
Applying Molecular Modeling to Predict Novel FmlH-Binding Glycomimetics with Improved Pharmacokinetic Properties for the Prevention of Urinary Tract Infections



Abstract
1. Introduction
2. Methods and Materials
2.1. Generation of the Protein 3D Structure
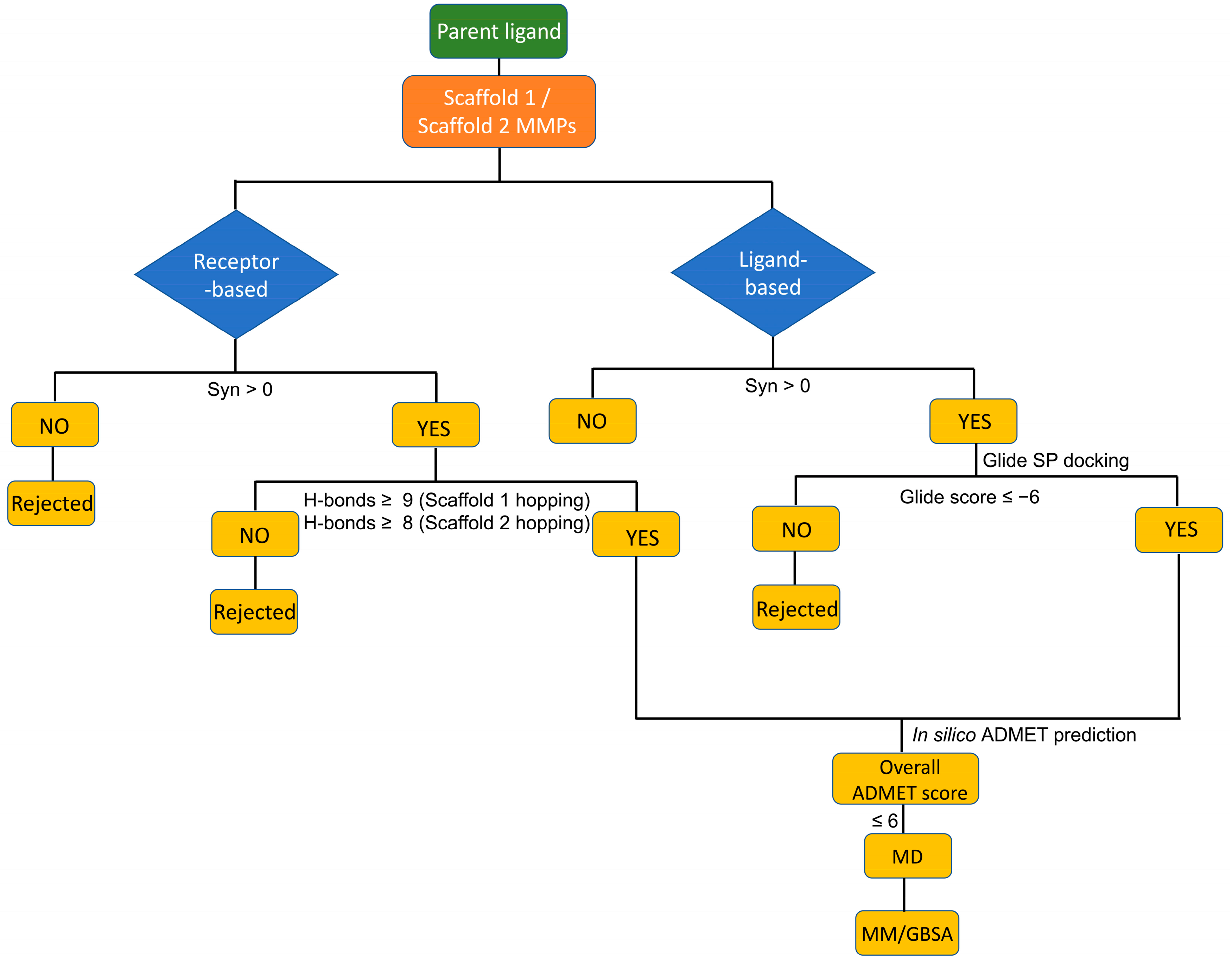
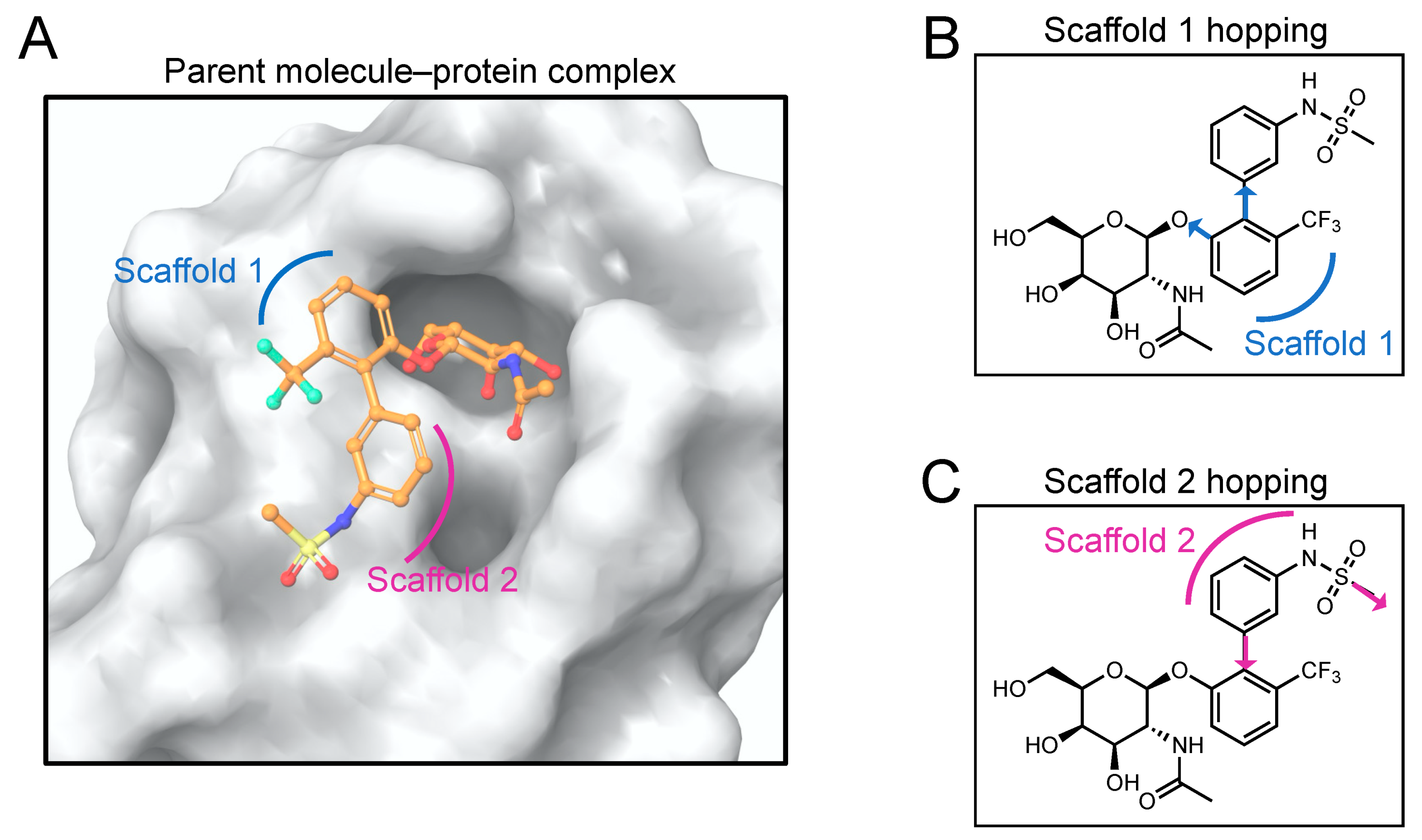
2.2. Scaffold Hopping
2.2.1. Scaffold 1 Hopping
2.2.2. Scaffold 2 Hopping
2.3. Molecular Docking
2.4. ADMET Prediction
2.5. Molecular Dynamics Simulations
2.6. Prime MM-GBSA Calculations
3. Results and Discussion
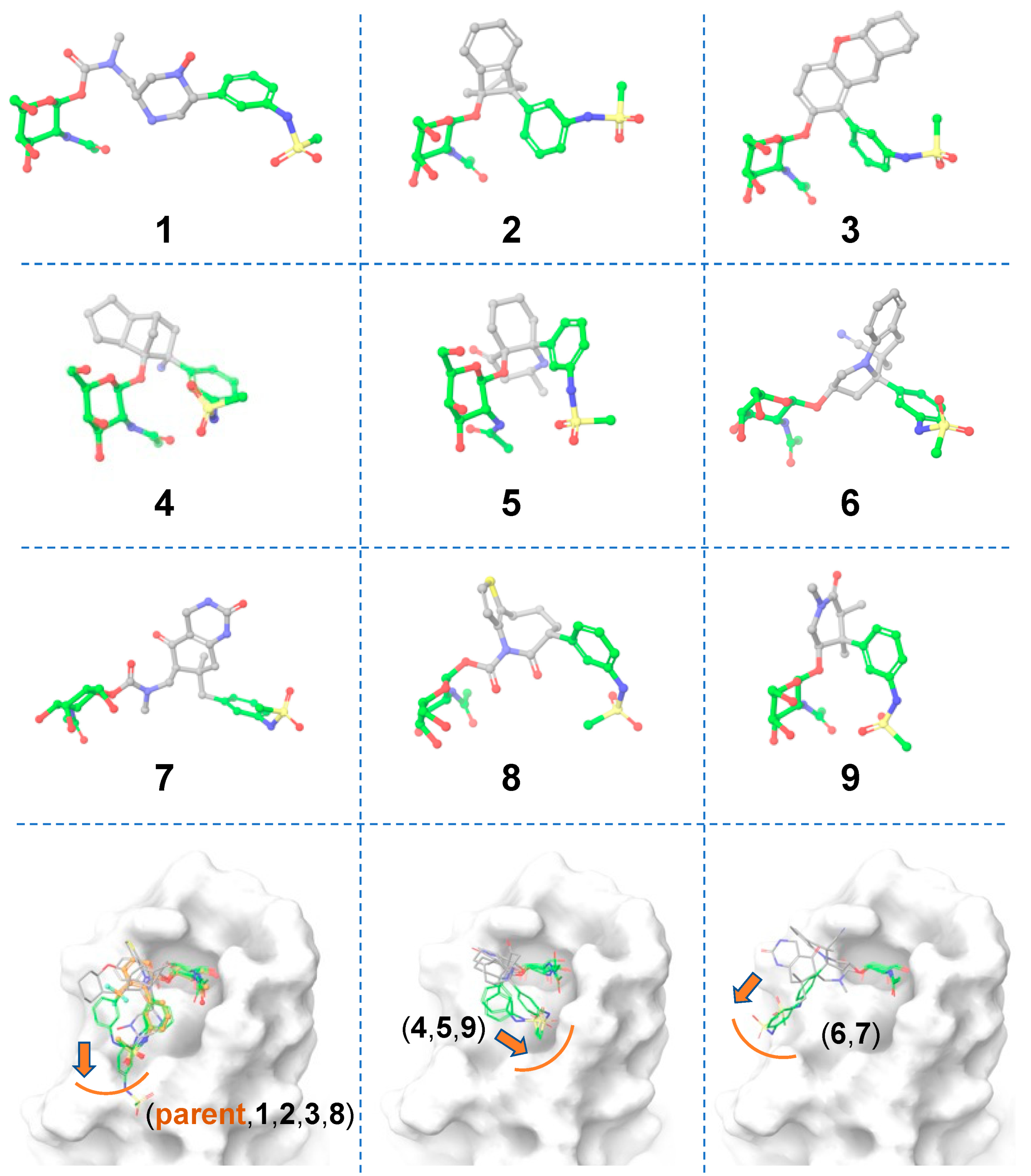
3.1. Scaffold 1
3.1.1. Ligand-Based Scaffold Hopping
3.1.2. Receptor-Based Scaffold Hopping
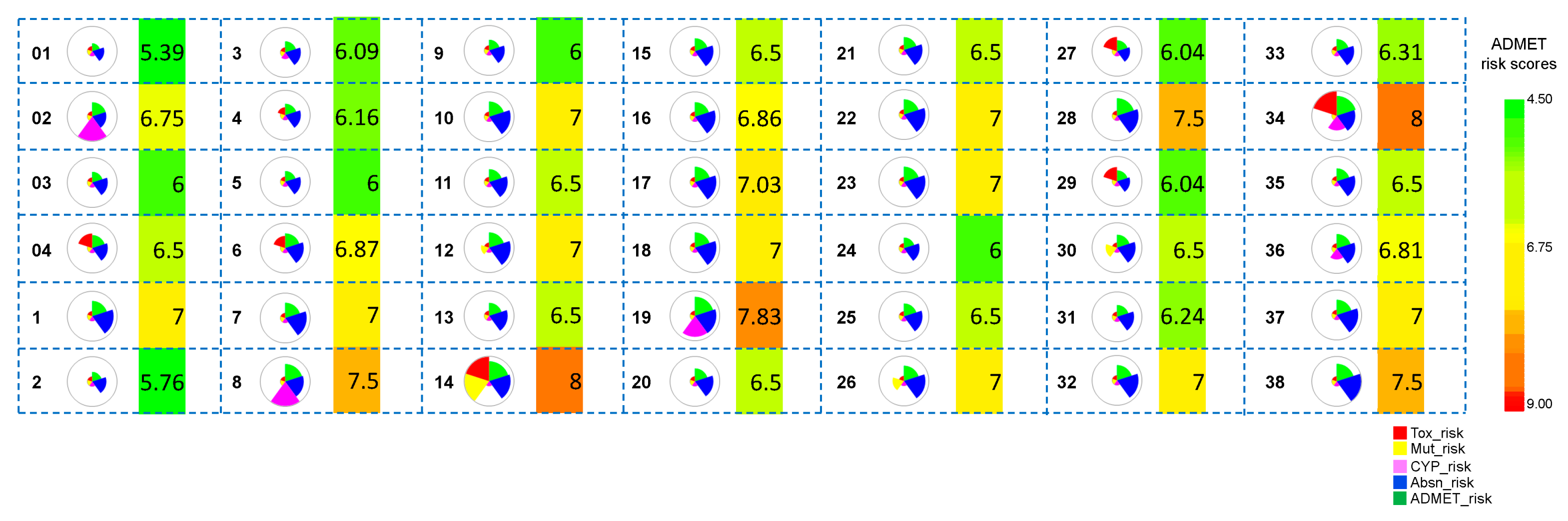
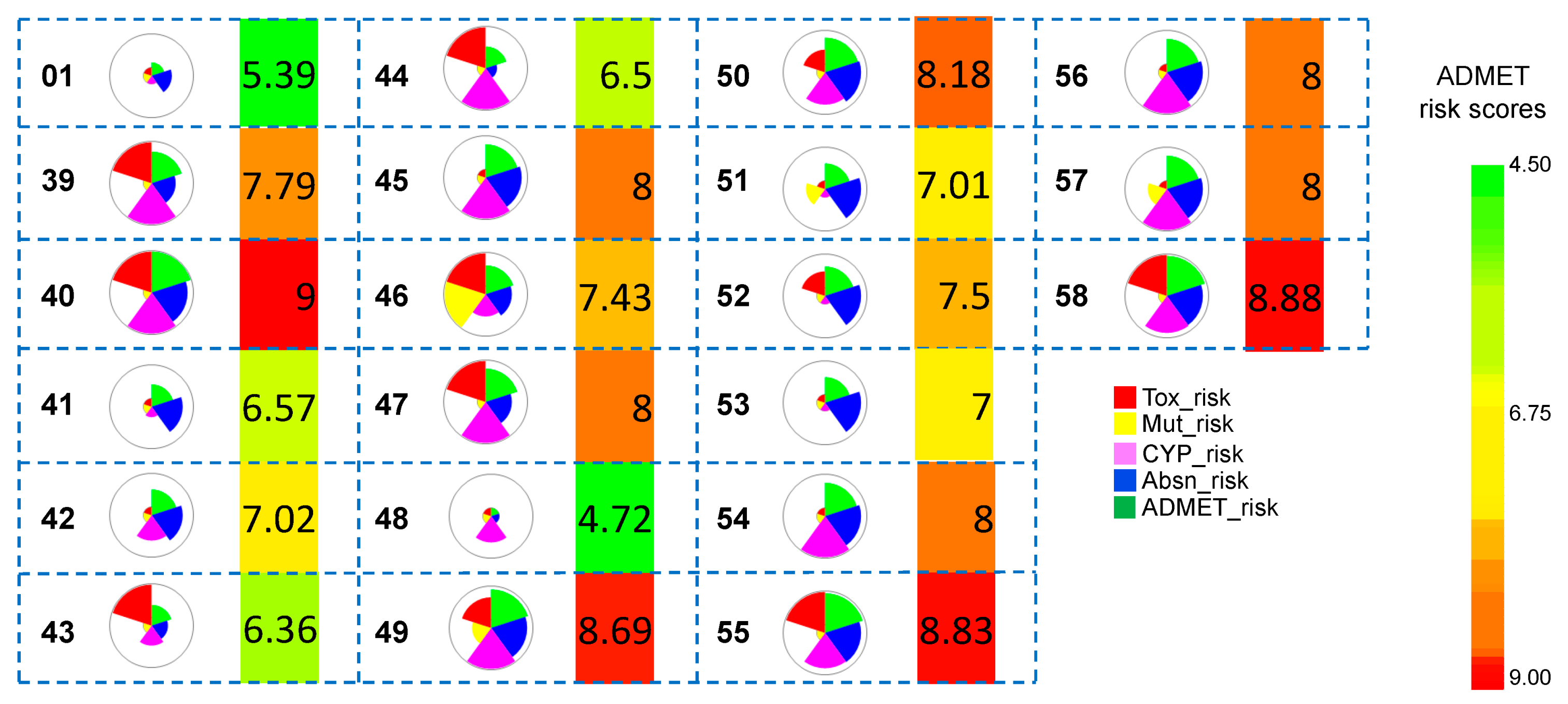
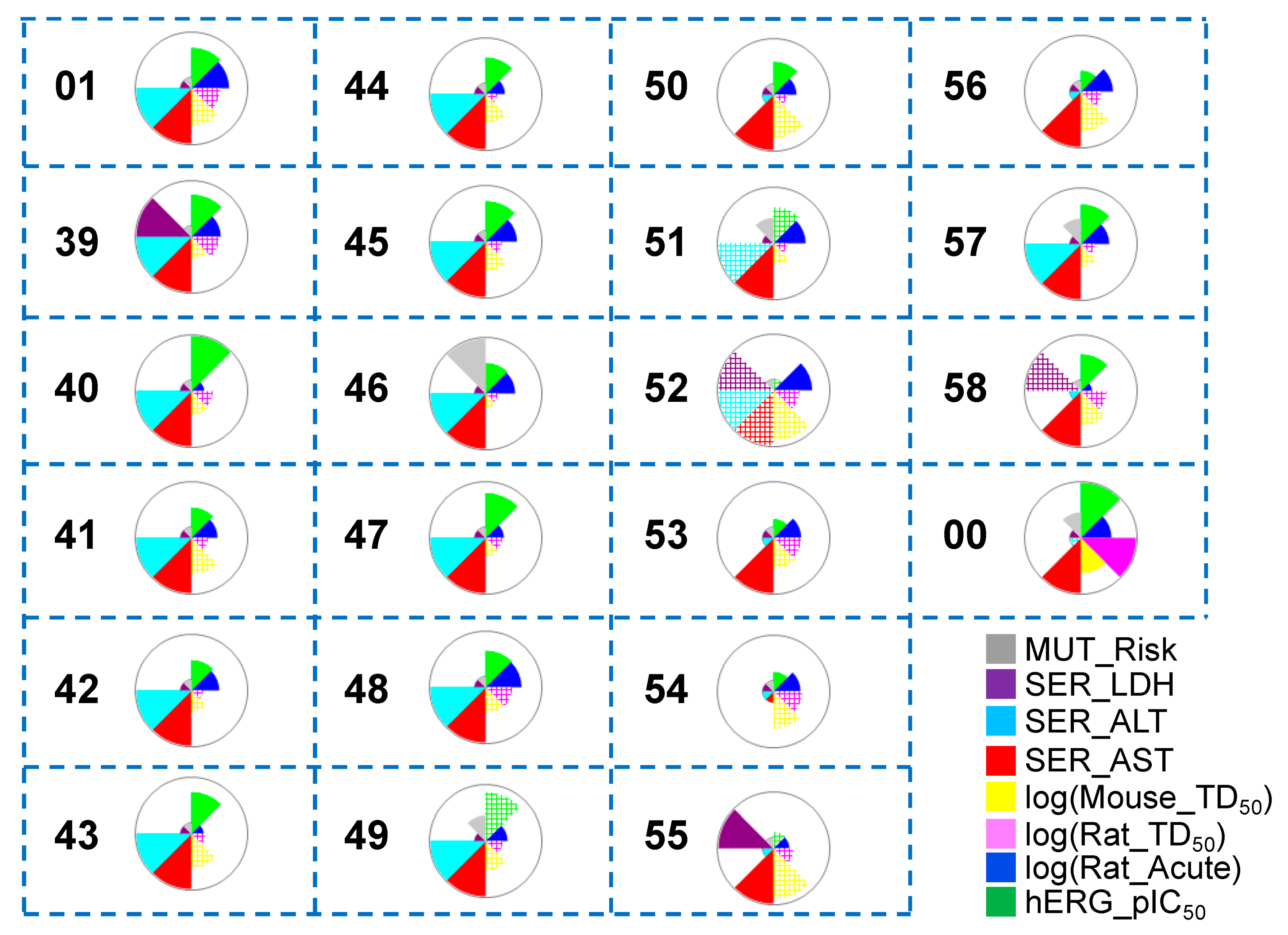
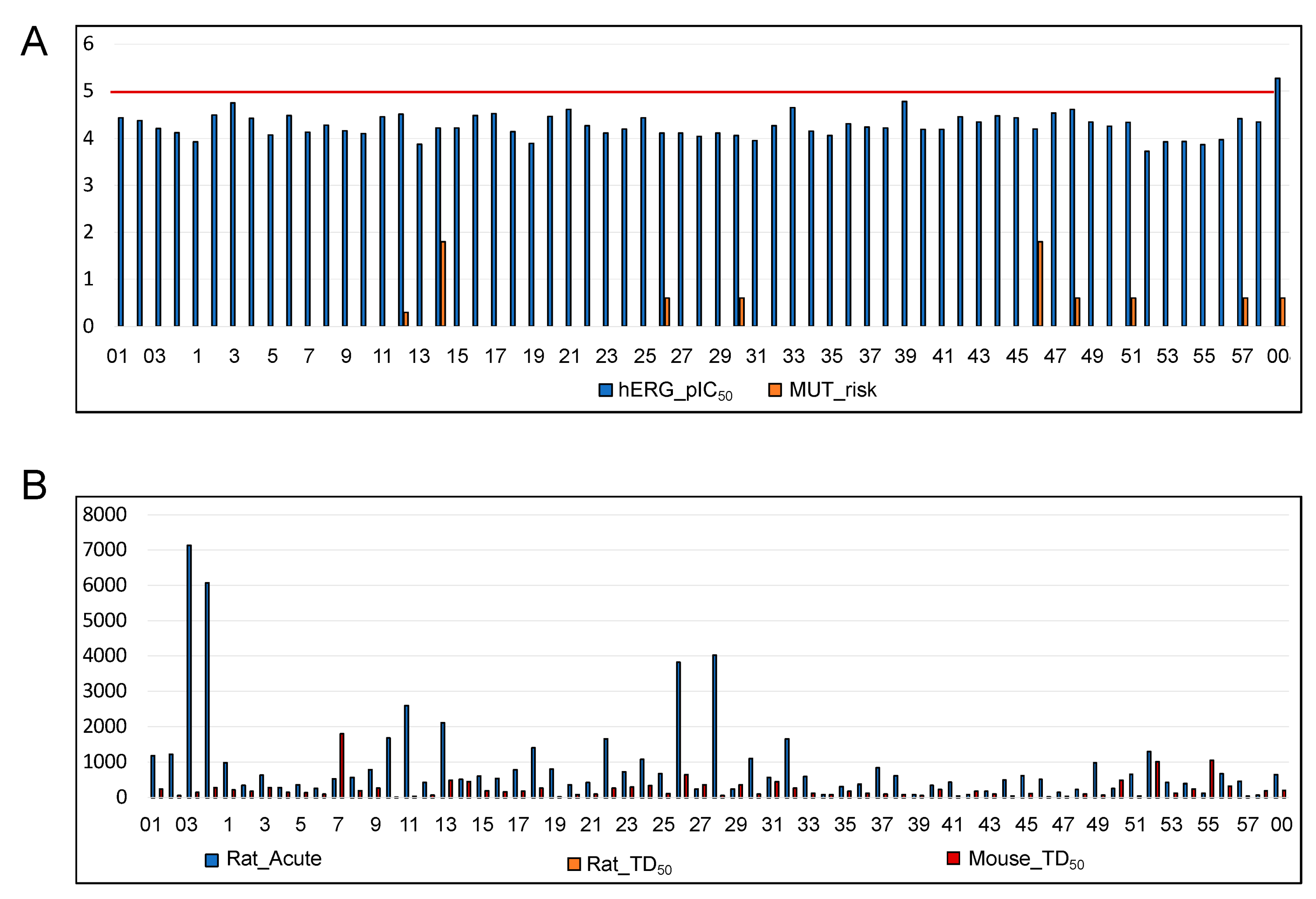
3.1.3. ADMET Predictions
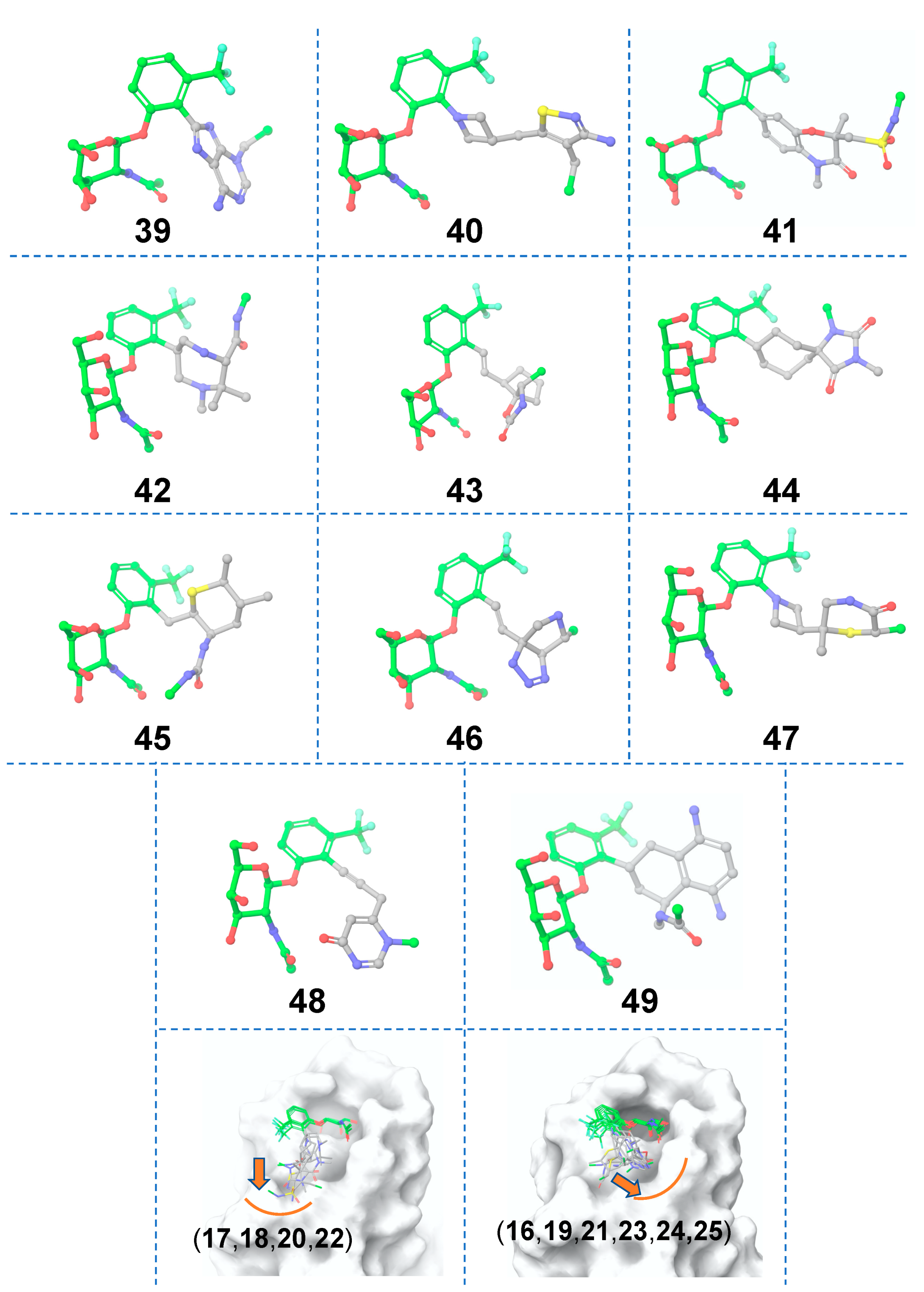
3.2. Scaffold 2
3.2.1. Ligand-Based Scaffold Hopping
3.2.2. Receptor-Based Scaffold Hopping
3.2.3. ADMET Predictions
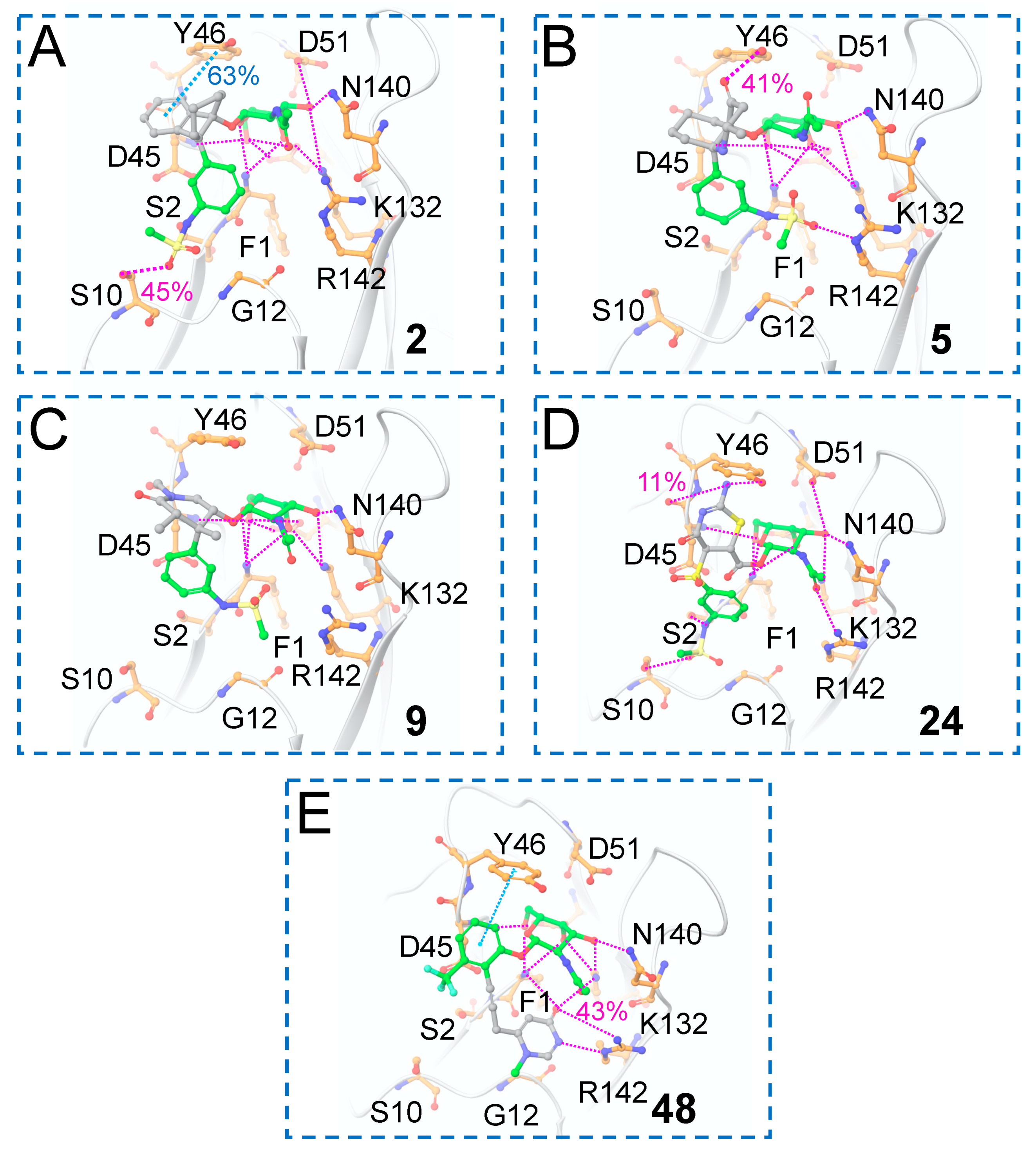
3.3. Molecular Dynamics Simulations
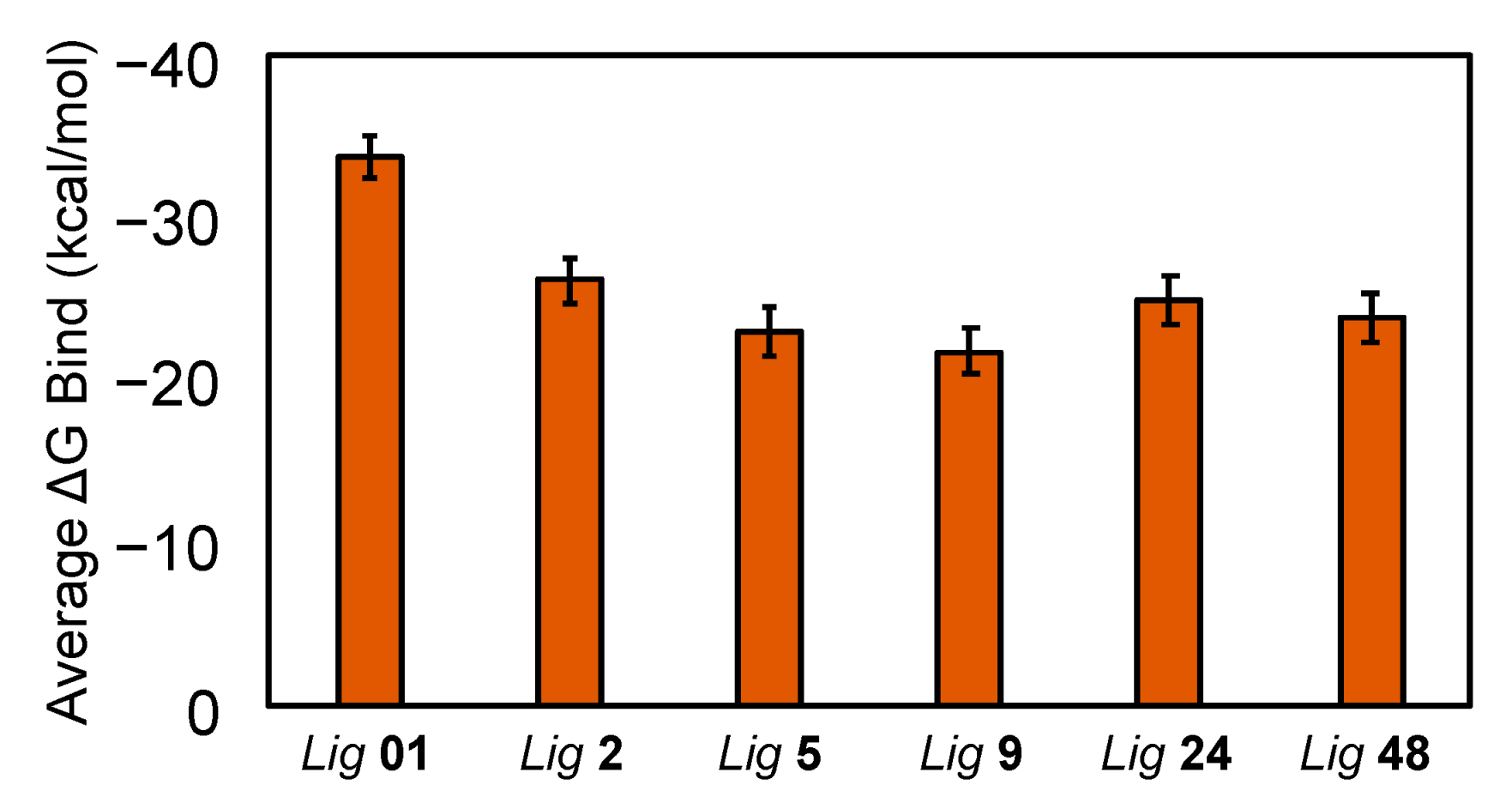
3.4. Binding Free Energy Calculations from the MD Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Sarshar, M.; Behzadi, P.; Ambrosi, C.; Zagaglia, C.; Palamara, A.T.; Scribano, D. FimH and Anti-Adhesive Therapeutics: A Disarming Strategy Against Uropathogens. Antibiotics 2020, 9, 397. [Google Scholar] [CrossRef]
- Yang, X.; Chen, H.; Zheng, Y.; Qu, S.; Wang, H.; Yi, F. Disease burden and long-term trends of urinary tract infections: A worldwide report. Front. Public Health 2022, 10, 888205. [Google Scholar] [CrossRef]
- Flores-Mireles, A.L.; Walker, J.N.; Caparon, M.; Hultgren, S.J. Urinary tract infections: Epidemiology, mechanisms of infection and treatment options. Nat. Rev. Microbiol. 2015, 13, 269–284. [Google Scholar] [CrossRef]
- Behzadi, P.; Behzadi, E.; Pawlak-Adamska, E.A. Urinary tract infections (UTIs) or genital tract infections (GTIs)? It’s the diagnostics that count. GMS Hyg. Infect. Control. 2019, 14, 1–12. [Google Scholar]
- Chockalingam, A.; Stewart, S.; Xu, L.; Gandhi, A.; Matta, M.K.; Patel, V.; Sacks, L.; Rouse, R. Evaluation of Immunocompetent Urinary Tract Infected Balb/C Mouse Model For the Study of Antibiotic Resistance Development Using Escherichia coli CFT073 infecyions. Antibiotics 2019, 8, 170. [Google Scholar] [CrossRef]
- Issakhanian, L.; Behzadi, P. Antimicrobial agents and urinary tract infections. Curr. Pharm. Des. 2019, 25, 1409–1423. [Google Scholar] [CrossRef]
- Behzadi, P. Classical chaperone-usher (CU) adhesive fimbriome: Uropathogenic Escherichia coli (UPEC) and urinary tract infections (UTIs). Folia Microbiol. 2020, 65, 45–65. [Google Scholar] [CrossRef]
- Hozzari, A.; Behzadi, P.; Kerishchi Khiabani, P.; Sholeh, M.; Sabokroo, N. Clinical cases, drug resistance, and virulence genes profiling in Uropathogenic Escherichia coli. J. Appl. Genet. 2020, 61, 265–273. [Google Scholar] [CrossRef]
- Kleeb, S.; Jiang, X.; Frei, P.; Sigl, A.; Bezençon, J.; Bamberger, K.; Schwardt, O.; Ernst, B. FimH Antagonists: Phosphate Prodrugs Improve Oral Bioavailability. J. Med. Chem. 2016, 59, 3163–3182. [Google Scholar] [CrossRef]
- Hooton, T.M.; Stamm, W.E. Diagnosis and treatment of uncomplicated urinary tract infection. Infect. Dis. Clin. 1997, 11, 551–581. [Google Scholar] [CrossRef]
- Svanborg, C.; Godaly, G. Bacterial virulence in urinary tract infection. Infect. Dis. Clin. North Am. 1997, 11, 513–529. [Google Scholar] [CrossRef]
- Zalewska-Piątek, B.M.; Piątek, R.J. Alternative treatment approaches of urinary tract infections caused by uropathogenic Escherichia coli strains. Acta Biochim. Pol. 2019, 66, 129–138. [Google Scholar] [CrossRef]
- Day, C.J.; Lo, A.W.; Hartley-Tassell, L.E.; Argente, M.P.; Poole, J.; King, N.P.; Tiralongo, J.; Jennings, M.P.; Schembri, M.A. Discovery of Bacterial Fimbria–Glycan Interactions Using Whole-Cell Recombinant Escherichia coli Expression. mBio 2021, 12, e03664-20. [Google Scholar] [CrossRef]
- Samanta, P.; Doerksen, R.J. Identifying FmlH lectin-binding small molecules for the prevention of Escherichia coli-induced urinary tract infections using hybrid fragment-based design and molecular docking. Comput Biol Med 2023, 163, 107072. [Google Scholar] [CrossRef]
- Mulvey, M.A.; Lopez-Boado, Y.S.; Wilson, C.L.; Roth, R.; Parks, W.C.; Heuser, J.; Hultgren, S.J. Induction and evasion of host defenses by type 1-piliated uropathogenic Escherichia coli. Science 1998, 282, 1494–1497. [Google Scholar] [CrossRef]
- Jones, C.H.; Pinkner, J.S.; Roth, R.; Heuser, J.; Nicholes, A.V.; Abraham, S.N.; Hultgren, S.J. FimH adhesin of type 1 pili is assembled into a fibrillar tip structure in the Enterobacteriaceae. Proc. Natl. Acad. Sci. USA 1995, 92, 2081–2085. [Google Scholar] [CrossRef]
- Kalas, V.; Hibbing, M.E.; Maddirala, A.R.; Chugani, R.; Pinkner, J.S.; Mydock-McGrane, L.K.; Conover, M.S.; Janetka, J.W.; Hultgren, S.J. Structure-based discovery of glycomimetic FmlH ligands as inhibitors of bacterial adhesion during urinary tract infection. Proc. Natl. Acad. Sci. USA 2018, 115, E2819–E2828. [Google Scholar] [CrossRef]
- Maddirala, A.R.; Klein, R.; Pinkner, J.S.; Kalas, V.; Hultgren, S.J.; Janetka, J.W. Biphenyl Gal and GalNAc FmlH Lectin Antagonists of Uropathogenic E. coli (UPEC): Optimization through Iterative Rational Drug Design. J Med Chem 2019, 62, 467–479. [Google Scholar] [CrossRef]
- Wurpel, D.J.; Totsika, M.; Allsopp, L.P.; Hartley-Tassell, L.E.; Day, C.J.; Peters, K.M.; Sarkar, S.; Ulett, G.C.; Yang, J.; Tiralongo, J.; et al. F9 fimbriae of uropathogenic Escherichia coli are expressed at low temperature and recognise Galβ1-3GlcNAc-containing glycans. PLoS ONE 2014, 9, e93177. [Google Scholar] [CrossRef]
- Conover, M.S.; Ruer, S.; Taganna, J.; Kalas, V.; De Greve, H.; Pinkner, J.S.; Dodson, K.W.; Remaut, H.; Hultgren, S.J. Inflammation-induced adhesin-receptor interaction provides a fitness advantage to uropathogenic E. coli during chronic infection. Cell Host Microbe 2016, 20, 482–492. [Google Scholar] [CrossRef]
- Mydock-McGrane, L.K.; Hannan, T.J.; Janetka, J.W. Rational design strategies for FimH antagonists: New drugs on the horizon for urinary tract infection and Crohn’s disease. Expert Opin. Drug Discov. 2017, 12, 711–731. [Google Scholar] [CrossRef]
- Jarvis, C.; Han, Z.; Kalas, V.; Klein, R.; Pinkner, J.S.; Ford, B.; Binkley, J.; Cusumano, C.K.; Cusumano, Z.; Mydock-McGrane, L.; et al. Antivirulence Isoquinolone Mannosides: Optimization of the Biaryl Aglycone for FimH Lectin Binding Affinity and Efficacy in the Treatment of Chronic UTI. ChemMedChem 2016, 11, 367–373. [Google Scholar] [CrossRef]
- Mydock-McGrane, L.; Cusumano, Z.; Han, Z.; Binkley, J.; Kostakioti, M.; Hannan, T.; Pinkner, J.S.; Klein, R.; Kalas, V.; Crowley, J.; et al. Antivirulence C-Mannosides as Antibiotic-Sparing, Oral Therapeutics for Urinary Tract Infections. J. Med. Chem. 2016, 59, 9390–9408. [Google Scholar] [CrossRef]
- Han, Z.; Pinkner, J.S.; Ford, B.; Obermann, R.; Nolan, W.; Wildman, S.A.; Hobbs, D.; Ellenberger, T.; Cusumano, C.K.; Hultgren, S.J.; et al. Structure-based drug design and optimization of mannoside bacterial FimH antagonists. J. Med. Chem. 2010, 53, 4779–4792. [Google Scholar] [CrossRef]
- Cusumano, C.K.; Pinkner, J.S.; Han, Z.; Greene, S.E.; Ford, B.A.; Crowley, J.R.; Henderson, J.P.; Janetka, J.W.; Hultgren, S.J. Treatment and prevention of urinary tract infection with orally active FimH inhibitors. Sci. Transl. Med. 2011, 3, 109ra115. [Google Scholar] [CrossRef]
- Grabosch, C.; Hartmann, M.; Schmidt-Lassen, J.; Lindhorst, T.K. Squaric acid monoamide mannosides as ligands for the bacterial lectin FimH: Covalent inhibition or not? ChemBioChem 2011, 12, 1066–1074. [Google Scholar] [CrossRef]
- Kleeb, S.; Pang, L.; Mayer, K.; Eris, D.; Sigl, A.; Preston, R.C.; Zihlmann, P.; Sharpe, T.; Jakob, R.P.; Abgottspon, D.; et al. FimH antagonists: Bioisosteres to improve the in vitro and in vivo PK/PD profile. J. Med. Chem. 2015, 58, 2221–2239. [Google Scholar] [CrossRef]
- Chalopin, T.; Alvarez Dorta, D.; Sivignon, A.; Caudan, M.; Dumych, T.I.; Bilyy, R.O.; Deniaud, D.; Barnich, N.; Bouckaert, J.; Gouin, S.G. Second generation of thiazolylmannosides, FimH antagonists for E. coli-induced Crohn’s disease. Org. Biomol. Chem. 2016, 14, 3913–3925. [Google Scholar] [CrossRef]
- Mydock-McGrane, L.K.; Cusumano, Z.T.; Janetka, J.W. Mannose-derived FimH antagonists: A promising anti-virulence therapeutic strategy for urinary tract infections and Crohn’s disease. Expert Opin. Ther. Pat. 2016, 26, 175–197. [Google Scholar] [CrossRef]
- Ofek, I.; Hasty, D.L.; Sharon, N. Anti-adhesion therapy of bacterial diseases: Prospects and problems. FEMS Immunol. Med. Microbiol. 2003, 38, 181–191. [Google Scholar] [CrossRef]
- Hu, Y.; Stumpfe, D.; Bajorath, J. Recent Advances in Scaffold Hopping. J. Med. Chem. 2017, 60, 1238–1246. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, S.Q.; Xu, W.R.; Wang, R.L.; Chou, K.C. Design novel dual agonists for treating type-2 diabetes by targeting peroxisome proliferator-activated receptors with core hopping approach. PLoS ONE 2012, 7, e38546. [Google Scholar] [CrossRef]
- Schrödinger Release 2021-2: Core Hopping; Schrödinger, LLC: New York, NY, USA, 2021.
- Pasala, C.; Katari, S.K.; Nalamolu, R.M.; Aparna, R.B.; Amineni, U. Integration of core hopping, quantum-mechanics, molecular mechanics coupled binding-energy estimations and dynamic simulations for fragment-based novel therapeutic scaffolds against Helicobacter pylori strains. Comput. Biol. Chem. 2019, 83, 107126. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Schrödinger Release 2021-2: Glide; Schrödinger, LLC: New York, NY, USA, 2021.
- Samanta, P.; Mishra, S.K.; Pomin, V.H.; Doerksen, R.J. Docking and Molecular Dynamics Simulations Clarify Binding Sites for Interactions of Novel Marine Sulfated Glycans with SARS-CoV-2 Spike Glycoprotein. Molecules 2023, 28, 6413. [Google Scholar] [CrossRef]
- Maurya, A.K.; Sharma, P.; Samanta, P.; Shami, A.A.; Misra, S.K.; Zhang, F.; Thara, R.; Kumar, D.; Shi, D.; Linhardt, R.J.; et al. Structure, anti-SARS-CoV-2, and anticoagulant effects of two sulfated galactans from the red alga Botryocladia occidentalis. Int. J. Biol. Macromol. 2023, 238, 124168. [Google Scholar] [CrossRef]
- Samanta, P.; Doerksen, R.J. Identifying p56lck SH2 Domain Inhibitors Using Molecular Docking and In Silico Scaffold Hopping. Appl. Sci. 2024, 14, 4277. [Google Scholar] [CrossRef]
- Simulations Plus, Inc. Available online: http://www.simulations-plus.com (accessed on 3 October 2024).
- Vlachakis, D.; Bencurova, E.; Papangelopoulos, N.; Kossida, S. Chapter Seven–Current State-of-the-Art Molecular Dynamics Methods and Applications. Adv. Protein Chem. Struct. Biol. 2014, 94, 269–313. [Google Scholar]
- Tomasulo, P. ChemIDplus-super source for chemical and drug information. Med. Ref. Serv. Q 2002, 21, 53–59. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the SC’06: Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 84-es. [Google Scholar]
- Schrödinger Release 2021-2: Desmond Molecular Dynamics System; D. E. Shaw Research: New York, NY, USA, 2021; Maestro-Desmond Interoperability Tools; Schrödinger: New York, NY, USA, 2021. Cf. Available online: https://www.schrodinger.com/citations/ (accessed on 9 October 2024).
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins 2011, 79, 2794–2812. [Google Scholar] [CrossRef]
- Ludwig, S.G.; Kiyohara, C.L.; Carlucci, L.A.; Kisiela, D.; Sokurenko, E.V.; Thomas, W.E. FimH as a scaffold for regulated molecular recognition. J Biol Eng 2021, 15, 3. [Google Scholar] [CrossRef]
- Nilsson, L.M.; Thomas, W.E.; Sokurenko, E.V.; Vogel, V. Beyond induced-fit receptor-ligand interactions: Structural changes that can significantly extend bond lifetimes. Structure 2008, 16, 1047–1058. [Google Scholar] [CrossRef]
- Nilsson, L.M.; Thomas, W.E.; Trintchina, E.; Vogel, V.; Sokurenko, E.V. Catch bond-mediated adhesion without a shear threshold: Trimannose versus monomannose interactions with the FimH adhesin of Escherichia coli. J. Biol. Chem. 2006, 281, 16656–16663. [Google Scholar] [CrossRef]
- Schönemann, W.; Cramer, J.; Mühlethaler, T.; Fiege, B.; Silbermann, M.; Rabbani, S.; Dätwyler, P.; Zihlmann, P.; Jakob, R.P.; Sager, C.P.; et al. Improvement of Aglycone π-Stacking Yields Nanomolar to Sub-nanomolar FimH Antagonists. ChemMedChem 2019, 14, 749–757. [Google Scholar] [CrossRef]
- Roos, G.; Wellens, A.; Touaibia, M.; Yamakawa, N.; Geerlings, P.; Roy, R.; Wyns, L.; Bouckaert, J. Validation of Reactivity Descriptors to Assess the Aromatic Stacking within the Tyrosine Gate of FimH. ACS Med. Chem. Lett. 2013, 4, 1085–1090. [Google Scholar] [CrossRef]
- Fiege, B.; Rabbani, S.; Preston, R.C.; Jakob, R.P.; Zihlmann, P.; Schwardt, O.; Jiang, X.; Maier, T.; Ernst, B. The tyrosine gate of the bacterial lectin FimH: A conformational analysis by NMR spectroscopy and X-ray crystallography. ChemBioChem 2015, 16, 1235–1246. [Google Scholar] [CrossRef]
- Vanwetswinkel, S.; Volkov, A.N.; Sterckx, Y.G.; Garcia-Pino, A.; Buts, L.; Vranken, W.F.; Bouckaert, J.; Roy, R.; Wyns, L.; van Nuland, N.A. Study of the structural and dynamic effects in the FimH adhesin upon α-d-heptyl mannose binding. J. Med. Chem. 2014, 57, 1416–1427. [Google Scholar] [CrossRef]
- Singh, K.D.; Kirubakaran, P.; Nagarajan, S.; Sakkiah, S.; Muthusamy, K.; Velmurgan, D.; Jeyakanthan, J. Homology modeling, molecular dynamics, e-pharmacophore mapping and docking study of Chikungunya virus nsP2 protease. J. Mol. Model. 2012, 18, 39–51. [Google Scholar] [CrossRef]
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef]
- Halgren, T. New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des. 2007, 69, 146–148. [Google Scholar] [CrossRef]
- Schrödinger Release 2021-2: SiteMap; Schrödinger, LLC: New York, NY, USA, 2021.
- Alonso-Caballero, A.; Schönfelder, J.; Poly, S.; Corsetti, F.; De Sancho, D.; Artacho, E.; Perez-Jimenez, R. Mechanical architecture and folding of E. coli type 1 pilus domains. Nat. Commun. 2018, 9, 2758. [Google Scholar] [CrossRef]
- Sivignon, A.; Bouckaert, J.; Bernard, J.; Gouin, S.G.; Barnich, N. The potential of FimH as a novel therapeutic target for the treatment of Crohn’s disease. Expert Opin. Ther. Targets 2017, 21, 837–847. [Google Scholar] [CrossRef]
- Segall, M.D. Multi-parameter optimization: Identifying high quality compounds with a balance of properties. Curr Pharm Des 2012, 18, 1292–1310. [Google Scholar] [CrossRef]
- Schwardt, O.; Rabbani, S.; Hartmann, M.; Abgottspon, D.; Wittwer, M.; Kleeb, S.; Zalewski, A.; Smieško, M.; Cutting, B.; Ernst, B. Design, synthesis and biological evaluation of mannosyl triazoles as FimH antagonists. Bioorg. Med. Chem. 2011, 19, 6454–6473. [Google Scholar] [CrossRef]
- Klein, T.; Abgottspon, D.; Wittwer, M.; Rabbani, S.; Herold, J.; Jiang, X.; Kleeb, S.; Lüthi, C.; Scharenberg, M.; Bezençon, J.; et al. FimH antagonists for the oral treatment of urinary tract infections: From design and synthesis to in vitro and in vivo evaluation. J. Med. Chem. 2010, 53, 8627–8641. [Google Scholar] [CrossRef]
- Renner, S.; Schneider, G. Scaffold-hopping potential of ligand-based similarity concepts. ChemMedChem 2006, 1, 181–185. [Google Scholar] [CrossRef]
- Franke, L.; Schwarz, O.; Müller-Kuhrt, L.; Hoernig, C.; Fischer, L.; George, S.; Tanrikulu, Y.; Schneider, P.; Werz, O.; Steinhilber, D.; et al. Identification of natural-product-derived inhibitors of 5-lipoxygenase activity by ligand-based virtual screening. J. Med. Chem. 2007, 50, 2640–2646. [Google Scholar] [CrossRef]
- Maeda, I.; Tamura, S.; Ogura, Y.; Serizawa, T.; Shimada, T.; Kunimoto, R.; Miyao, T. Scaffold-Hopped Compound Identification by Ligand-Based Approaches with a Prospective Affinity Test. J. Chem. Inf. Model. 2024, 64, 5557–5569. [Google Scholar] [CrossRef]
- Aparoy, P.; Reddy, K.K.; Reddanna, P. Structure and ligand based drug design strategies in the development of novel 5- LOX inhibitors. Curr. Med. Chem. 2012, 19, 3763–3778. [Google Scholar] [CrossRef]
- Fontaine, F.; Cross, S.; Plasencia, G.; Pastor, M.; Zamora, I. SHOP: A method for structure-based fragment and scaffold hopping. ChemMedChem 2009, 4, 427–439. [Google Scholar] [CrossRef]
- Johansson, E.; Caraballo, R.; Hurdiss, D.L.; Mistry, N.; Andersson, C.D.; Thompson, R.F.; Ranson, N.A.; Zocher, G.; Stehle, T.; Arnberg, N.; et al. Exploring the Effect of Structure-Based Scaffold Hopping on the Inhibition of Coxsackievirus A24v Transduction by Pentavalent N-Acetylneuraminic Acid Conjugates. Int. J. Mol. Sci. 2021, 22, 8418. [Google Scholar] [CrossRef]
- Bajorath, J. Computational Scaffold Hopping: Cornerstone for the Future of Drug Design? Future Med. Chem. 2017, 629–631. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Glide Score | Synth Score |
|---|---|---|
| 01 * | −5.71 | n/a |
| 02 * | −6.16 | n/a |
| 03 * | −6.31 | n/a |
| 04 * | −6.42 | n/a |
| 1 | −6.34 | 1.000 |
| 2 | −6.33 | 0.250 |
| 3 | −6.32 | 0.250 |
| 4 | −6.27 | 1.500 |
| 5 | −6.26 | 0.250 |
| 6 | −6.16 | 0.250 |
| 7 | −6.10 | 2.000 |
| 8 | −6.10 | 2.188 |
| 9 | −6.08 | 0.250 |
| 10 | n/a | 1.500 |
| 11 | n/a | 0.496 |
| 12 | n/a | 1.000 |
| 13 | n/a | 0.500 |
| 14 | n/a | 0.500 |
| 15 | n/a | 0.500 |
| 16 | n/a | 0.500 |
| 17 | n/a | 0.500 |
| 18 | n/a | 0.498 |
| 19 | n/a | 0.492 |
| 20 | n/a | 0.492 |
| 21 | n/a | 0.469 |
| 22 | n/a | 0.469 |
| 23 | n/a | 0.438 |
| 24 | n/a | 0.598 |
| 25 | n/a | 0.375 |
| 26 | n/a | 0.375 |
| 27 | n/a | 0.250 |
| 28 | n/a | 0.250 |
| 29 | n/a | 0.250 |
| 30 | n/a | 0.250 |
| 31 | n/a | 0.250 |
| 32 | n/a | 0.250 |
| 33 | n/a | 0.250 |
| 34 | n/a | 0.250 |
| 35 | n/a | 0.250 |
| 36 | n/a | 0.250 |
| 37 | n/a | 0.250 |
| 38 | n/a | 0.250 |
| Ligands | Glide Score | Synth Score |
|---|---|---|
| 01 * | −5.710 | n/a |
| 39 | −6.506 | 1.498 |
| 40 | −6.465 | 1.000 |
| 41 | −6.402 | 2.875 |
| 42 | −6.216 | 0.500 |
| 43 | −6.168 | 0.250 |
| 44 | −6.118 | 0.500 |
| 45 | −6.093 | 0.250 |
| 46 | −6.093 | 1.500 |
| 47 | −6.076 | 2.250 |
| 48 | −6.045 | 2.500 |
| 49 | −6.023 | 1.000 |
| 50 | n/a | 2.961 |
| 51 | n/a | 2.000 |
| 52 | n/a | 2.000 |
| 53 | n/a | 2.000 |
| 54 | n/a | 2.000 |
| 55 | n/a | 0.500 |
| 56 | n/a | 0.498 |
| 57 | n/a | 0.375 |
| 58 | n/a | 0.375 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samanta, P.; Doerksen, R.J. Applying Molecular Modeling to Predict Novel FmlH-Binding Glycomimetics with Improved Pharmacokinetic Properties for the Prevention of Urinary Tract Infections. Appl. Sci. 2024, 14, 9496. https://doi.org/10.3390/app14209496
Samanta P, Doerksen RJ. Applying Molecular Modeling to Predict Novel FmlH-Binding Glycomimetics with Improved Pharmacokinetic Properties for the Prevention of Urinary Tract Infections. Applied Sciences. 2024; 14(20):9496. https://doi.org/10.3390/app14209496
Chicago/Turabian StyleSamanta, Priyanka, and Robert J. Doerksen. 2024. "Applying Molecular Modeling to Predict Novel FmlH-Binding Glycomimetics with Improved Pharmacokinetic Properties for the Prevention of Urinary Tract Infections" Applied Sciences 14, no. 20: 9496. https://doi.org/10.3390/app14209496
APA StyleSamanta, P., & Doerksen, R. J. (2024). Applying Molecular Modeling to Predict Novel FmlH-Binding Glycomimetics with Improved Pharmacokinetic Properties for the Prevention of Urinary Tract Infections. Applied Sciences, 14(20), 9496. https://doi.org/10.3390/app14209496