Abstract
The emergence of drug resistance among pathogens has become a major challenge to human health on a global scale. Among them, antibiotic resistance is already a critical issue, and finding new therapeutic agents to address this problem is therefore urgent. One of the most promising alternatives to antibiotics are antibacterial peptides (ABPs), i.e., short peptides with antibacterial activity. In this study, we propose a novel ABP recognition method, called ABPCaps. It integrates a convolutional neural network (CNN), a long short-term memory (LSTM), and a new type of neural network named the capsule network. The capsule network can extract critical features automatically from both positive and negative samples, leading to superior performance of ABPCaps over all baseline models built on hand-crafted peptide descriptors. Evaluated on independent test sets, ABPCaps achieves an accuracy of 93.33% and an F1-score of 91.34%, and consistently outperforms the baseline models in other extensive experiments as well. Our study demonstrates that the proposed ABPCaps, built on the capsule network method, is a valuable addition to the current state-of-the-art in the field of ABP recognition and has significant potential for further development.
1. Introduction
Antibiotic resistance has become one of the most urgent health concerns worldwide. Despite significant progress in antibiotic research since the discovery of the first antibiotic in 1928, the emergence of drug-resistant bacteria continues to be a challenge for infectious disease researchers [1]. The alarming increase in antibiotic resistance has led to the rise of superbugs, such as methicillin-resistant Staphylococcus aureus (MRSA), which poses a severe threat in both hospital and community settings [2]. Even the most commonly used antibiotics, such as amoxicillin and cefuroxime, have demonstrated high rates of resistance in strains of Escherichia coli [2]. The misuse of antibiotics has contributed significantly to the rise of drug-resistant bacteria, highlighting the urgent need for alternative treatments. Antimicrobial peptides (AMPs) have emerged as promising alternatives to antibiotics, potentially solving the antibiotic resistance problem [3,4]. Among them, antibacterial peptides (ABPs) have been the focus of considerable attention due to their ability to inhibit various types of bacterial pathogens and treat infections. Their broad-spectrum activity and low propensity to induce resistance make them an attractive alternative to conventional antibiotics. With the growing challenge of antibiotic resistance, the potential of AMPs and ABPs to develop new treatments and therapies cannot be overstated.
Over the past few years, the development and identification of AMPs has received considerable attention from researchers. Several databases and identification tools have been developed to facilitate this research. ANTIMIC, which integrates a number of tools and contains 1700 peptides, is one of the earliest and most prominent AMP databases [5]. Another prominent AMP database is LAMP2, which contains over 23,000 peptide sequences classified according to their source: experimental, predicted or patent [6]. The latest version of DRAMP, on the other hand, contains over 22,000 entries, including 181 stapled AMPs [7]. Finally, the latest version of dbAMP contains over 26,000 AMPs and antimicrobial proteins from more than 3000 organisms [8]. Several machine learning-based methods have been developed for identification of AMPs, as assessing the activity of AMPs is crucial for determining their potential as drugs [9,10,11,12,13,14,15,16]. Table 1 summarizes existing AMP prediction tools, with random forests (RF) and support vector machines (SVM) showing particular success [9]. In addition, Xiao et al. developed iAMP-2L, a two-stage multi-label classifier that can predict whether a given peptide is an AMP and identify its functional target [10]. More recently, deep learning-based models have been used to identify AMPs, with the development of the Deep-AmPEP30 method based on a convolutional neural network (CNN) [11]. To build a predictive model for AMP identification, Veltri et al. integrated CNN and long short-term memory (LSTM) layers [12,17]. Additionally, a bidirectional LSTM (biLSTM) was used for this task [13].

Table 1.
Related works on predicting AMPs. This table provides a comparison of different methods used for AMP prediction, including their model architectures, features used, and evaluation metrics.
Although CNNs have proven to be a powerful tool for a wide range of machine learning tasks, they are not without their limitations. One such limitation is the problem of invariance caused by pooling, which can lead to the loss of important spatial relationships between features [18,19]. In addition, CNNs are often unable to understand the relationships between different features. To address these limitations, researchers have developed a new type of neural network called a capsule network [20]. Capsule networks differ from CNNs in several ways. First, the inputs and outputs of a capsule network are represented in vector form, where each element represents a particular feature of the entity. The length of the vector indicates the probability of that feature being present [21]. Secondly, the use of dynamic routing algorithms between different capsules allows the network to have a better understanding of the relationships between different features. These differences have enabled capsule networks to outperform CNNs in a wide range of tasks in computer vision [22,23,24] and natural language processing [25,26]. In recent years, capsule networks have also been applied to bioinformatics with great success [27,28,29,30]. For example, researchers have used capsule networks to predict protein post-translational modification (PTM) sites [27], to detect protein lysine crotonylation sites [29], and to predict drug-drug interactions [30]. In each of these cases, capsule networks were able to effectively compensate for the shortcomings of CNNs and improve on previous methods by using high-throughput data.
With the unprecedented growth of data generated by high-throughput sequencing technologies, the field of bioinformatics has become flooded with massive amounts of data, waiting to be analyzed to reveal the underlying principles [31,32,33,34,35,36,37]. Traditional approaches of sequence analysis have limitations that hinder their ability to accurately predict ABPs, which are essential for the discovery of novel therapies for disease. To address this critical challenge, this study develops a capsule network-based model, ABPCaps, to identify ABPs. By taking advantage of the unique architecture of capsule networks, the ABPCaps model overcomes the limitations of traditional neural networks and shows superior performance when compared to conventional baseline models. First, is the hierarchical representation. Capsule networks capture the hierarchical relationship between features. Unlike traditional neural networks, which use scalar output units, capsules are vector-valued units that encode properties of entities along with their instantiation parameters. This hierarchical representation allows capsules to capture more nuanced information. Second, is the dynamic routing mechanism. Dynamic routing mechanism can be viewed as a parallel attention mechanism. Capsule networks use a dynamic routing algorithm to determine the coupling coefficients between capsules in adjacent layers. This routing mechanism enables the network to learn to assign appropriate weights to different capsules based on their agreement, improving the network’s ability to handle complex relationships and variations in data, ultimately achieving better classification performance. As such, the ABPCaps model has the potential to revolutionize the field of bioinformatics and contribute to significant advances in the understanding of biological processes and the development of novel therapies.
2. Materials and Methods
2.1. Dataset Preparation
ABPs were retrieved from GRAMPA, a comprehensive public database containing a large collection of ABPs against different bacteria and their activity levels [38]. After retrieving the ABP sequences from GRAMPA, we performed a pre-processing step to eliminate redundant records and to retain only those peptides with a length between 10 and 150 amino acids. The resulting ABPs dataset, which was used as positive samples for the study, contained 6017 non-replicated peptides targeting different bacteria. For accurate identification of ABPs in a wide range of sequences, we also collected peptides with antimicrobial functions but without antibacterial activity (non-ABPs) as well as normal peptides without antimicrobial activity (non-AMPs) as negative samples. The non-ABP data sets were obtained from various general AMP databases such as dbAMP [8], DRAMP [7], DBAASP [39], AVPdb [40], AntiCP 2.0 [41], and AntiFP [42]. To build the negative dataset, the researchers excluded entries with UniProt keywords such as “antimicrobial”, “antibiotic”, “antiviral”, “antifungal”, “secreted”, “excreted”, or “effector”. The peptides were then selected at random to create a negative set of samples with the same number of sequences as the positive set. The length distribution of the non-AMPs was matched to that of the ABPs to ensure unbiased results. We ended up using 80% of the positive and negative datasets to generate training sets, while the remaining 20% was used to generate independent test sets.
2.2. Development of ABPCaps
In various fields such as natural language processing, computer vision, and bioinformatics, deep learning algorithms such as CNNs and recurrent neural networks (RNNs) have become ubiquitous. Although CNNs have been successful in many tasks, they are still limited by pooling invariance and the inability to understand spatial relationships between features. Capsule networks, which have shown excellent performance on many datasets [43,44,45], have been developed to overcome these limitations. In this study, we attempted to develop a novel model that integrates 1D convolution (Conv1D), LSTM, and capsule layers to improve the feature extraction process. Conv1D was used to extract local features, which were then passed to the LSTM layer responsible for capturing long-range dependencies. The high-level features extracted by the LSTM layer were then passed to the capsule layer. An overview of the proposed model is shown in Figure 1.
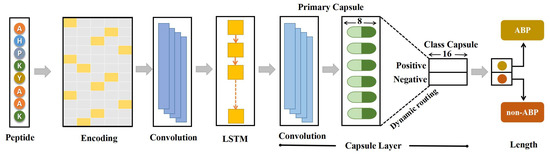
Figure 1.
The architecture of ABPCaps. The proposed model, ABPCaps, is a fusion of Conv1D, LSTM, and a capsule network. The input sequences are encoded using the one-hot encoding method, and the Conv1D and LSTM layers are used to extract crucial features. The hybrid features are passed to the capsule layer, which encapsulates the relationships between different features and provides interpretable results for downstream tasks.
A one-dimensional protein sequence with 20 types of amino acids was first converted into two-dimensional data using the popular binary one-hot encoding method to input peptides into the network. The transformation converted the input protein sequence into a 20 matrix, where k is the length of the input sequence and each row represents an amino acid. Due to the variable length of peptides, this matrix was converted to a zero-padded numerical matrix of size 150 × 20 to fit the data set. The protein sequence was ready for input to the network after encoding. The first layer of the network was Conv1D, which had 64 filters, and the step was set to 1. The activation function of the rectified linear units (ReLU) was chosen for this layer. The local features extracted by Conv1D were passed to the RNN layer, which can do long-range dependency capture. However, RNNs suffered from slope disappearance, which would affect performance. Therefore, in this study, we used an LSTM layer, which could avoid the gradient vanishing problem and had shown superior performance compared to other RNN variants such as gated recurrent units (GRU) [46]. Each LSTM unit consists of a forget gate, an input gate, and an output gate, which were calculated using the following equations:
, , , denote the forget gate, input gate, and output gate, respectively. The forget gate and input gate are responsible for updating the cell state. In this study, using GRU instead of LSTM was tried, but the performance of GRU was not satisfactory.
A capsule, consisting of a set of neurons in the form of a vector, is a fundamental component of capsule networks. In contrast to traditional neural networks, capsule networks use vectors as both the input and the output units. Each capsule represents a pattern, with each dimension representing unique characteristics. In this study, the primary capsule layer used a conv1D for further extraction of relevant features, with the resultant outputs being transformed into multiple 8-dimensional vectors. According to the concept of capsule networks, the length of a vector refers to the probability of the represented pattern. A non-linear function called squash is applied to these 8-dimensional vectors and is given below.
This function compresses the length of the vector to a value between 0 and 1, while maintaining the direction of the vector. If the magnitude of the input vector is relatively large, the norm of the output vector will approach unity; conversely, if the magnitude of the input vector is very small, the norm of the output vector will approach zero. The primary capsule layer outputs are the compressed scaled vectors.
In a binary classification task, the class capsule layer consists of two capsules-a positive capsule and a negative capsule-each represented by a 16-dimension vector. Each capsule represents a different class of input sequence. The computational process between the primary capsule layer and the class capsule layer is illustrated in Supplementary Figure S1. To obtain the prediction vectors, the outputs of the primary capsule layer, , are first multiplied by the corresponding weight matrix, , given below, is the weighted sum of all computed using coupling coefficients, , determined by the dynamic routing process.
These coefficients describe the probabilities of primary capsules to be coupled to the class capsules activated by specific input sequences. L is the total number of primary capsules.
The process of dynamic routing, as shown in Supplementary Materials—Algorithm S1, is a key feature of capsule networks. The log prior probability between primary capsule i and class capsule j, denoted by , was computed to determine the coupling coefficients . These coefficients were computed using a softmax function on that ensures that the sum of the coupling coefficients of class capsule i to all primary capsules is equal to one. The number of iterations r is a hyperparameter that must be set prior to model training. Two output vectors , each representing one of the two classification categories, ABP or non-ABP, were generated by the class capsule layer during dynamic routing. The elements of encode the features, and their length, determined by the L2 norm , denotes the probability distribution () of the two classes. In the last step of the network, the length of these output vectors was calculated and shown below.
To train the model, this study used the margin loss introduced by Sabour et al. [21], which considered the classification category and an indicator function.
c and I represent the classification category and the indicator function, respectively. Specifically, = 1 if the current sample belongs to class c; otherwise, = 0. The , and are hyperparameters in this function, with suggested values of 0.9, 0.1, and 0.5, respectively.
The Keras framework (https://keras.io/ (accessed on 27 March 2015)) and the TensorFlow [47] library backend were used to build the model in this study. To avoid overfitting, an early termination strategy was implemented during training. This strategy was the termination of the training process if there was no improvement in accuracy within several epochs. The Adam [48] optimizer was used with learning rate and batch size set to 0.001 and 128, respectively. This study applied the training process with 4×Nvidia 2080 Ti graphics processing units (GPUs), which significantly reduced the computational time.
2.3. Evaluation Metrics
Evaluating different models is critical to determining their effectiveness in solving the problem presented. In this study, a number of widely accepted evaluation metrics have been used to evaluate the performance of the proposed model. These included accuracy, precision, recall, F1-score, and the area under the receiver operating characteristic (AUROC), the latter defined as the area under the receiver operating characteristic (ROC) curve. Other metrics are given as follows:
TP, TN, FP, FN denote the number of true positives, true negatives, false positives, and false negatives.
3. Results and Discussion
3.1. Comparisons of Baseline Models
A detailed analysis of the results was performed and compared with baseline models and existing identification tools. Selecting appropriate baselines is critical to evaluating the effectiveness of proposed models. In this study, machine learning-based models with sequence features were carefully selected as baselines. This was done to ensure a comprehensive comparison. These baselines include widely used features such as amino acid composition (AAC), dipeptide composition (DPC), composition of k-spaced amino acid pairs (CKSAAGP), pseudo amino acid composition (PAAC), and eight different physicochemical features (PHYC). The baseline models were developed using different machine learning algorithms, k-nearest neighbor (KNN) and RF. In addition, to capture more complex sequence patterns, CNN-based and RNN-based models, such as LSTM, GRU, bidirectional LSTM, and bidirectional GRU (biGRU), were also applied as baseline classifiers. The architecture of these baseline models is shown in Supplementary Figure S2, providing a clear overview of their design and complexity. The performance of the models was evaluated using an independent test set and the results are presented in Table 2. In terms of accuracy, F1-score, AUROC and recall, the proposed model outperformed all the baseline models. Specifically, the proposed model achieved an impressive AUROC of 0.9289, an F1-score of 0.9134, a recall of 90.95%, and an accuracy of 93.33%. In comparison to other baseline models, these results demonstrate the superior predictive power of the proposed capsule network for the identification of ABPs.
Table 2.
Performance comparison between ABPCaps and baselines on the independent test set This table provides a comprehensive evaluation of the proposed model and the baselines, including their accuracy, F1-score, and AUROC on an independent test set. The best performance in each category is highlighted in bold.
To further visualize the effectiveness of the proposed model, ROC curves were plotted to compare the performance of the proposed model with the baseline models, and the curves are shown in Figure 2. The ROC curve of the proposed model was at the outermost point of all the curves, wrapping around the ROC curves of the baseline models. This suggests that the proposed model has excellent discriminative power to distinguish between ABPs and non-ABPs. In addition, the AUROC value of the proposed model for the independent test set was higher than that of the baseline models. This further supports the superior performance of the proposed model.
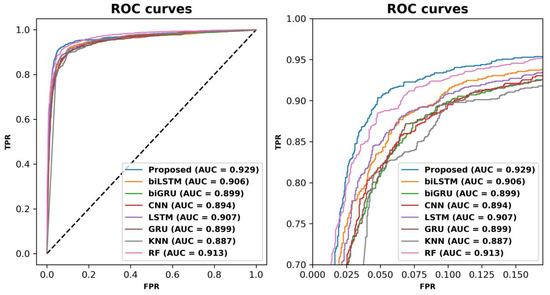
Figure 2.
Receiver operating characteristic (ROC) curves of ABPCaps and baselines on an independent test set. The left panel shows the ROC curves of ABPCaps and baselines, while the right panel is an enlarged view of the same graph for better visibility.
As shown in Table 3, the proposed model was also compared with existing identification tools, including iAMPpred [49], ClassAMP [9], and AMPfun [50]. The results showed that the proposed capsule network-based method outperformed all existing tools and provided the most accurate prediction of ABPs. These results highlight the potential of capsule networks for sequence analysis tasks. The excellent performance of ABPCaps can be attributed to the design of the network, which can be broadly attributed to the following factors. Firstly, the initial modeling of peptide sequences is performed by the CNN and LSTM network, with the former being used to extract local features and the latter capturing the long-range dependencies of the sequence. Secondly, the instantiation parameters of the capsule layer allow for diverse feature extraction and representation. Each instantiation parameter represents a specific feature of the peptide sequence, while the length of each capsule represents the probability of a certain entity occurring. This diverse feature representation provides a guarantee for communication between different capsule layers. Finally, the dynamic routing mechanism can be viewed as a parallel attention mechanism [51]. Capsule networks use a dynamic routing algorithm to compute the coupling coefficients between capsules in adjacent layers. This allows for more accurate representation of the spatial relationships between features in the input data, while directing the network’s attention towards internal capsules that are relevant for prediction. Therefore, it is not surprising that ABPCaps achieved excellent predictive performance.
Table 3.
Comparisons of previous work on ABP prediction. The table summarizes the performance of different ABP prediction models proposed in previous studies in terms of accuracy, recall, precision, F1-score, and AUROC. The evaluation metrics are calculated on the independent test set. The best performance in each category is highlighted in bold.
3.2. Comparison of Capsule Network-Based Features and Hand-Crafted Features
ABPCaps automatically generates and extracts critical features from peptide sequences. Compared to a number of hand-crafted features, the capsule network has a more powerful feature learning capability. For a more intuitive demonstration of the effectiveness of ABPCaps, we applied the dimension reduction technique based on t-distributed stochastic neighbor embedding (t-SNE) [52] to visualize the differences in amino acid coding between ABPCaps and traditional handcrafted features on test sets. The t-SNE plots in Figure 3 show that ABP and non-ABP can be easily separated by the features based on ABPCaps, but they cannot be well distinguished by the hand-crafted features. This result suggests that ABPCaps features can better capture the characteristic features of ABP sequences.
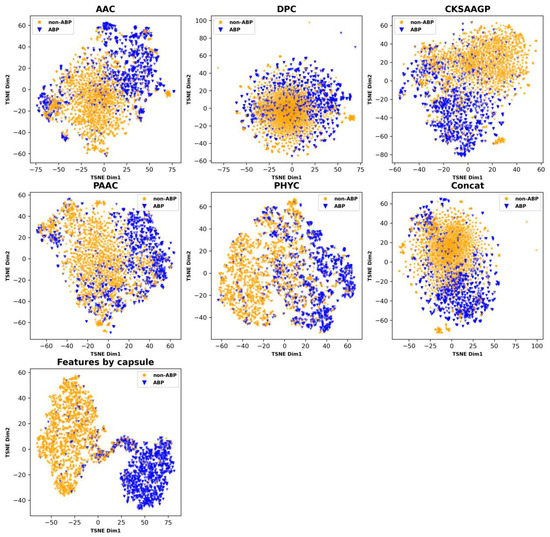
Figure 3.
Visualization of five hand-crafted features and features extracted by ABPCaps. The figure shows a comparison of five hand-crafted features and features extracted by ABPCaps, with ABP and non-ABP shown in blue and yellow colors, respectively. This visualization highlights the superior discriminative ability of the ABPCaps model in identifying ABP, thereby aiding in the diagnosis and treatment of related medical conditions.
To further evaluate the performance of ABPCaps, four basic machine learning-based classifiers, including KNN, RF, SVM, and Gaussian naive bayes (GNB), were used to compare the performance of hand-crafted features and features extracted by ABPCaps on five hand-crafted features, ACC, DPC, PAAC, CKSAAGP, and PHYC. Accuracy, F1-score and AUROC were chosen as evaluation metrics. The results in Figure 4 show that the features extracted by ABPCaps outperform the other five hand-crafted features for all four classifiers, especially for KNN and GNB. This suggests that the features learned by ABPCaps would be more effective for the identification of ABPs, as well as being more suitable for general classification. In short, ABPCaps is a powerful tool that can automatically generate and extract critical features from peptide sequences using a capsule network.
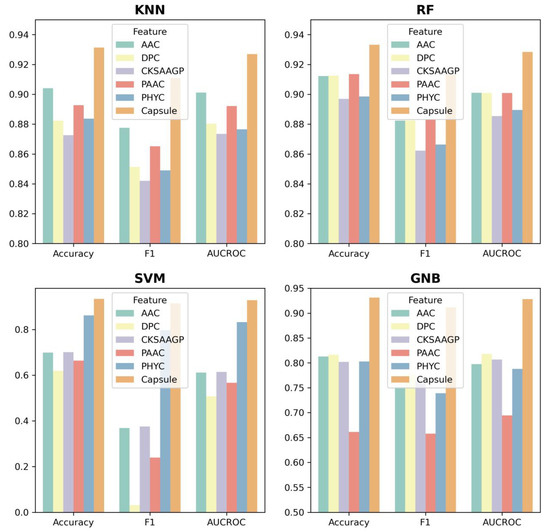
Figure 4.
Feature comparison between five hand-crafted features and features extracted by ABPCaps using four basic classifiers (KNN, RF, SVM, and GNB). Models are trained with these features and accuracy, F1-score and AUCROC are chosen as evaluation metrics. Based on the features extracted by ABPCaps and the hand-crafted features, the graph shows the performance of the different classifiers in distinguishing between ABP and non-ABP.
3.3. Evaluation of ABPCaps on the Subsets with Functions against Different Bacteria
Further experiments were performed to verify the accuracy and effectiveness of the APCaps. Based on the functions against different bacteria such as Escherichia coli and Staphylococcus aureus, the ABP dataset was divided into several subsets. A negative set of non-AMPs was also created for each subset using UniProt. Then, in an 8:2 ratio, the subsets were randomly divided into training and test sets. Table 4 shows the number of sequences for each subset.
Table 4.
The number of peptide sequences in ten subsets. This table shows the number of peptide sequences used in each of the ten subsets used for evaluation.
For each subset, the performance of the ABPCaps was compared to the baseline models. Table 5 shows the average performance metrics over the ten subsets, showing that ABPCaps outperformed all baseline models in terms of precision, accuracy, F1-score, AUROC, and recall. The full evaluation metrics of ABPCaps in each subset are summarized in Supplementary Table S1. Furthermore, ROC curves were drawn for the models trained on every subgroup, as shown in Supplementary Figure S3, and the trained models achieved the best AUROC on every subgroup. Through extensive experiments on multiple datasets, ABPCaps outperformed all baselines and demonstrated excellent predictive abilities on various ABP datasets of different sizes and function against different bacteria. Overall, the results of the experiments demonstrated the accuracy and effectiveness of the ABPCaps and their ability to predict the antibacterial activity of peptides against a wide variety of bacteria.
Table 5.
Evaluation metrics averaged over ten subsets. This table shows the average evaluation metrics for the proposed model and the baselines across the ten subsets. The best performance in each category is highlighted in bold.
4. Conclusions
In this study, we have proposed a novel classification scheme, ABPCaps, to distinguish ABPs from non-ABPs. Harnessing the ability to automatically extract essential features simultaneously from both positive and negative samples, the capsule network-based model (ABPCaps) achieved state-of-the-art performance on the elaborate dataset for identifying antibacterial peptides with an AUROC of 0.9289, an F1-score of 0.9134, a recall of 90.95% and an accuracy of 93.33%. Furthermore, ABPCaps captured the characteristic features of ABP sequences better and outperformed the other five hand-crafted features for all four classifiers. In particular, the t-SNE plots showed that ABPCaps can easily distinguish ABPs from non-ABPs, while the models built with traditional hand-crafted features failed to do so. Our results demonstrate that the capsule network-based model can indeed overcome some limitations encountered in the current sequence analysis approaches, in terms of predicting ABPs. To sum up, ABPCaps can identify ABPs with a high precision and simultaneously with a high accuracy. It is an important step forward in predicting ABPs, and hence contributes to the development of new therapies based on ABPs. In future research, we will fully exploit the characteristics of capsule networks to enhance the interpretability of ABPCaps and investigate the intrinsic features of ABPs and non-ABPs.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/app13126965/s1, Algorithm S1: Dynamic Routing; Figure S1: The computation process between primary capsule layer and class capsule layer in ABPCaps; Figure S2: The architectures of the baseline models used for comparison in this study; Figure S3: Receiver operating characteristic (ROC) curves of the proposed model and baselines on ten subsets; Table S1: Performance comparison of ABPCaps and baseline models on subsets with the functions against different bacteria.
Author Contributions
L.Y. and T.-Y.L. presented the idea. L.Y., Y.P. and J.W. implemented the model and built the pipeline. J.G. and J.Y. collected the data. L.Y., C.-R.C. and C.L. analyzed the results. T.-Y.L. and Y.-C.C. supervised the research project. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Guangdong Province Basic and Applied Basic Research Fund (2021A1515012447), National Natural Science Foundation of China (32070659), the Science, Technology and Innovation Commission of Shenzhen Municipality (JCYJ20200109150003938), Ganghong Young Scholar Development Fund (2021E007), Shenzhen-Hong Kong Cooperation Zone for Technology and Innovation (HZQB-KCZYB-2020056). This work was also financially supported by the Center for Intelligent Drug Systems and Smart Bio-devices (IDS2B) from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan. Y.-C.C. was also partially supported by the Chinese University of Hong Kong, Shenzhen. This work was also supported by the Kobilka Institute of Innovative Drug Discovery, The Chinese University of Hong Kong, Shenzhen, China.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The datasets of this study are available at https://github.com/lantianyao/ABPCaps/ (accessed on 5 June 2023).
Acknowledgments
The authors sincerely appreciate Kobilka Institute of Innovative Drug Discovery and The Chinese University of Hong Kong (Shenzhen) for financially supporting this research.
Conflicts of Interest
The authors declare no competing interest.
References
- Fair, R.J.; Tor, Y. Antibiotics and bacterial resistance in the 21st century. Perspect. Med. Chem. 2014, 6, PMC-S14459. [Google Scholar] [CrossRef] [PubMed]
- Chambers, H.F.; DeLeo, F.R. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol. 2009, 7, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Kosikowska, P.; Lesner, A. Antimicrobial peptides (AMPs) as drug candidates: A patent review (2003–2015). Expert Opin. Ther. Patents 2016, 26, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Dou, X.; Song, J.; Lyu, Y.; Zhu, X.; Xu, L.; Li, W.; Shan, A. Antimicrobial peptides: Promising alternatives in the post feeding antibiotic era. Med. Res. Rev. 2019, 39, 831–859. [Google Scholar] [CrossRef] [PubMed]
- Brahmachary, M.; Krishnan, S.; Koh, J.L.Y.; Khan, A.M.; Seah, S.H.; Tan, T.W.; Brusic, V.; Bajic, V.B. ANTIMIC: A database of antimicrobial sequences. Nucleic Acids Res. 2004, 32, D586–D589. [Google Scholar] [CrossRef]
- Ye, G.; Wu, H.; Huang, J.; Wang, W.; Ge, K.; Li, G.; Zhong, J.; Huang, Q. LAMP2: A major update of the database linking antimicrobial peptides. Database 2020, 2020, baaa061. [Google Scholar] [CrossRef]
- Shi, G.; Kang, X.; Dong, F.; Liu, Y.; Zhu, N.; Hu, Y.; Xu, H.; Lao, X.; Zheng, H. DRAMP 3.0: An enhanced comprehensive data repository of antimicrobial peptides. Nucleic Acids Res. 2022, 50, D488–D496. [Google Scholar] [CrossRef]
- Jhong, J.H.; Yao, L.; Pang, Y.; Li, Z.; Chung, C.R.; Wang, R.; Li, S.; Li, W.; Luo, M.; Ma, R.; et al. dbAMP 2.0: Updated resource for antimicrobial peptides with an enhanced scanning method for genomic and proteomic data. Nucleic Acids Res. 2022, 50, D460–D470. [Google Scholar] [CrossRef]
- Joseph, S.; Karnik, S.; Nilawe, P.; Jayaraman, V.K.; Idicula-Thomas, S. ClassAMP: A prediction tool for classification of antimicrobial peptides. IEEE/ACM Trans. Comput. Biol. Bioinform. 2012, 9, 1535–1538. [Google Scholar] [CrossRef]
- Xiao, X.; Wang, P.; Lin, W.Z.; Jia, J.H.; Chou, K.C. iAMP-2L: A two-level multi-label classifier for identifying antimicrobial peptides and their functional types. Anal. Biochem. 2013, 436, 168–177. [Google Scholar] [CrossRef]
- Yan, J.; Bhadra, P.; Li, A.; Sethiya, P.; Qin, L.; Tai, H.K.; Wong, K.H.; Siu, S.W. Deep-AmPEP30: Improve short antimicrobial peptides prediction with deep learning. Mol. Ther.-Nucleic Acids 2020, 20, 882–894. [Google Scholar] [CrossRef] [PubMed]
- Veltri, D.; Kamath, U.; Shehu, A. Deep learning improves antimicrobial peptide recognition. Bioinformatics 2018, 34, 2740–2747. [Google Scholar] [CrossRef] [PubMed]
- Youmans, M.; Spainhour, C.; Qiu, P. Long short-term memory recurrent neural networks for antibacterial peptide identification. In Proceedings of the 2017 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Kansas City, MO, USA, 13–16 November 2017; pp. 498–502. [Google Scholar]
- Singh, V.; Shrivastava, S.; Kumar Singh, S.; Kumar, A.; Saxena, S. StaBle-ABPpred: A stacked ensemble predictor based on biLSTM and attention mechanism for accelerated discovery of antibacterial peptides. Briefings Bioinform. 2022, 23, bbab439. [Google Scholar] [CrossRef]
- Sharma, R.; Shrivastava, S.; Kumar Singh, S.; Kumar, A.; Saxena, S.; Kumar Singh, R. Deep-ABPpred: Identifying antibacterial peptides in protein sequences using bidirectional LSTM with word2vec. Briefings Bioinform. 2021, 22, bbab065. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lin, J.; Zhao, L.; Zeng, X.; Liu, X. A novel antibacterial peptide recognition algorithm based on BERT. Briefings Bioinform. 2021, 22, bbab200. [Google Scholar] [CrossRef]
- Hochreiter, S.; Schmidhuber, J. Long short-term memory. Neural Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef]
- LaLonde, R.; Bagci, U. Capsules for object segmentation. arXiv 2018, arXiv:1804.04241. [Google Scholar]
- Dong, Z.; Lin, S. Research on image classification based on capsnet. In Proceedings of the 2019 IEEE 4th Advanced Information Technology, Electronic and Automation Control Conference (IAEAC), Chengdu, China, 20–22 December 2019; Volume 1, pp. 1023–1026. [Google Scholar]
- Hinton, G.E.; Krizhevsky, A.; Wang, S.D. Transforming auto-encoders. In Proceedings of the Artificial Neural Networks and Machine Learning–ICANN 2011: 21st International Conference on Artificial Neural Networks, Espoo, Finland, 14–17 June 2011, Proceedings, Part I 21; Springer: Berlin, Germany, 2011; pp. 44–51. [Google Scholar]
- Sabour, S.; Frosst, N.; Hinton, G.E. Dynamic routing between capsules. Adv. Neural Inf. Process. Syst. 2017, 30, 3859–3869. [Google Scholar]
- Nguyen, H.H.; Yamagishi, J.; Echizen, I. Use of a capsule network to detect fake images and videos. arXiv 2019, arXiv:1910.12467. [Google Scholar]
- Pan, C.; Velipasalar, S. PT-CapsNet: A novel prediction-tuning capsule network suitable for deeper architectures. In Proceedings of the IEEE/CVF International Conference on Computer Vision, Montreal, BC, Canada, 11–17 October 2021; pp. 11996–12005. [Google Scholar]
- Zhang, Z.; Ye, S.; Liao, P.; Liu, Y.; Su, G.; Sun, Y. Enhanced capsule network for medical image classification. In Proceedings of the 2020 42nd Annual International Conference of the IEEE Engineering in Medicine & Biology Society (EMBC), Montreal, QC, Canada, 20–24 July 2020; pp. 1544–1547. [Google Scholar]
- Zhao, W.; Ye, J.; Yang, M.; Lei, Z.; Zhang, S.; Zhao, Z. Investigating capsule networks with dynamic routing for text classification. arXiv 2018, arXiv:1804.00538. [Google Scholar]
- Hu, J.; Liao, J.; Liu, L.; Ma, W. RCapsNet: A recurrent capsule network for text classification. In Proceedings of the 2020 International Joint Conference on Neural Networks (IJCNN), Glasgow, UK, 19–24 July 2020; pp. 1–8. [Google Scholar]
- Wang, D.; Liang, Y.; Xu, D. Capsule network for protein post-translational modification site prediction. Bioinformatics 2019, 35, 2386–2394. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yu, W.; Han, K.; Nandi, A.K.; Huang, D.S. Multi-scale capsule network for predicting DNA-protein binding sites. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 18, 1793–1800. [Google Scholar] [CrossRef] [PubMed]
- Khanal, J.; Tayara, H.; Zou, Q.; To Chong, K. DeepCap-Kcr: Accurate identification and investigation of protein lysine crotonylation sites based on capsule network. Briefings Bioinform. 2022, 23, bbab492. [Google Scholar] [CrossRef]
- Su, X.; You, Z.H.; Huang, D.s.; Wang, L.; Wong, L.; Ji, B.; Zhao, B. Biomedical knowledge graph embedding with capsule network for multi-label drug-drug interaction prediction. IEEE Trans. Knowl. Data Eng. 2022, 35, 5640–5651. [Google Scholar] [CrossRef]
- Li, Z.; Li, S.; Luo, M.; Jhong, J.H.; Li, W.; Yao, L.; Pang, Y.; Wang, Z.; Wang, R.; Ma, R.; et al. dbPTM in 2022: An updated database for exploring regulatory networks and functional associations of protein post-translational modifications. Nucleic Acids Res. 2022, 50, D471–D479. [Google Scholar] [CrossRef]
- Kao, H.J.; Huang, C.H.; Bretaña, N.A.; Lu, C.T.; Huang, K.Y.; Weng, S.L.; Lee, T.Y. A two-layered machine learning method to identify protein O-GlcNAcylation sites with O-GlcNAc transferase substrate motifs. BMC Bioinform. 2015, 16, S10. [Google Scholar] [CrossRef] [PubMed]
- Bretana, N.A.; Lu, C.T.; Chiang, C.Y.; Su, M.G.; Huang, K.Y.; Lee, T.Y.; Weng, S.L. Identifying protein phosphorylation sites with kinase substrate specificity on human viruses. PLoS ONE 2012, 7, e40694. [Google Scholar] [CrossRef]
- Lee, T.Y.; Huang, K.Y.; Chuang, C.H.; Lee, C.Y.; Chang, T.H. Incorporating deep learning and multi-omics autoencoding for analysis of lung adenocarcinoma prognostication. Comput. Biol. Chem. 2020, 87, 107277. [Google Scholar] [CrossRef]
- Bui, V.M.; Weng, S.L.; Lu, C.T.; Chang, T.H.; Weng, J.T.Y.; Lee, T.Y. SOHSite: Incorporating evolutionary information and physicochemical properties to identify protein S-sulfenylation sites. BMC Genom. 2016, 17, 9. [Google Scholar] [CrossRef]
- Chen, S.A.; Lee, T.Y.; Ou, Y.Y. Incorporating significant amino acid pairs to identify O-linked glycosylation sites on transmembrane proteins and non-transmembrane proteins. BMC Bioinform. 2010, 11, 536. [Google Scholar] [CrossRef]
- Yao, L.; Li, W.; Zhang, Y.; Deng, J.; Pang, Y.; Huang, Y.; Chung, C.R.; Yu, J.; Chiang, Y.C.; Lee, T.Y. Accelerating the Discovery of Anticancer Peptides through Deep Forest Architecture with Deep Graphical Representation. Int. J. Mol. Sci. 2023, 24, 4328. [Google Scholar] [CrossRef] [PubMed]
- Witten, J.; Witten, Z. Deep learning regression model for antimicrobial peptide design. BioRxiv 2019, 692681. [Google Scholar] [CrossRef]
- Pirtskhalava, M.; Amstrong, A.A.; Grigolava, M.; Chubinidze, M.; Alimbarashvili, E.; Vishnepolsky, B.; Gabrielian, A.; Rosenthal, A.; Hurt, D.E.; Tartakovsky, M. DBAASP v3: Database of antimicrobial/cytotoxic activity and structure of peptides as a resource for development of new therapeutics. Nucleic Acids Res. 2021, 49, D288–D297. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, A.; Thakur, N.; Tandon, H.; Kumar, M. AVPdb: A database of experimentally validated antiviral peptides targeting medically important viruses. Nucleic Acids Res. 2014, 42, D1147–D1153. [Google Scholar] [CrossRef]
- Agrawal, P.; Bhagat, D.; Mahalwal, M.; Sharma, N.; Raghava, G.P. AntiCP 2.0: An updated model for predicting anticancer peptides. Briefings Bioinform. 2021, 22, bbaa153. [Google Scholar] [CrossRef]
- Agrawal, P.; Bhalla, S.; Chaudhary, K.; Kumar, R.; Sharma, M.; Raghava, G.P. In silico approach for prediction of antifungal peptides. Front. Microbiol. 2018, 9, 323. [Google Scholar] [CrossRef]
- Afshar, P.; Heidarian, S.; Naderkhani, F.; Oikonomou, A.; Plataniotis, K.N.; Mohammadi, A. COVID-CAPS: A capsule network-based framework for identification of COVID-19 cases from X-ray images. Pattern Recognit. Lett. 2020, 138, 638–643. [Google Scholar] [CrossRef]
- Zhang, K.; Jiao, M.; Chen, X.; Wang, Z.; Liu, B.; Liu, L. SC-BiCapsNet: A Sentiment Classification Model Based on Bi-Channel Capsule Network. IEEE Access 2019, 7, 171801–171813. [Google Scholar] [CrossRef]
- Li, C.; Quan, C.; Peng, L.; Qi, Y.; Deng, Y.; Wu, L. A Capsule Network for Recommendation and Explaining What You Like and Dislike. In Proceedings of the 42nd International ACM SIGIR Conference on Research and Development in Information Retrieval, SIGIR’19, Paris, France, 21–25 July 2019; Association for Computing Machinery: New York, NY, USA, 2019; pp. 275–284. [Google Scholar] [CrossRef]
- Chung, J.; Gulcehre, C.; Cho, K.; Bengio, Y. Empirical evaluation of gated recurrent neural networks on sequence modeling. arXiv 2014, arXiv:1412.3555. [Google Scholar]
- Abadi, M.; Barham, P.; Chen, J.; Chen, Z.; Davis, A.; Dean, J.; Devin, M.; Ghemawat, S.; Irving, G.; Isard, M.; et al. Tensorflow: A system for large-scale machine learning. In Proceedings of the Osdi, Savannah, GA, USA, 2–4 November 2016; Volume 16, pp. 265–283. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Meher, P.K.; Sahu, T.K.; Saini, V.; Rao, A.R. Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou’s general PseAAC. Sci. Rep. 2017, 7, 42362. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.R.; Kuo, T.R.; Wu, L.C.; Lee, T.Y.; Horng, J.T. Characterization and identification of antimicrobial peptides with different functional activities. Briefings Bioinform. 2020, 21, 1098–1114. [Google Scholar] [CrossRef] [PubMed]
- Bahdanau, D.; Cho, K.; Bengio, Y. Neural machine translation by jointly learning to align and translate. arXiv 2014, arXiv:1409.0473. [Google Scholar]
- Van der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).