pre-trained ProtBERT weights. Therefore, the pre-trained weights of the ProtBERT model are already capable of extracting useful contextual data.
The indoor airborne fungal species predicted by our algorithm to harbor potential toxic and/or virulent proteins includes Alternaria alternata, Aspergillus niger, Chaetomium globosum, Fusarium equiseti, Fusarium proliferatum, Neurospora tetrasperma, Penicillium brasilianum, Penicillium chrysogenum, Penicillium oxalicum, Phanerochaete sordida, Schizophyllum commune, Trichoderma harzianum, and they exhibited toxic activity in the two human cell lines used in this study. Known as plant pathogens, they are subjects of ongoing research in various areas, including basic research or biotechnology related to enzymes and genomics. While the fungal spores that they produce, like those of most fungi, can induce allergic reactions in humans, their pathogenic potential in humans remains to be further explored. Our in vitro cellular viability results closely align with in silico predictions, suggesting a potential methodology for evaluating the in vitro cytotoxicity of fungi present in indoor air, combining in silico prediction with experimental assays. Despite limited data on fungal protein sequences for training transformer models, this pilot study successfully developed an in silico prediction module, running in parallel with in vitro cytotoxicity evaluation. These efforts contribute to the advancement of technology development for a swift understanding of unidentified fungi floating indoors, which might pose threats or exacerbate human health.
5. Conclusions
In this study, we improved the in silco model performance and assessed the reliability of using ProtBERT for fungi toxicity prediction. In improving model performance, we applied LoRA to the ProtBERT model.The in silico experimental results showed that ProtBERT with LoRA outperformed the normal fine-tuning method. Using the trained in silico model, we compared the toxicity prediction of fungal species with our in vitro experimental results. The results of the toxicity prediction of the fungal species using the in silico model showed that there may be possible protein sequences whose functions are unknown that may present fungal toxicity. In vitro experiments reveal A. alternata, F. proliferatum, and P. brasilianum as possible toxic fungal species.
By comparing the possibly toxic proteins of these fungal species with those in our in silico results, we presume that certain unknown proteins predicted to be either toxic or virulent may be the cause. In the future, we plan to confirm this hypothesis by performing additional in vitro and in vivo experiments.
Supplementary Materials
The training data for the in silico model and prediction results of the 15 species of fungi are made available online at https://github.com/sungyoonahn/Comparing-in-silico-Fungi-Toxicity-Prediction-with-in-vitro-MTT-Assay (accessed 14 November 2023).
Author Contributions
Data curation, M.K. and H.-W.J.; Funding acquisition, I.-S.B. and S.-W.L.; Methodology, S.-Y.A., M.K., H.-W.J., W.Y., I.-S.B. and S.-W.L.; Software, S.-Y.A.; Supervision, I.-S.B. and S.-W.L.; Writing—original draft, S.-Y.A., M.K., H.-W.J. and W.Y.; Writing—review and editing, I.-S.B. and S.-W.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Korea Environment Industry Technology Institute (KEITI) through the Technology Development Project for Biological Hazards Management in Indoor Air Program (or Project) funded by the Korea Ministry of Environment (MOE) (2021003380003), and by a research fund from Chosun University, 2017.
Data Availability Statement
The MTT assay data shown in this research cannot be shared publicly for now. Any inquiries about the MTT assay data can be requested by email to the corresponding author. The protein sequence data used in the in silico model research are available online at https://github.com/sungyoonahn/Comparing-in-silico-Fungi-Toxicity-Prediction-with-in-vitro-MTT-Assay (accessed 14 November 2023).
Acknowledgments
We would like to acknowledge and thank Joohee Oh and Yunyoung Chang for aiding in data collection for the training of the in silico model.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- World Health Organization. Household Air Pollution. Available online: https://www.who.int/news-room/fact-sheets/detail/household-air-pollution-and-health?gclid=CjwKCAiAxreqBhAxEiwAfGfndH3VhED1dNR75_bIo6hEOggDPIgR8zHskVOAo9fITvo-TfUuZwd--xoCSlcQAvD_BwE (accessed on 11 February 2023).
- Pillarisetti, A.; Ye, W.; Chowdhury, S. Indoor air pollution and health: Bridging perspectives from developing and developed countries. Annu. Rev. Environ. Resour. 2022, 47, 197–229. [Google Scholar] [CrossRef]
- Tran, V.V.; Park, D.; Lee, Y.C. Indoor air pollution, related human diseases, and recent trends in the control and improvement of indoor air quality. Int. J. Environ. Res. Public Health 2020, 17, 2927. [Google Scholar] [CrossRef]
- Raju, S.; Siddharthan, T.; McCormack, M.C. Indoor air pollution and respiratory health. Clin. Chest Med. 2020, 41, 825–843. [Google Scholar] [CrossRef]
- Park, J.E.; Jung, S.; Kim, A.; Park, J.E. MERS transmission and risk factors: A systematic review. BMC Public Health 2018, 18, 1–15. [Google Scholar] [CrossRef]
- Mackay, I.M.; Arden, K.E. MERS coronavirus: Diagnostics, epidemiology and transmission. Virol. J. 2015, 12, 1–21. [Google Scholar] [CrossRef]
- De Wit, E.; Van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, L. SARS-CoV-2: Virus dynamics and host response. Lancet Infect. Dis. 2020, 20, 515–516. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Song, Y.; Chen, Y.; Wu, N.; Xu, J.; Sun, C.; Zhang, J.; Weng, T.; Zhang, Z.; Wu, Z.; et al. Molecular architecture of the SARS-CoV-2 virus. Cell 2020, 183, 730–738. [Google Scholar] [CrossRef]
- Platto, S.; Xue, T.; Carafoli, E. COVID19: An announced pandemic. Cell Death Dis. 2020, 11, 799. [Google Scholar] [CrossRef] [PubMed]
- Segal, B.H. Aspergillosis. N. Engl. J. Med. 2009, 360, 1870–1884. [Google Scholar] [CrossRef] [PubMed]
- Cadena, J.; Thompson, G.R.; Patterson, T.F. Aspergillosis: Epidemiology, diagnosis, and treatment. Infect. Dis. Clin. 2021, 35, 415–434. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.; Lundgren, J.D.; Masur, H.; Walzer, P.D.; Hanson, D.L.; Frederick, T.; Huang, L.; Beard, C.B.; Kaplan, J.E. Current epidemiology of Pneumocystis pneumonia. Emerg. Infect. Dis. 2004, 10, 1713. [Google Scholar] [CrossRef]
- Thomas, C.F., Jr.; Limper, A.H. Pneumocystis pneumonia. N. Engl. J. Med. 2004, 350, 2487–2498. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Impact of Fungal Diseases in the United States. Available online: https://www.cdc.gov/fungal/cdc-and-fungal/burden.html (accessed on 3 February 2023).
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł.; Polosukhin, I. Attention is all you need. In Proceedings of the Advances in Neural Information Processing Systems 30: Annual Conference on Neural Information Processing Systems 2017, Long Beach, CA, USA, 4–9 December 2017. [Google Scholar]
- Devlin, J.; Chang, M.W.; Lee, K.; Toutanova, K. Bert: Pre-training of deep bidirectional transformers for language understanding. arXiv 2018, arXiv:1810.04805. [Google Scholar]
- Liu, Y.; Ott, M.; Goyal, N.; Du, J.; Joshi, M.; Chen, D.; Levy, O.; Lewis, M.; Zettlemoyer, L.; Stoyanov, V. Roberta: A robustly optimized bert pretraining approach. arXiv 2019, arXiv:1907.11692. [Google Scholar]
- Sanh, V.; Debut, L.; Chaumond, J.; Wolf, T. DistilBERT, a distilled version of BERT: Smaller, faster, cheaper and lighter. arXiv 2019, arXiv:1910.01108. [Google Scholar]
- Radford, A.; Narasimhan, K.; Salimans, T.; Sutskever, I. Improving Language Understanding by Generative Pre-Training; OpenAI: San Francisco, CA, USA, 2018. [Google Scholar]
- OpenAI. GPT-4 Technical Report. arXiv 2023, arXiv:2303.08774. [Google Scholar]
- Chithrananda, S.; Grand, G.; Ramsundar, B. ChemBERTa: Large-scale self-supervised pretraining for molecular property prediction. arXiv 2020, arXiv:2010.09885. [Google Scholar]
- Fabian, B.; Edlich, T.; Gaspar, H.; Segler, M.; Meyers, J.; Fiscato, M.; Ahmed, M. Molecular representation learning with language models and domain-relevant auxiliary tasks. arXiv 2020, arXiv:2011.13230. [Google Scholar]
- Yu, J.; Zhang, C.; Cheng, Y.; Yang, Y.F.; She, Y.B.; Liu, F.; Su, W.; Su, A. SolvBERT for solvation free energy and solubility prediction: A demonstration of an NLP model for predicting the properties of molecular complexes. Digit. Discov. 2023, 2, 409–421. [Google Scholar] [CrossRef]
- Ofer, D.; Brandes, N.; Linial, M. The language of proteins: NLP, machine learning & protein sequences. Comput. Struct. Biotechnol. J. 2021, 19, 1750–1758. [Google Scholar]
- Elnaggar, A.; Heinzinger, M.; Dallago, C.; Rehawi, G.; Wang, Y.; Jones, L.; Gibbs, T.; Feher, T.; Angerer, C.; Steinegger, M.; et al. Prottrans: Toward understanding the language of life through self-supervised learning. IEEE Trans. Pattern Anal. Mach. Intell. 2021, 44, 7112–7127. [Google Scholar] [CrossRef]
- Elnaggar, A.; Essam, H.; Salah-Eldin, W.; Moustafa, W.; Elkerdawy, M.; Rochereau, C.; Rost, B. Ankh☥: Optimized protein language model unlocks general-purpose modeling. bioRxiv 2023. [Google Scholar] [CrossRef]
- Houlsby, N.; Giurgiu, A.; Jastrzebski, S.; Morrone, B.; De Laroussilhe, Q.; Gesmundo, A.; Attariyan, M.; Gelly, S. Parameter-efficient transfer learning for NLP. In Proceedings of the International Conference on Machine Learning, PMLR, Long Beach, CA, USA, 9–15 June 2019; pp. 2790–2799. [Google Scholar]
- Hu, E.J.; Shen, Y.; Wallis, P.; Allen-Zhu, Z.; Li, Y.; Wang, S.; Wang, L.; Chen, W. Lora: Low-rank adaptation of large language models. arXiv 2021, arXiv:2106.09685. [Google Scholar]
- Ahn, S.Y.; Kim, M.; Bae, J.E.; Bang, I.S.; Lee, S.W. Reliability of the In Silico Prediction Approachm to In Vitro Evaluation of Bacterial Toxicity. Sensors 2022, 22, 6557. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G.O. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004, 32, D258–D261. [Google Scholar] [CrossRef]
- Wolf, T.; Debut, L.; Sanh, V.; Chaumond, J.; Delangue, C.; Moi, A.; Cistac, P.; Rault, T.; Louf, R.; Funtowicz, M.; et al. Huggingface’s transformers: State-of-the-art natural language processing. arXiv 2019, arXiv:1910.03771. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).