
Prediction of Linear Cationic Antimicrobial Peptides Active against Gram-Negative and Gram-Positive Bacteria Based on Machine Learning Models

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Dataset and Data Preprocessing
2.1.1. Feature Generation
Generation of Physico-Chemical Features (Descriptors)
Generation of Sequence-Based, Structure-Based, and Linguistic-Based Features
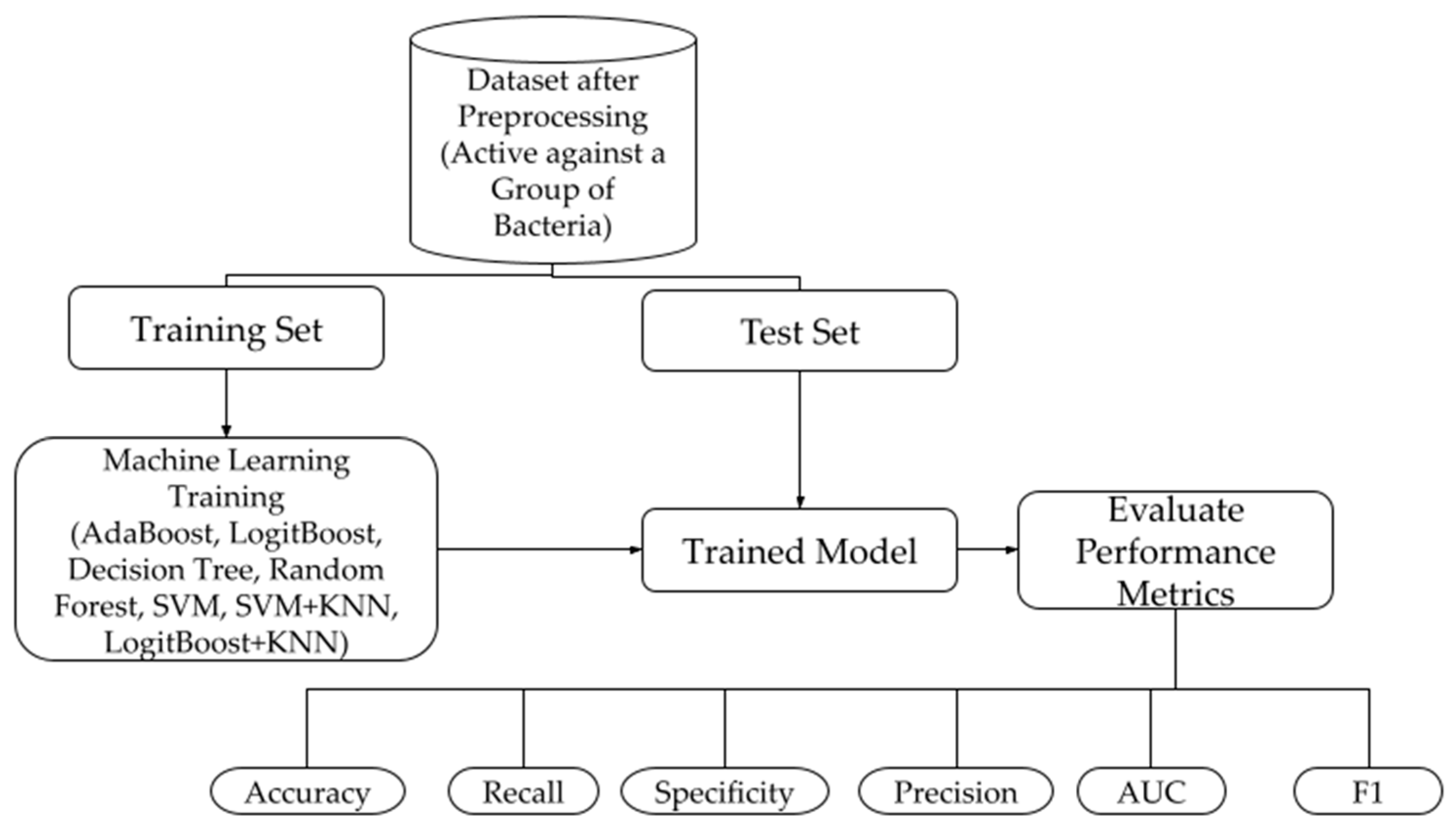
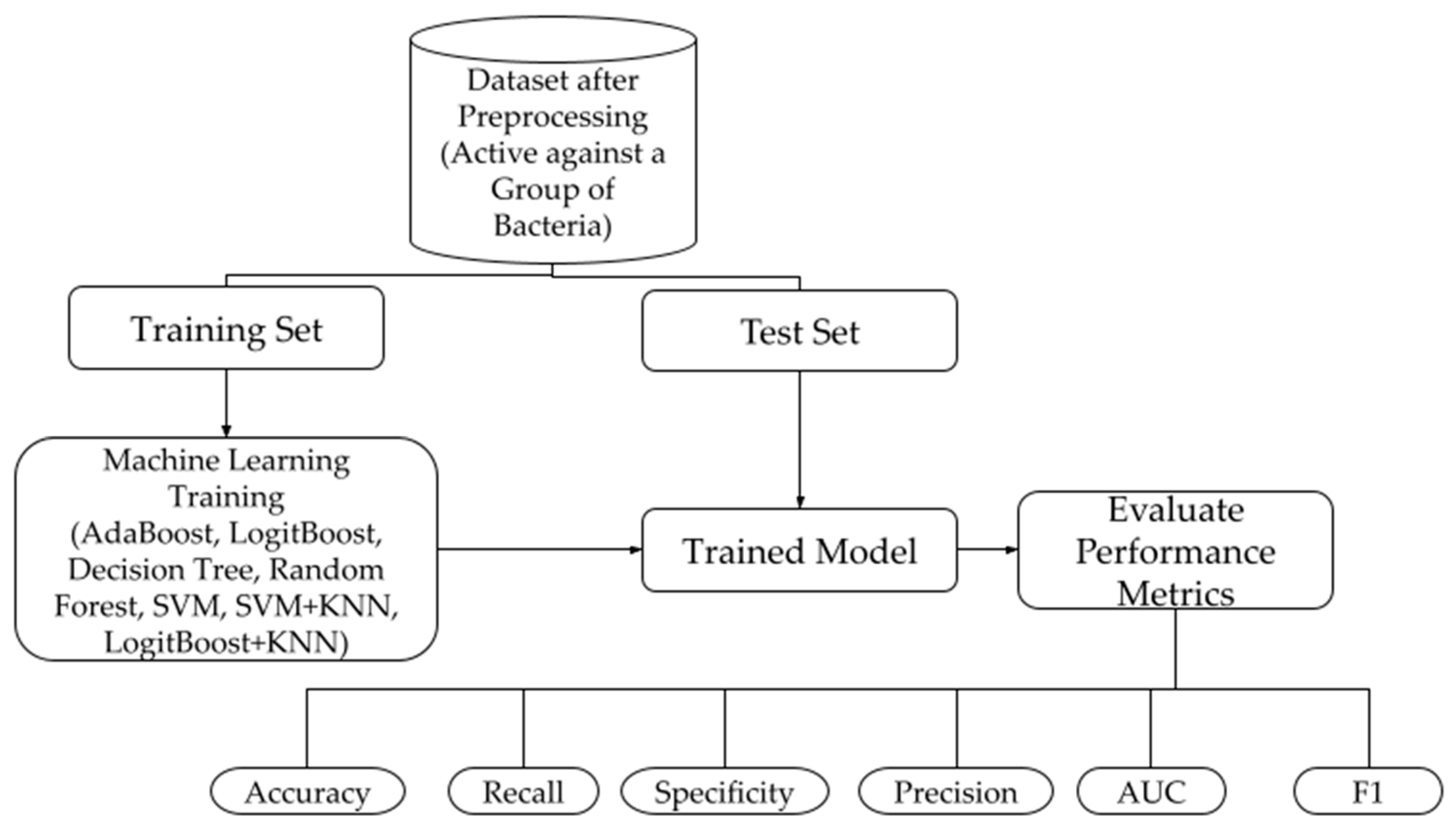
2.2. Machine Learning Models
2.2.1. Model Construction
2.2.2. Performance Metrics
3. Results
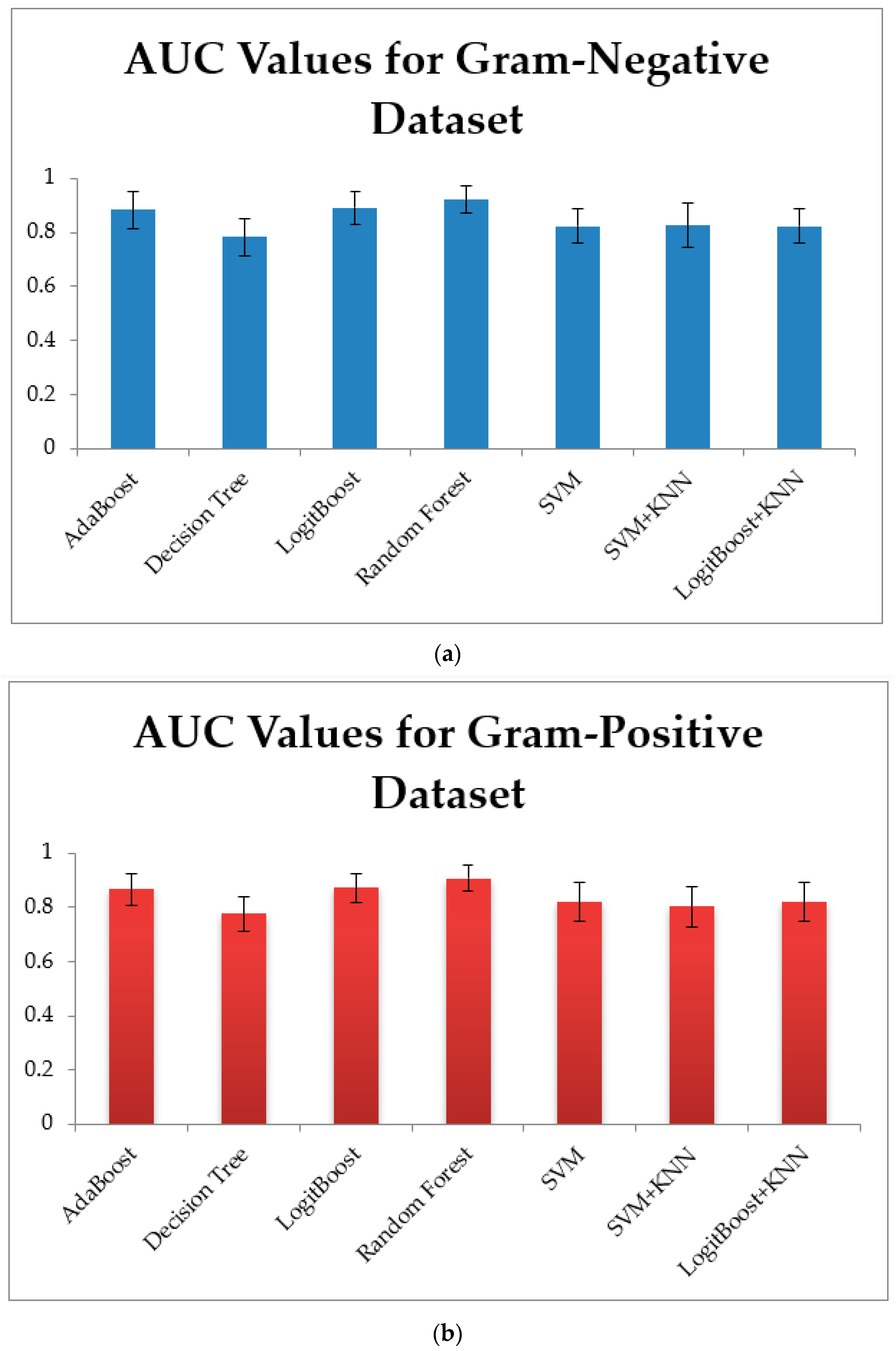
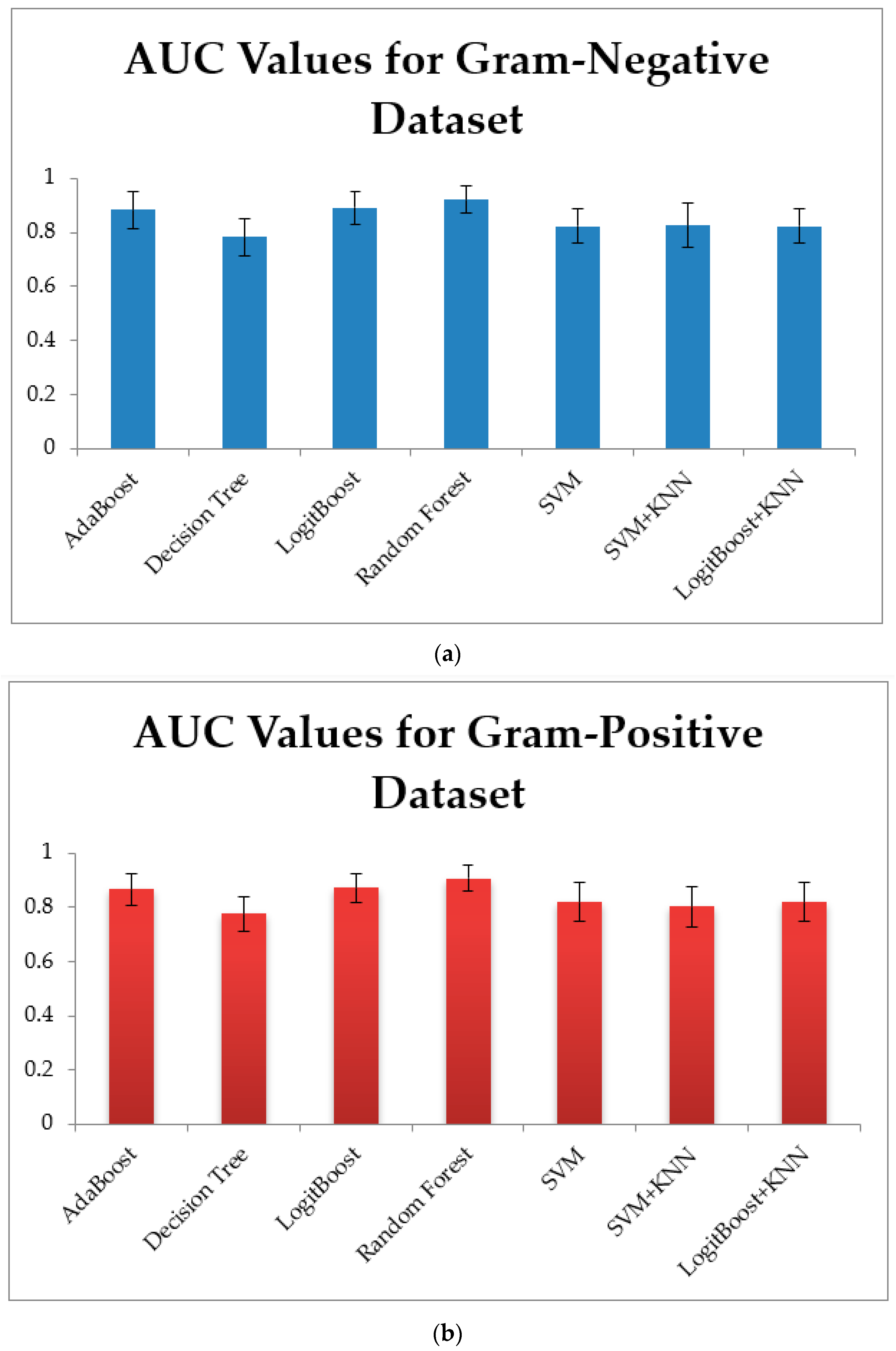
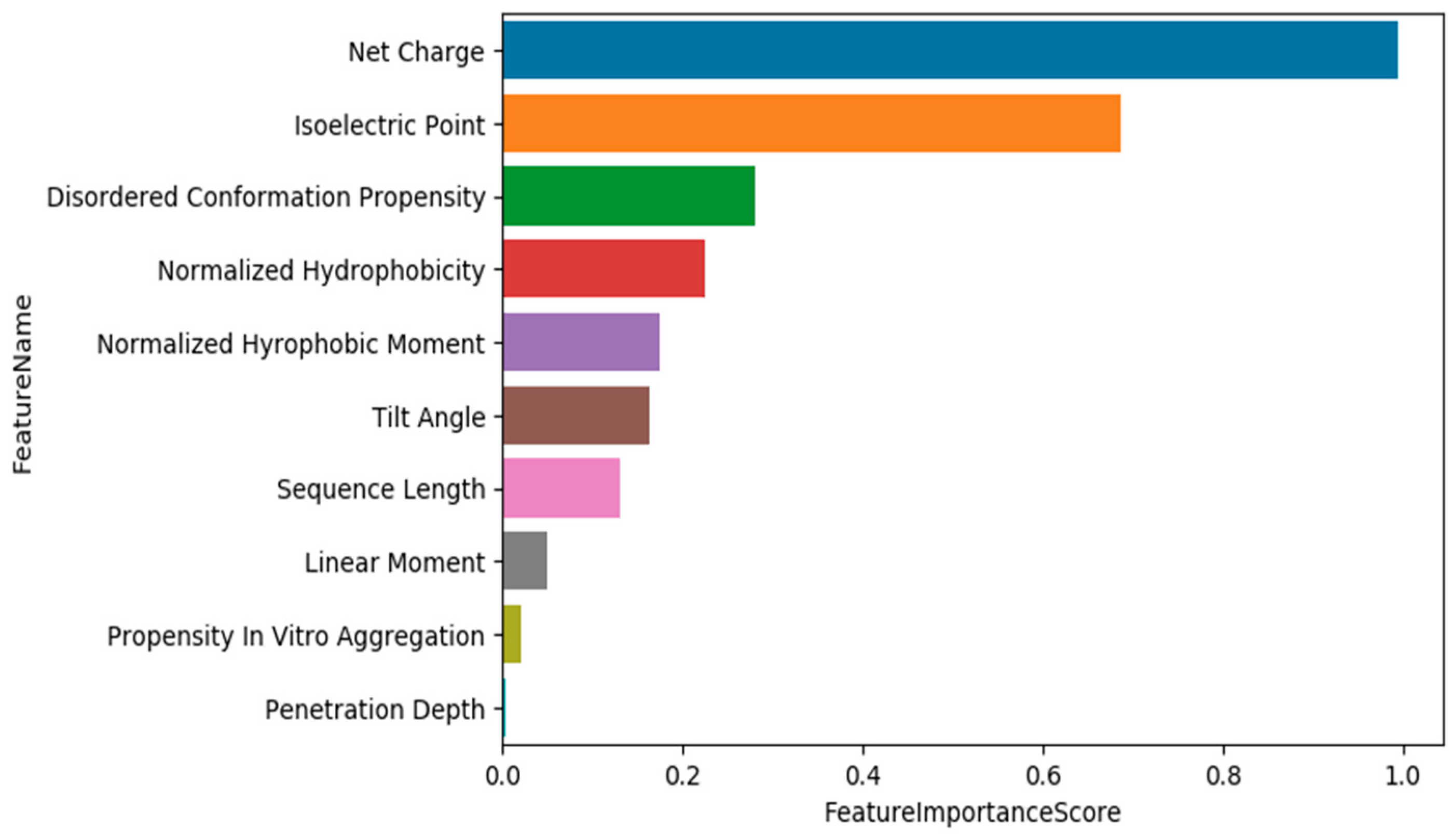
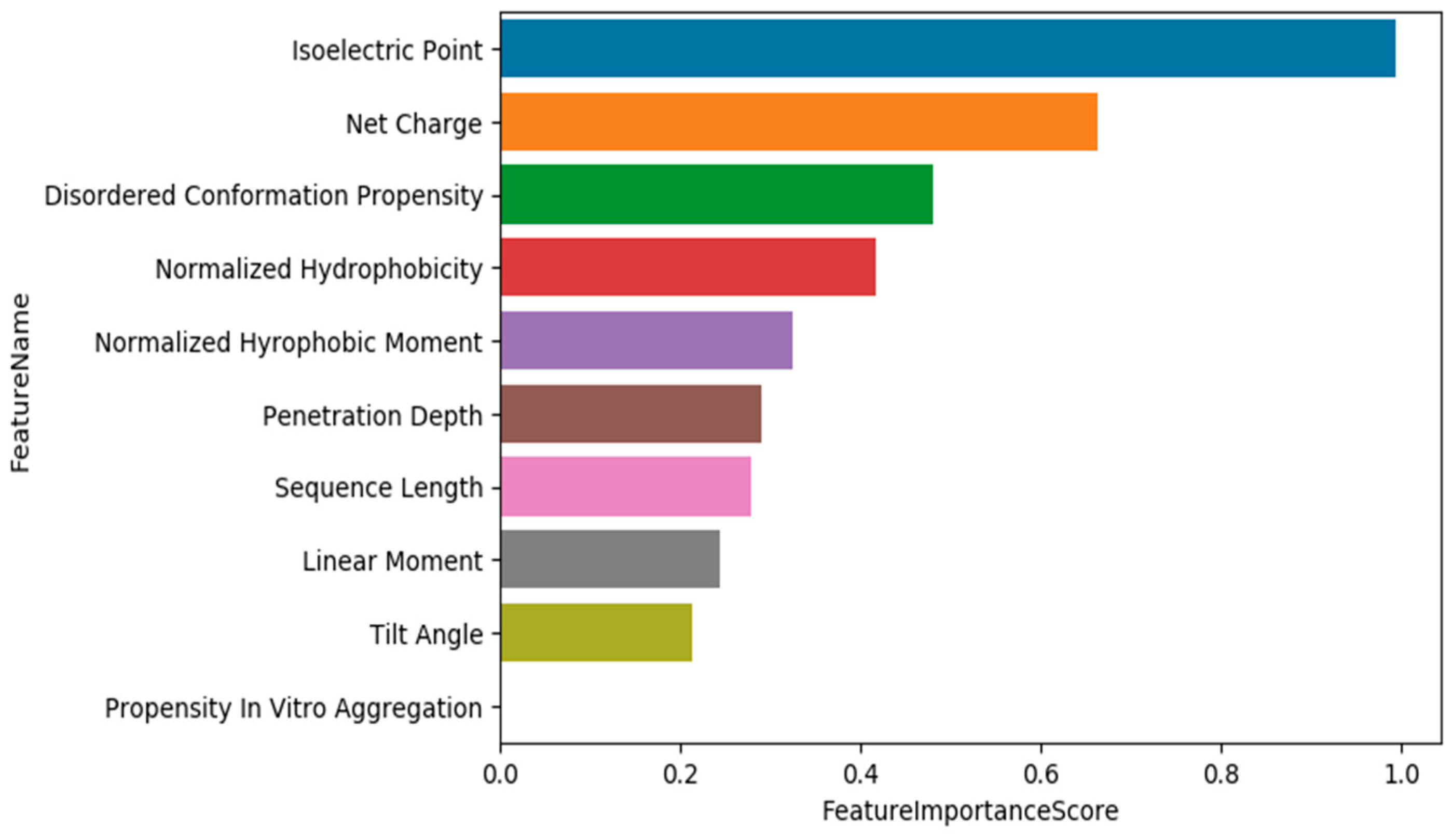
3.1. Training Models Using Physico-Chemical Features
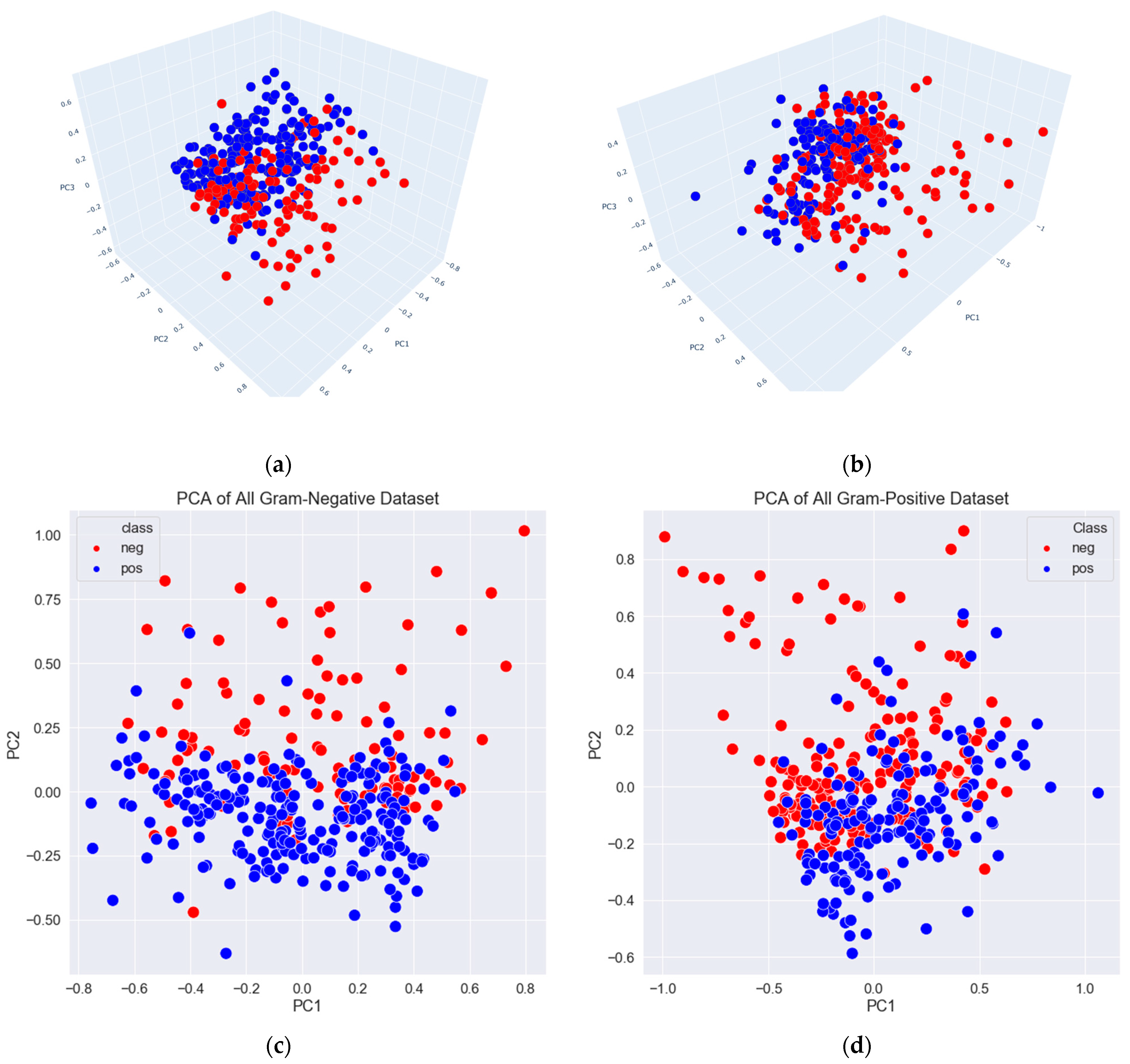
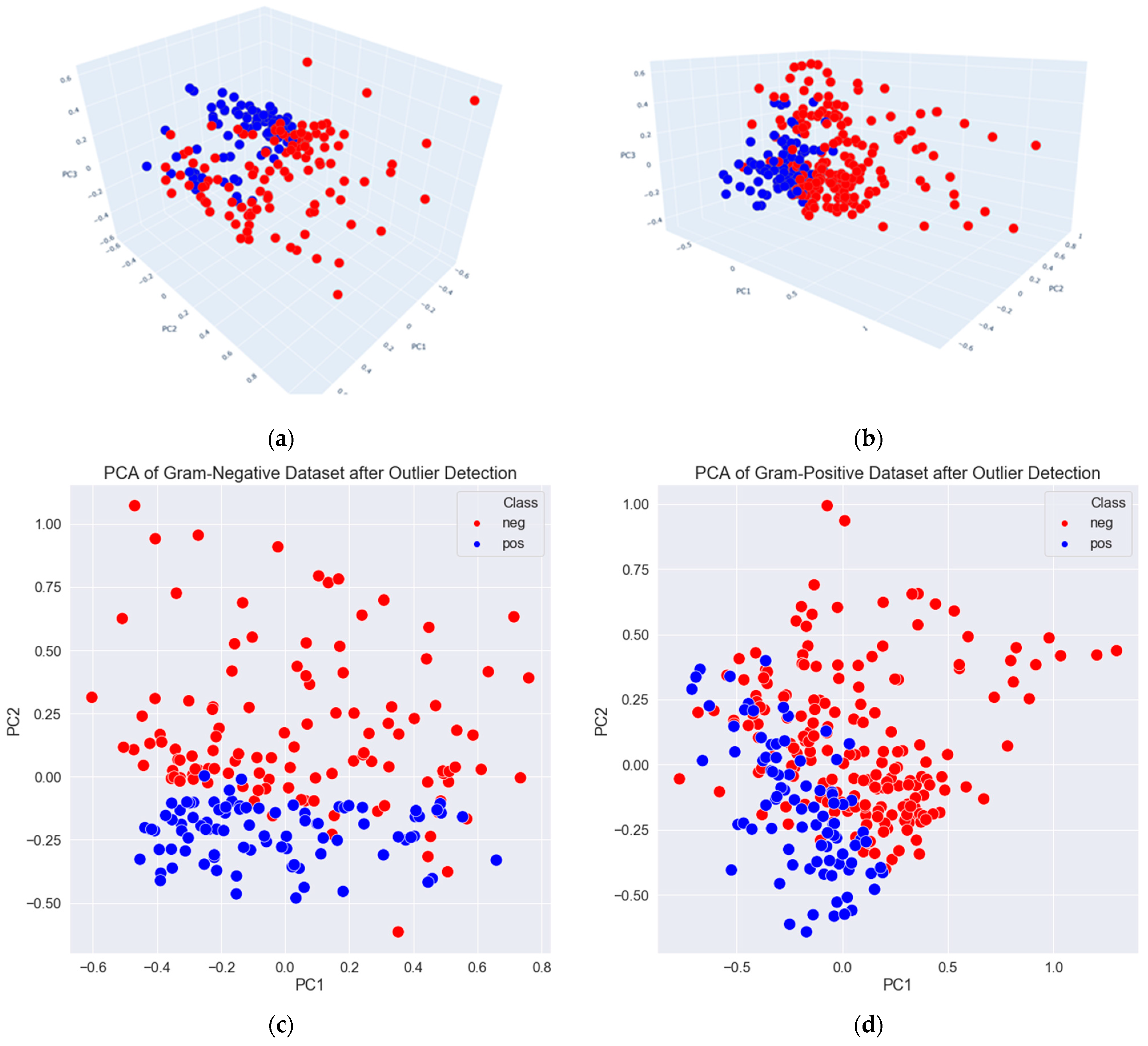
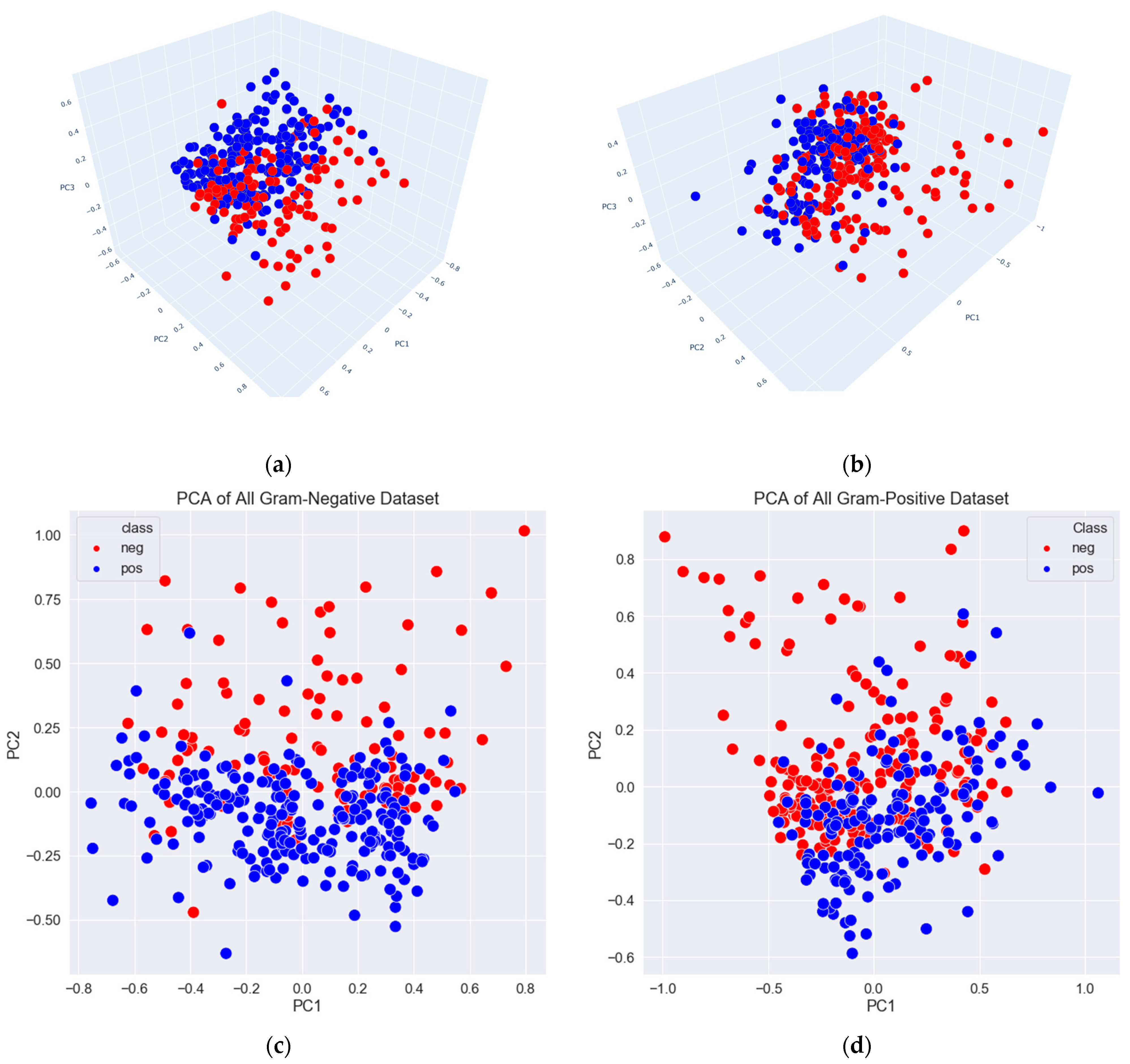
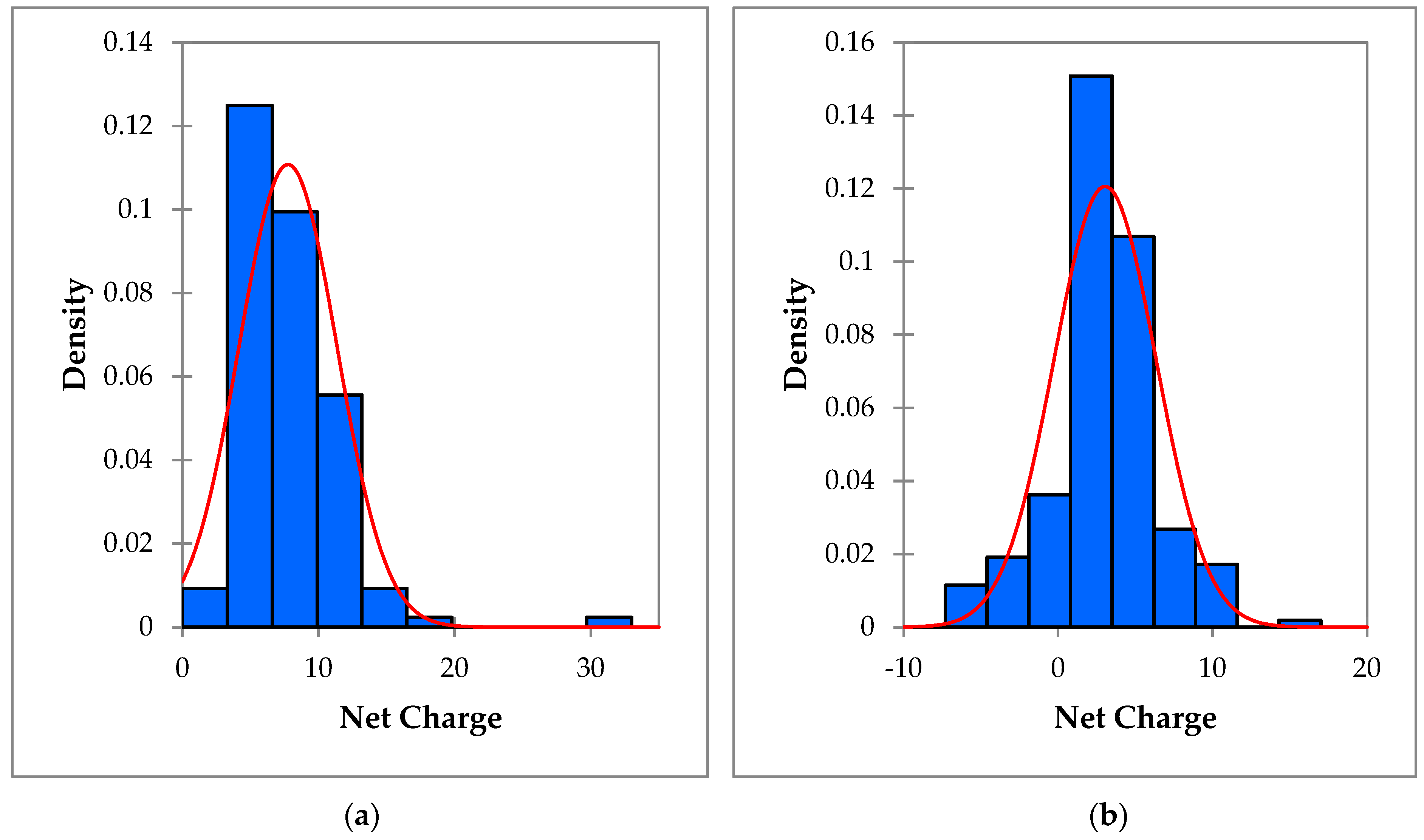
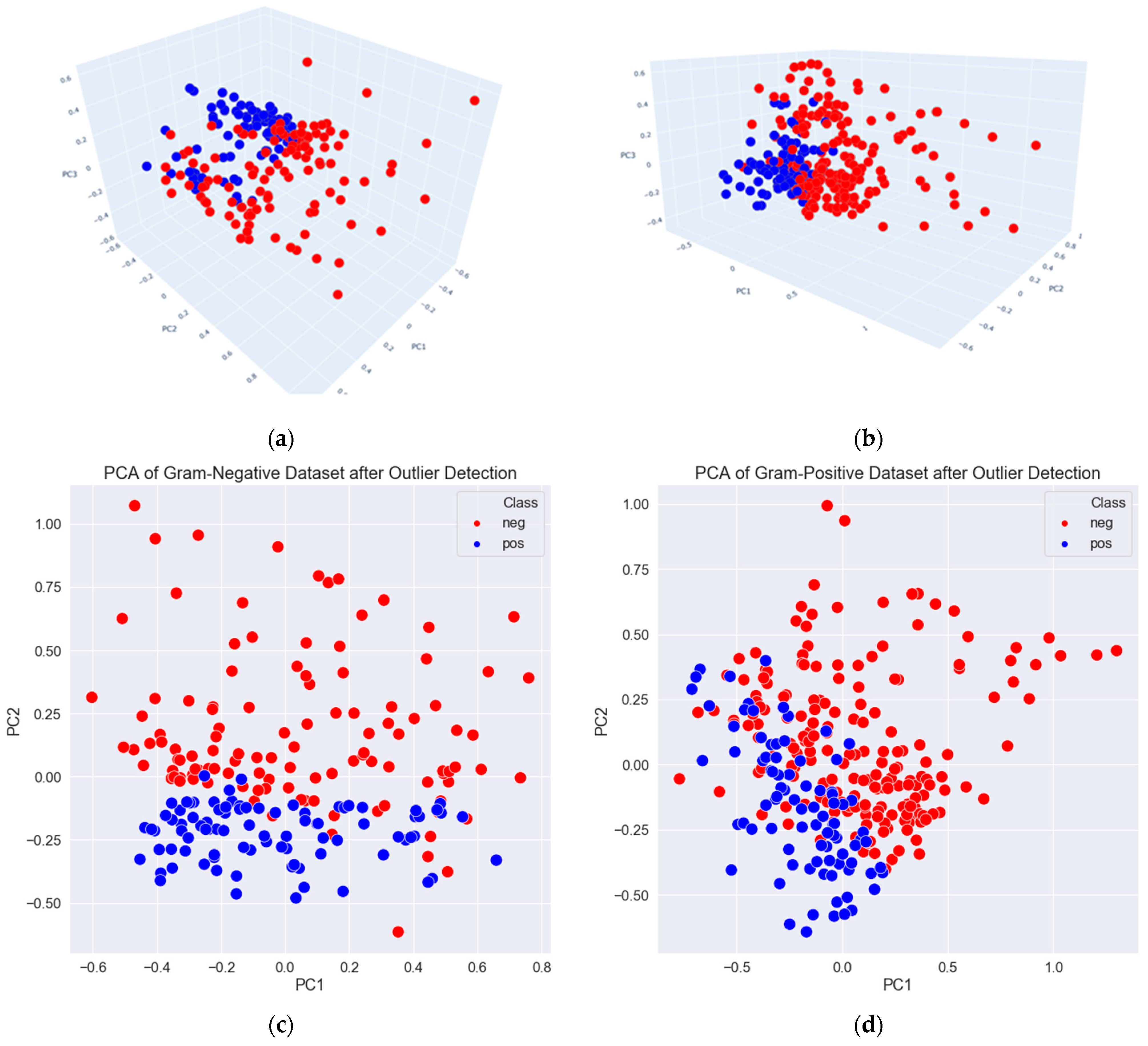
3.2. Data Exploration, Outlier Detection, and Elimination
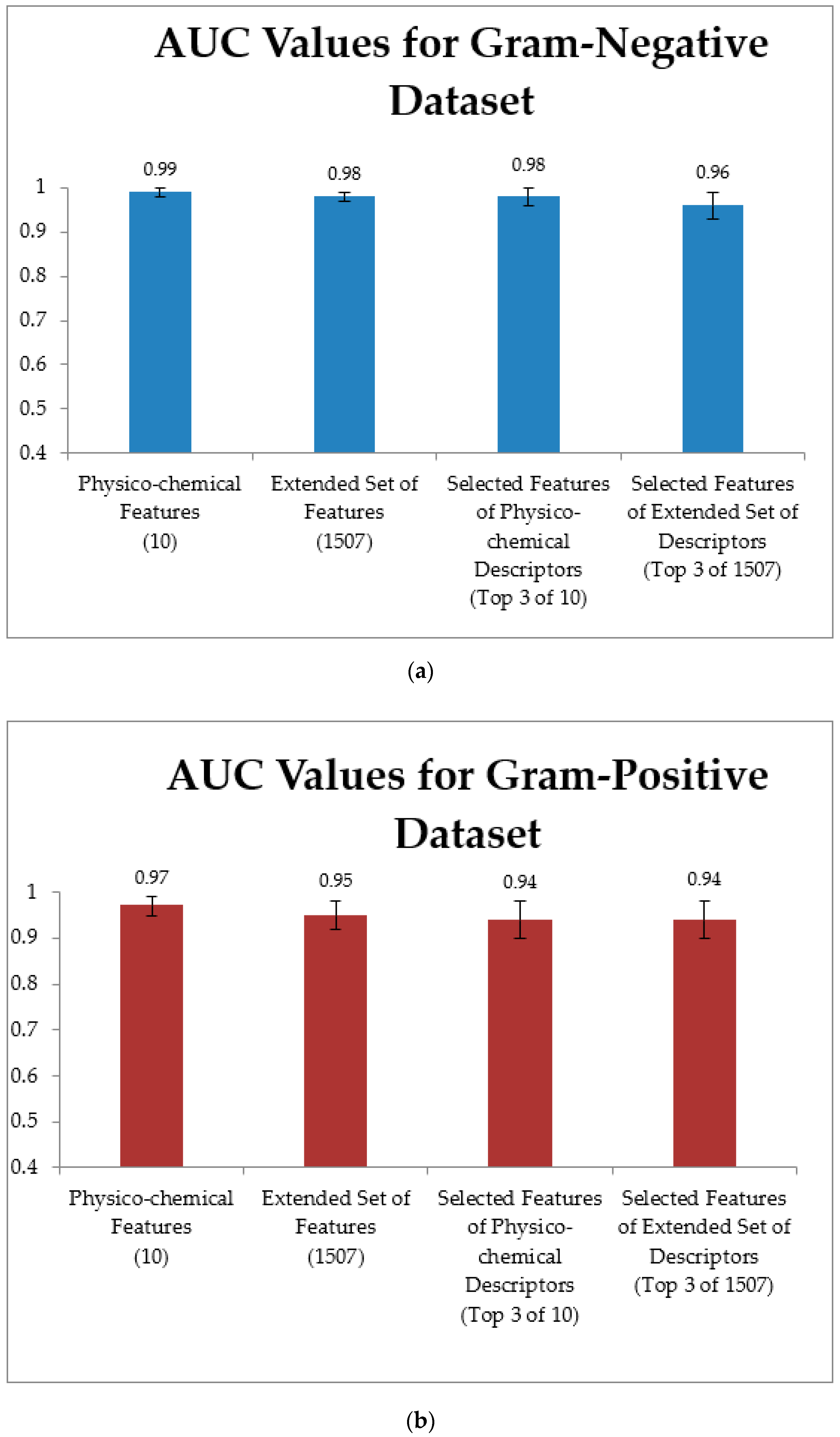
3.3. Training Models Using an Extended Set of Features
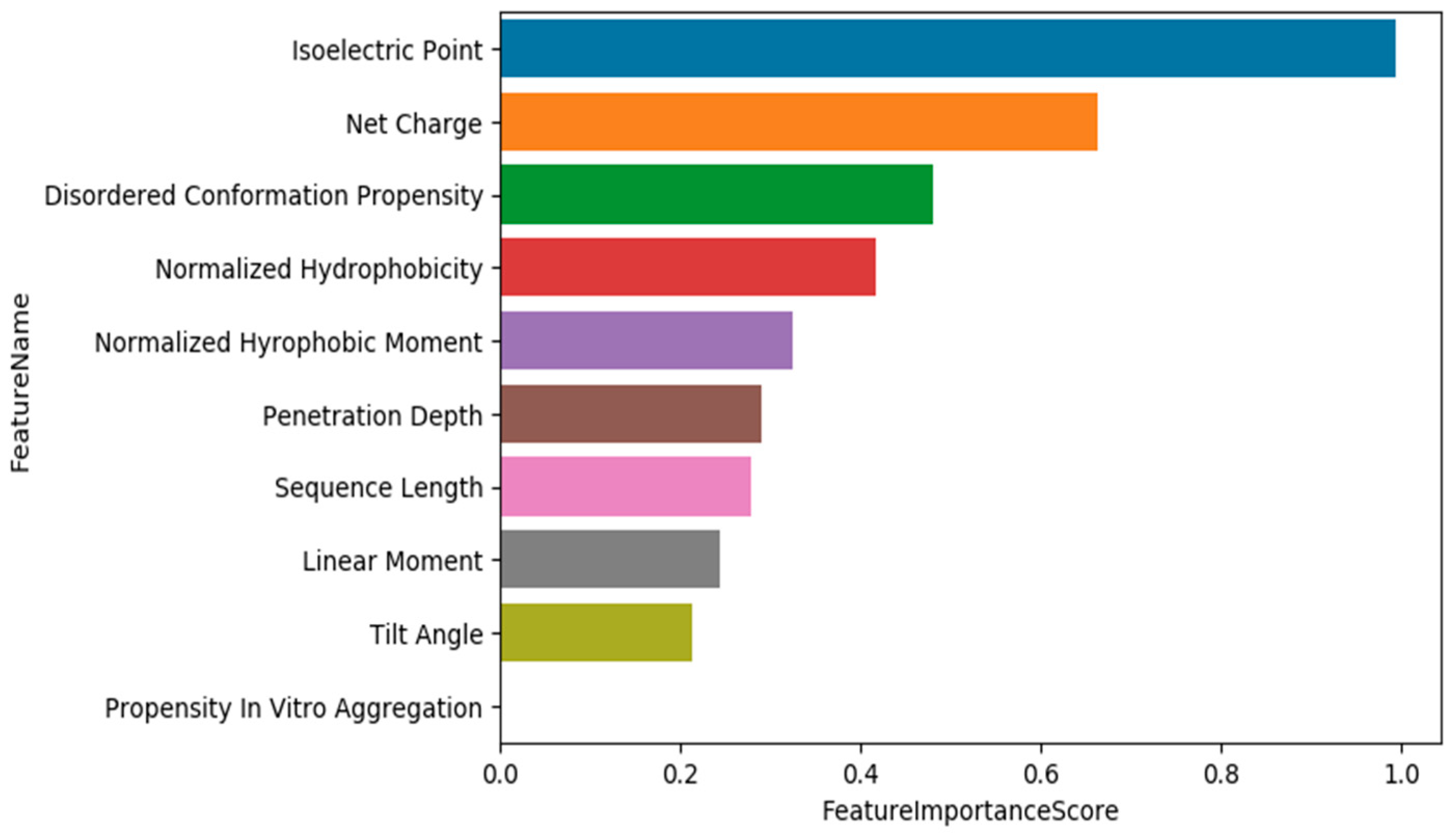
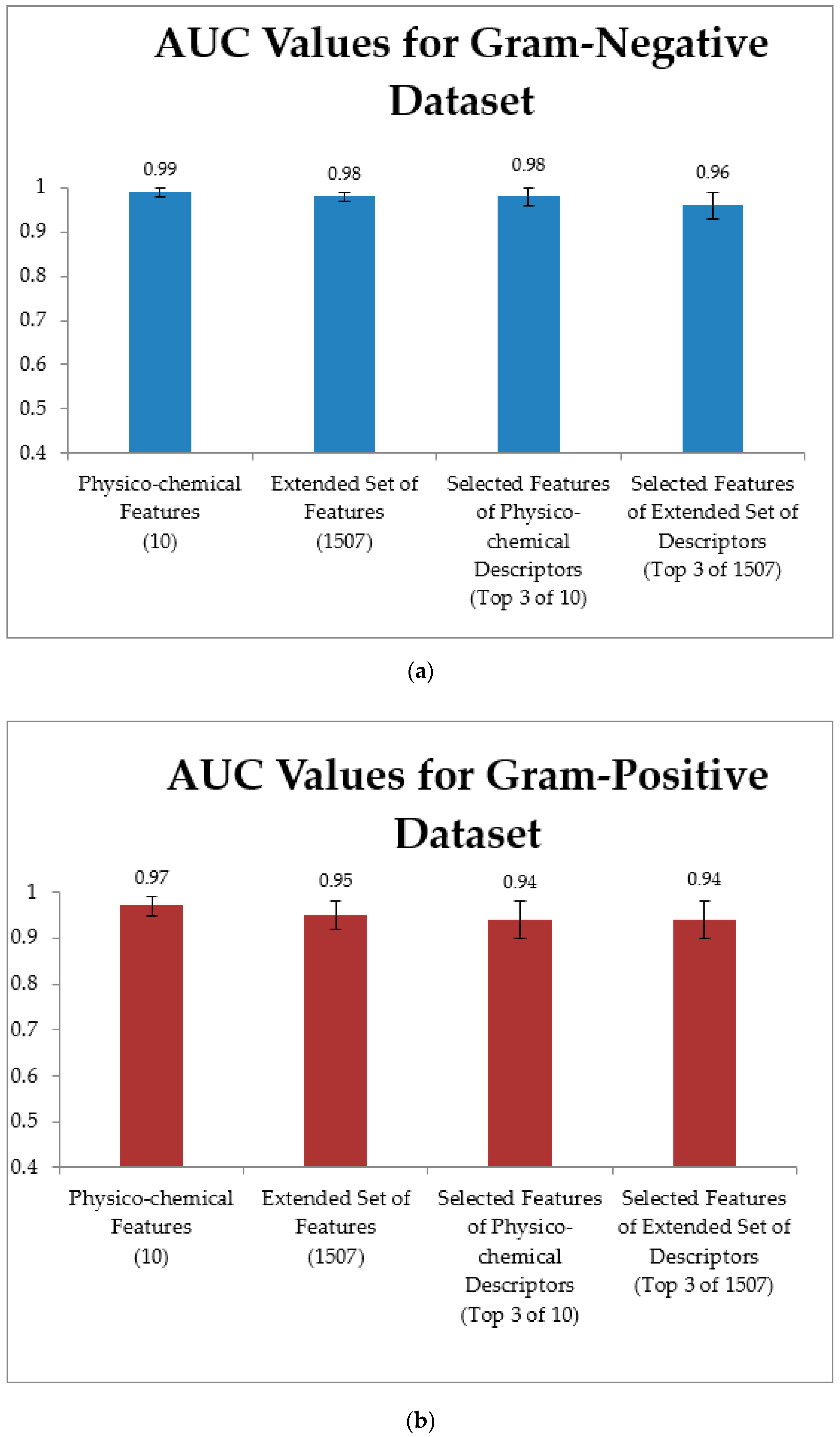
3.4. Training Models Using an Extended Set of Features and Applying Feature Selection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carnicelli, V.; Lizzi, A.R.; Ponzi, A.; Amicosante, G.; Bozzi, A.; Di Giulio, A. Interaction between antimicrobial peptides (AMPs) and their primary target, the biomembranes. In Microbial Pathogens and Strategies for Combating Them: Science, Technology and Education; Formatex Research Center: Badajoz, Spain, 2013; Volume 2, pp. 1123–1134. [Google Scholar]
- Brogden, N.K.; Brogden, K.A. Will new generations of modified antimicrobial peptides improve their potential as pharmaceuticals? Int. J. Antimicrob. Agents 2011, 38, 217–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, F.; Wang, Y.; Li, G.; Liu, S.; Cui, N.; Liu, S.; Langford, P.R.; Wang, C. The SapA Protein Is Involved in Resistance to Antimicrobial Peptide PR-39 and Virulence of Actinobacillus pleuropneumoniae. Front. Microbiol. 2017, 8, 811. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, D.; Jaśkiewicz, M.; Migoń, D.; Bauer, M.; Sikora, K.; Sikorska, E.; Kamysz, E.; Kamysz, W. Retro analog concept: Comparative study on physico-chemical and biological properties of selected antimicrobial peptides. Amino Acids 2017, 49, 1755–1771. [Google Scholar] [CrossRef] [Green Version]
- Büyükkiraz, M.E.; Kesmen, Z. Antimicrobial peptides (AMPs): A promising class of antimicrobial compounds. J. Appl. Microbiol. 2021, 132, 1573–1596. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.; Wang, G. Ab Initio Design of Potent Anti-MRSA Peptides Based on Database Filtering Technology. J. Am. Chem. Soc. 2012, 134, 12426–12429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faccone, D.; Veliz, O.; Corso, A.; Noguera, M.; Martínez, M.; Payes, C.; Semorile, L.; Maffía, P.C. Antimicrobial activity of de novo designed cationic peptides against multi-resistant clinical isolates. Eur. J. Med. Chem. 2014, 71, 31–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.H.; Starr, C.G.; Troendle, E.P.; Wiedman, G.; Wimley, W.C.; Ulmschneider, J.P.; Ulmschneider, M.B. Simulation-Guided Rational de Novo Design of a Small Pore-Forming Antimicrobial Peptide. J. Am. Chem. Soc. 2019, 141, 4839–4848. [Google Scholar] [CrossRef]
- Vishnepolsky, B.; Zaalishvili, G.; Karapetian, M.; Nasrashvili, T.; Kuljanishvili, N.; Gabrielian, A.; Rosenthal, A.; Hurt, D.E.; Tartakovsky, M.; Grigolava, M.; et al. De Novo Design and In Vitro Testing of Antimicrobial Peptides against Gram-Negative Bacteria. Pharmaceuticals 2019, 12, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loose, C.; Jensen, K.; Rigoutsos, I.; Stephanopoulos, G. A linguistic model for the rational design of antimicrobial peptides. Nature 2006, 443, 867–869. [Google Scholar] [CrossRef]
- Nagarajan, D.; Nagarajan, T.; Roy, N.; Kulkarni, O.; Ravichandran, S.; Mishra, M.; Chakravortty, D.; Chandra, N. Computational antimicrobial peptide design and evaluation against multidrug-resistant clinical isolates of bacteria. J. Biol. Chem. 2018, 293, 3492–3509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, M.H.; Cândido, E.S.; Chan, L.Y.; Torres, M.D.T.; Oshiro, K.G.N.; Rezende, S.B.; Porto, W.F.; Lu, T.K.; de la Fuente-Nunez, C.; Craik, D.J.; et al. A Computationally Designed Peptide Derived from Escherichia coli as a Potential Drug Template for Antibacterial and Antibiofilm Therapies. ACS Infect. Dis. 2018, 4, 1727–1736. [Google Scholar] [CrossRef] [PubMed]
- Cândido, E.D.S.; Cardoso, M.H.; Chan, L.Y.; Torres, M.; Oshiro, K.G.N.; Porto, W.F.; Ribeiro, S.; Haney, E.F.; Hancock, R.; Lu, T.K.; et al. Short Cationic Peptide Derived from Archaea with Dual Antibacterial Properties and Anti-Infective Potential. ACS Infect. Dis. 2019, 5, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Fensterseifer, I.C.; Felício, M.R.; Alves, E.S.; Cardoso, M.; Torres, M.; Matos, C.O.; Silva, O.N.; Lu, T.K.; Freire, M.V.; Neves, N.C.; et al. Selective antibacterial activity of the cationic peptide PaDBS1R6 against Gram-negative bacteria. Biochim. Biophys. Acta (BBA)—Biomembr. 2019, 1861, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
- Oshiro, K.G.N.; Cândido, E.S.; Chan, L.Y.; Torres, M.D.T.; Monges, B.E.D.; Rodrigues, S.G.; Porto, W.F.; Ribeiro, S.M.; Henriques, S.T.; Lu, T.K.; et al. Computer-Aided Design of Mastoparan-like Peptides Enables the Generation of Nontoxic Variants with Extended Antibacterial Properties. J. Med. Chem. 2019, 62, 8140–8151. [Google Scholar] [CrossRef] [PubMed]
- Fjell, C.D.; Jenssen, H.; Cheung, W.; Hancock, R.; Cherkasov, A. Optimization of Antibacterial Peptides by Genetic Algorithms and Cheminformatics. Chem. Biol. Drug Des. 2010, 77, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Maccari, G.; Di Luca, M.; Nifosì, R.; Cardarelli, F.; Signore, G.; Boccardi, C.; Bifone, A. Antimicrobial Peptides Design by Evolutionary Multiobjective Optimization. PLoS Comput. Biol. 2013, 9, e1003212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porto, W.F.; Irazazabal, L.; Alves, E.S.F.; Ribeiro, S.M.; Matos, C.O.; Pires, Á.S.; Fensterseifer, I.C.M.; Miranda, V.J.; Haney, E.F.; Humblot, V.; et al. In silico optimization of a guava antimicrobial peptide enables combinatorial exploration for peptide design. Nat. Commun. 2018, 9, 1490. [Google Scholar] [CrossRef]
- Yoshida, M.; Hinkley, T.; Tsuda, S.; Abul-Haija, Y.; McBurney, R.T.; Kulikov, V.; Mathieson, J.S.; Reyes, S.G.; Castro, M.D.; Cronin, L. Using Evolutionary Algorithms and Machine Learning to Explore Sequence Space for the Discovery of Antimicrobial Peptides. Chem 2018, 4, 533–543. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Fan, L.; Sun, J.; Lao, X.; Zheng, H. Computational resources and tools for antimicrobial peptides. J. Pept. Sci. 2017, 23, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Wang, P.; Lin, W.-Z.; Jia, J.-H.; Chou, K.-C. iAMP-2L: A two-level multi-label classifier for identifying antimicrobial peptides and their functional types. Anal. Biochem. 2013, 436, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Schierz, A.C. Virtual screening of bioassay data. J. Cheminform. 2009, 1, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhadra, P.; Yan, J.; Li, J.; Fong, S.; Siu, S.W.I. AmPEP: Sequence-based prediction of antimicrobial peptides using distribution patterns of amino acid properties and random forest. Sci. Rep. 2018, 8, 1697. [Google Scholar] [CrossRef] [Green Version]
- Lata, S.; Mishra, N.K.; Raghava, G.P. AntiBP2: Improved version of antibacterial peptide prediction. BMC Bioinform. 2010, 11, S19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhall, D.; Kaur, R.; Juneja, M. Machine Learning: A Review of the Algorithms and Its Applications. In Proceedings of ICRIC 2019; Springer: Cham, Switzerland, 2020; pp. 47–63. [Google Scholar]
- Lee, E.Y.; Lee, M.W.; Fulan, B.M.; Ferguson, A.L.; Wong, G.C.L. What can machine learning do for antimicrobial peptides, and what can antimicrobial peptides do for machine learning? Interface Focus 2017, 7, 20160153. [Google Scholar] [CrossRef] [PubMed]
- Burdukiewicz, M.; Sidorczuk, K.; Rafacz, D.; Pietluch, F.; Chilimoniuk, J.; Rödiger, S.; Gagat, P. Proteomic Screening for Prediction and Design of Antimicrobial Peptides with AmpGram. Int. J. Mol. Sci. 2020, 21, 4310. [Google Scholar] [CrossRef]
- Chung, C.-R.; Jhong, J.-H.; Wang, Z.; Chen, S.; Wan, Y.; Horng, J.-T.; Lee, T.-Y. Characterization and Identification of Natural Antimicrobial Peptides on Different Organisms. Int. J. Mol. Sci. 2020, 21, 986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Hu, L.; Liu, G.; Jiang, N.; Chen, X.; Xu, J.; Zheng, W.; Li, L.; Tan, M.; Chen, Z.; et al. Prediction of Antimicrobial Peptides Based on Sequence Alignment and Feature Selection Methods. PLoS ONE 2011, 6, e18476. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, P.; Raghava, G.P.S. Prediction of Antimicrobial Potential of a Chemically Modified Peptide From Its Tertiary Structure. Front. Microbiol. 2018, 9, 2551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gull, S.; Shamim, N.; Minhas, F. AMAP: Hierarchical multi-label prediction of biologically active and antimicrobial peptides. Comput. Biol. Med. 2019, 107, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Torrent, M.; Nogués, V.M.; Boix, E. A theoretical approach to spot active regions in antimicrobial proteins. BMC Bioinform. 2009, 10, 373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waghu, F.H.; Barai, R.S.; Gurung, P.; Idicula-Thomas, S. CAMP R3: A database on sequences, structures and signatures of antimicrobial peptides: Table 1. Nucleic Acids Res. 2016, 44, D1094–D1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.; Xu, D. Imbalanced multi-label learning for identifying antimicrobial peptides and their functional types. Bioinformatics 2016, 32, 3745–3752. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Bhadra, P.; Li, A.; Sethiya, P.; Qin, L.; Tai, H.K.; Wong, K.H.; Siu, S.W. Deep-AmPEP30: Improve Short Antimicrobial Peptides Prediction with Deep Learning. Mol. Ther.-Nucleic Acids 2020, 20, 882–894. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Xu, J.; Yin, Y.; Quan, X.; Zhang, H. Antimicrobial peptide identification using multi-scale convolutional network. BMC Bioinform. 2019, 20, 730. [Google Scholar] [CrossRef] [Green Version]
- Schneider, P.; Müller, A.T.; Gabernet, G.; Button, A.L.; Posselt, G.; Wessler, S.; Hiss, J.A.; Schneider, G. Hybrid Network Model for “Deep Learning” of Chemical Data: Application to Antimicrobial Peptides. Mol. Inform. 2017, 36, 1600011. [Google Scholar] [CrossRef] [PubMed]
- Witten, J.; Witten, Z. Deep learning regression model for antimicrobial peptide design. bioRxiv 2019, 692681. [Google Scholar] [CrossRef] [Green Version]
- Beltran, J.A.; Aguilera-Mendoza, L.; Brizuela, C.A. Optimal selection of molecular descriptors for antimicrobial peptides classification: An evolutionary feature weighting approach. BMC Genom. 2018, 19, 672. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Cao, Z.; Li, M.; Wang, S. ACEP: Improving antimicrobial peptides recognition through automatic feature fusion and amino acid embedding. BMC Genom. 2020, 21, 597. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.T.; Kaymaz, A.C.; Gabernet, G.; Posselt, G.; Wessler, S.; Hiss, J.A.; Schneider, G. Sparse Neural Network Models of Antimicrobial Peptide-Activity Relationships. Mol. Inform. 2016, 35, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Hamid, M.-N.; Friedberg, I. Identifying antimicrobial peptides using word embedding with deep recurrent neural networks. Bioinformatics 2019, 35, 2009–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Sutherland, D.; Hammond, S.A.; Yang, C.; Taho, F.; Bergman, L.; Houston, S.; Warren, R.L.; Wong, T.; Hoang, L.M.N.; et al. AMPlify: Attentive deep learning model for discovery of novel antimicrobial peptides effective against WHO priority pathogens. BMC Genom. 2022, 23, 77. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Bao, J.; Lao, X.; Zheng, H. Novel 3D Structure Based Model for Activity Prediction and Design of Antimicrobial Peptides. Sci. Rep. 2018, 8, 11189. [Google Scholar] [CrossRef] [PubMed]
- Capecchi, A.; Cai, X.; Personne, H.; Köhler, T.; van Delden, C.; Reymond, J.-L. Machine learning designs non-hemolytic antimicrobial peptides. Chem. Sci. 2021, 12, 9221–9232. [Google Scholar] [CrossRef] [PubMed]
- Vishnepolsky, B.; Gabrielian, A.; Rosenthal, A.; Hurt, D.E.; Tartakovsky, M.; Managadze, G.; Grigolava, M.; Makhatadze, G.I.; Pirtskhalava, M. Predictive Model of Linear Antimicrobial Peptides Active against Gram-Negative Bacteria. J. Chem. Inf. Model. 2018, 58, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Vishnepolsky, B.; Grigolava, M.; Zaalishvili, G.; Karapetian, M.; Pirtskhalava, M. DBAASP Special prediction as a tool for the prediction of antimicrobial potency against particular target species. In Proceedings of the 4th International Electronic Conference on Medicinal Chemistry, Basel, Switzerland, 1–30 November 2018. [Google Scholar]
- Plisson, F.; Ramírez-Sánchez, O.; Martínez-Hernández, C. Machine learning-guided discovery and design of non-hemolytic peptides. Sci. Rep. 2020, 10, 16581. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuka, Y.; Inagaki, H. In silico identification and functional validation of linear cationic α-helical antimicrobial peptides in the ascidian Ciona intestinalis. Sci. Rep. 2020, 10, 12619. [Google Scholar] [CrossRef]
- Wiegand, I.; Hilpert, K.; Hancock, R.E.W. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatices 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Vishnepolsky, B.; Pirtskhalava, M. Comment on: ‘Empirical comparison of web-based antimicrobial peptide prediction tools’. Bioinformatics 2019, 35, 2692–2694. [Google Scholar] [CrossRef]
- Lee, J.H.; Chung, H.; Shin, Y.P.; Kim, I.-W.; Natarajan, S.; Veerappan, K.; Seo, M.; Park, J.; Hwang, J.S. Transcriptome Analysis of Psacothea hilaris: De Novo Assembly and Antimicrobial Peptide Prediction. Insects 2020, 11, 676. [Google Scholar] [CrossRef]
- Fernandes, F.C.; Rigden, D.; Franco, O.L. Prediction of antimicrobial peptides based on the adaptive neuro-fuzzy inference system application. Biopolymers 2012, 98, 280–287. [Google Scholar] [CrossRef]
- Veltri, D.; Kamath, U.; Shehu, A. Deep learning improves antimicrobial peptide recognition. Bioinformatics 2018, 34, 2740–2747. [Google Scholar] [CrossRef] [Green Version]
- Gautam, A.; Sharma, A.; Jaiswal, S.; Fatma, S.; Arora, V.; Iquebal, M.A.; Nandi, S.; Sundaray, J.K.; Jayasankar, P.; Rai, A.; et al. Development of Antimicrobial Peptide Prediction Tool for Aquaculture Industries. Probiotics Antimicrob. Proteins 2016, 8, 141–149. [Google Scholar] [CrossRef]
- Gabere, M.N.; Noble, W.S. Empirical comparison of web-based antimicrobial peptide prediction tools. Bioinformatics 2017, 33, 1921–1929. [Google Scholar] [CrossRef] [PubMed]
- Waghu, F.H.; Gopi, L.; Barai, R.S.; Ramteke, P.; Nizami, B.; Idicula-Thomas, S. CAMP: Collection of sequences and structures of antimicrobial peptides. Nucleic Acids Res. 2014, 42, D1154–D1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.-Y.; Fu, R.; Luo, P.-Y.; Hong, Y.; Huang, Y.-H. Construction and Prediction of Antimicrobial Peptide Predicition Model Based on BERT. Available online: https://jasonyanglu.github.io/files/lecture_notes/%E6%B7%B1%E5%BA%A6%E5%AD%A6%E4%B9%A0_2020/Project/Construction%20and%20Prediction%20of%20Antimicrobial%20Peptide.pdf (accessed on 16 December 2021).
- Spänig, S.; Heider, D. Encodings and models for antimicrobial peptide classification for multi-resistant pathogens. BioData Min. 2019, 12, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khabbaz, H.; Karimi-Jafari, M.H.; Saboury, A.A.; BabaAli, B. Prediction of antimicrobial peptides toxicity based on their physico-chemical properties using machine learning techniques. BMC Bioinform. 2021, 22, 549. [Google Scholar] [CrossRef] [PubMed]
- Moretta, A.; Salvia, R.; Scieuzo, C.; Di Somma, A.; Vogel, H.; Pucci, P.; Sgambato, A.; Wolff, M.; Falabella, P. A bioinformatic study of antimicrobial peptides identified in the Black Soldier Fly (BSF) Hermetia illucens (Diptera: Stratiomyidae). Sci. Rep. 2020, 10, 16875. [Google Scholar] [CrossRef] [PubMed]
- Vishnepolsky, B.; Pirtskhalava, M. Prediction of Linear Cationic Antimicrobial Peptides Based on Characteristics Responsible for Their Interaction with the Membranes. J. Chem. Inf. Model. 2014, 54, 1512–1523. [Google Scholar] [CrossRef] [PubMed]
- Thakur, N.; Qureshi, A.; Kumar, M. AVPpred: Collection and prediction of highly effective antiviral peptides. Nucleic Acids Res. 2012, 40, W199–W204. [Google Scholar] [CrossRef] [Green Version]
- Lira, F.; Perez, P.S.; Baranauskas, J.A.; Nozawa, S.R. Prediction of Antimicrobial Activity of Synthetic Peptides by a Decision Tree Model. Appl. Environ. Microbiol. 2013, 79, 3156–3159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pane, K.; Durante, L.; Crescenzi, O.; Cafaro, V.; Pizzo, E.; Varcamonti, M.; Zanfardino, A.; Izzo, V.; Di Donato, A.; Notomista, E. Antimicrobial potency of cationic antimicrobial peptides can be predicted from their amino acid composition: Application to the detection of “cryptic” antimicrobial peptides. J. Theor. Biol. 2017, 419, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhao, P.; Li, F.; Marquez-Lago, T.T.; Leier, A.; Revote, J.; Zhu, Y.; Powell, D.R.; Akutsu, T.; Webb, G.I.; et al. iLearn: An integrated platform and meta-learner for feature engineering, machine-learning analysis and modeling of DNA, RNA and protein sequence data. Briefings Bioinform. 2020, 21, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Muhammod, R.; Ahmed, S.; Farid, D.M.; Shatabda, S.; Sharma, A.; Dehzangi, A. PyFeat: A Python-based effective feature generation tool for DNA, RNA and protein sequences. Bioinformatics 2019, 35, 3831–3833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikam, R.; Gromiha, M.M. Seq2Feature: A comprehensive web-based feature extraction tool. Bioinformatics 2019, 35, 4797–4799. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.-S.; Xu, Q.-S.; Liang, Y.-Z. Propy: A tool to generate various modes of Chou’s PseAAC. Bioinformatics 2013, 29, 960–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Zhao, P.; Li, F.; Leier, A.; Marquez-Lago, T.T.; Wang, Y.; Webb, G.I.; Smith, A.I.; Daly, R.J.; Chou, K.-C.; et al. iFeature: A Python package and web server for features extraction and selection from protein and peptide sequences. Bioinformatics 2018, 34, 2499–2502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, J.; Yao, Z.-J.; Zhang, L.; Luo, F.; Lin, Q.; Lu, A.-P.; Chen, A.F.; Cao, D.-S. PyBioMed: A python library for various molecular representations of chemicals, proteins and DNAs and their interactions. J. Cheminform. 2018, 10, 16. [Google Scholar] [CrossRef] [Green Version]
- Mahmud, S.H.; Chen, W.; Meng, H.; Jahan, H.; Liu, Y.; Hasan, S.M. Prediction of drug-target interaction based on protein features using undersampling and feature selection techniques with boosting. Anal. Biochem. 2020, 589, 113507. [Google Scholar] [CrossRef] [PubMed]
- Yeh, S.-J.; Lin, J.-F.; Chen, B.-S. Multiple-Molecule Drug Design Based on Systems Biology Approaches and Deep Neural Network to Mitigate Human Skin Aging. Molecules 2021, 26, 3178. [Google Scholar] [CrossRef]
- Yeh, S.-J.; Chung, Y.-C.; Chen, B.-S. Investigating the Role of Obesity in Prostate Cancer and Identifying Biomarkers for Drug Discovery: Systems Biology and Deep Learning Approaches. Molecules 2022, 27, 900. [Google Scholar] [CrossRef]
- Wani, M.A.; Garg, P.; Roy, K.K. Machine learning-enabled predictive modeling to precisely identify the antimicrobial peptides. Med. Biol. Eng. Comput. 2021, 59, 2397–2408. [Google Scholar] [CrossRef] [PubMed]
- Freund, Y.; Schapire, R.E. A Short Introduction to Boosting. J. Jpn. Soc. Artif. Intell. 1999, 14, 771–780. [Google Scholar]
- Friedman, J.; Hastie, T.; Tibshirani, R. Additive logistic regression: A statistical view of boosting (With discussion and a rejoinder by the authors). Ann. Stat. 2000, 28, 337–407. [Google Scholar] [CrossRef]
- Breiman, L.; Friedman, J.H.; Olshen, R.A.; Stone, C.J. Classification and Regression Trees, 1st ed.; Routledge: Boca Raton, FL, USA, 2017. [Google Scholar] [CrossRef]
- Ho, T.K. Random Decision Forests. In Proceedings of the 3rd International Conference on Document Analysis and Recognition, Montreal, QC, Canada, 14–16 August 1995; pp. 278–282. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Fix, E.; Hodges, J.L. Discriminatory Analysis. Nonparametric Discrimination: Consistency Properties. Int. Stat. Rev. Rev. Int. Stat. 1989, 57, 238–247. [Google Scholar] [CrossRef]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Thiel, K.; Wiswedel, B. KNIME—The Konstanz information miner. ACM SIGKDD Explor. Newsl. 2009, 11, 26–31. [Google Scholar] [CrossRef] [Green Version]
- Randles, B.M.; Pasquetto, I.V.; Golshan, M.S.; Borgman, C.L. Using the Jupyter Notebook as a Tool for Open Science: An Empirical Study. In Proceedings of the 2017 ACM/IEEE Joint Conference on Digital Libraries (JCDL), Toronto, ON, Canada, 19–23 June 2017; pp. 1–2. [Google Scholar]
- Xu, Q.-S.; Liang, Y.-Z. Monte Carlo cross validation. Chemom. Intell. Lab. Syst. 2001, 56, 1–11. [Google Scholar] [CrossRef]
- Jovic, A.; Brkic, K.; Bogunovic, N. A review of feature selection methods with applications. In Proceedings of the 2015 38th International Convention on Information and Communication Technology, Electronics and Microelectronics (MIPRO), Opatija, Croatia, 25–29 May 2015; pp. 1200–1205. [Google Scholar]
- Yousef, M.; Jung, S.; Showe, L.C.; Showe, M.K. Recursive Cluster Elimination (RCE) for classification and feature selection from gene expression data. BMC Bioinform. 2007, 8, 144. [Google Scholar] [CrossRef] [Green Version]
- Yousef, M.; Bakir-Gungor, B.; Jabeer, A.; Goy, G.; Qureshi, R.; Showe, L.C. Recursive Cluster Elimination based Rank Function (SVM-RCE-R) implemented in KNIME. F1000Research 2021, 9, 1255. [Google Scholar] [CrossRef] [PubMed]
- Yousef, M.; Jabeer, A.; Bakir-Gungor, B. SVM-RCE-R-OPT: Optimization of Scoring Function for SVM-RCE-R. In Database and Expert Systems Applications—DEXA 2021 Workshops; Kotsis, G., Tjoa, A.M., Khalil, I., Moser, B., Mashkoor, A., Sametinger, J., Fensel, A., Martinez-Gil, J., Fischer, L., Czech, G., et al., Eds.; Springer International Publishing: Cham, Switzerland, 2021; Volume 1479, pp. 215–224. [Google Scholar] [CrossRef]
- Yousef, M.; Abdallah, L.; Allmer, J.; Abddallah, L. maTE: Discovering expressed interactions between microRNAs and their targets. Bioinformatics 2019, 35, 4020–4028. [Google Scholar] [CrossRef] [PubMed]
- Yousef, M.; Ülgen, E.; Sezerman, O.U. CogNet: Classification of gene expression data based on ranked active-subnetwork-oriented KEGG pathway enrichment analysis. PeerJ Comput. Sci. 2021, 7, e336. [Google Scholar] [CrossRef] [PubMed]
- Yousef, M.; Goy, G.; Mitra, R.; Eischen, C.M.; Jabeer, A.; Bakir-Gungor, B. miRcorrNet: Machine learning-based integration of miRNA and mRNA expression profiles, combined with feature grouping and ranking. PeerJ 2021, 9, e11458. [Google Scholar] [CrossRef] [PubMed]
- Yousef, M.; Goy, G.; Bakir-Gungor, B. miRModuleNet: Detecting miRNA-mRNA Regulatory Modules. Front. Genet. 2022, 13, 767455. [Google Scholar] [CrossRef]
- Yousef, M.; Sayıcı, A.; Bakir-Gungor, B. Integrating Gene Ontology Based Grouping and Ranking into the Machine Learning Algorithm for Gene Expression Data Analysis. In Database and Expert Systems Applications—DEXA 2021 Workshops; Kotsis, G., Tjoa, A.M., Khalil, I., Moser, B., Mashkoor, A., Sametinger, J., Fensel, A., Martinez-Gil, J., Fischer, L., Czech, G., et al., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 205–214. [Google Scholar] [CrossRef]
- Yousef, M.; Kumar, A.; Bakir-Gungor, B. Application of Biological Domain Knowledge Based Feature Selection on Gene Expression Data. Entropy 2020, 23, 2. [Google Scholar] [CrossRef] [PubMed]
- Pearson, K. On lines and planes of closest fit to systems of points in space. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1901, 2, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Porto, W.F.; Pires, Á.S.; Franco, O.L. CS-AMPPred: An Updated SVM Model for Antimicrobial Activity Prediction in Cysteine-Stabilized Peptides. PLoS ONE 2012, 7, e51444. [Google Scholar] [CrossRef] [PubMed]
- Shu, M.; Yu, R.; Zhang, Y.; Wang, J.; Yang, L.; Wang, L.; Lin, Z. Predicting the Activity of Antimicrobial Peptides with Amino Acid Topological Information. Med. Chem. 2013, 9, 32–44. [Google Scholar] [CrossRef] [PubMed]
- Moll, L.; Badosa, E.; Planas, M.; Feliu, L.; Montesinos, E.; Bonaterra, A. Antimicrobial Peptides with Antibiofilm Activity against Xylella fastidiosa. Front. Microbiol. 2021, 12, 753874. [Google Scholar] [CrossRef]
- Lin, H.; Yan, T.; Wang, L.; Guo, F.; Ning, G.; Xiong, M. Statistical design, structural analysis, andin vitrosusceptibility assay of antimicrobial peptoids to combat bacterial infections. J. Chemom. 2016, 30, 369–376. [Google Scholar] [CrossRef]
- Thudumu, S.; Branch, P.; Jin, J.; Singh, J. A comprehensive survey of anomaly detection techniques for high dimensional big data. J. Big Data 2020, 7, 1–30. [Google Scholar] [CrossRef]
- Manevitz, L.M.; Yousef, M. One-Class SVMs for Document Classification. J. Mach. Learn. Res. 2001, 2, 139–154. [Google Scholar]
- Manevitz, L.; Yousef, M. One-class document classification via Neural Networks. Neurocomputing 2007, 70, 1466–1481. [Google Scholar] [CrossRef]
- Abdallah, L.; Badarna, M.; Khalifa, W.; Yousef, M. MultiKOC: Multi-One-Class Classifier Based K-Means Clustering. Algorithms 2021, 14, 134. [Google Scholar] [CrossRef]
- Abedalla, L.; Badarna, M.; Khalifa, W.; Yousef, M. K-Means Based One-Class SVM Classifier. In Database and Expert Systems Applications; Anderst-Kotsis, G., Tjoa, A.M., Khalil, I., Elloumi, M., Mashkoor, A., Sametinger, J., Larrucea, X., Fensel, A., Martinez-Gil, J., Moser, B., et al., Eds.; Springer International Publishing: Cham, Switzerland, 2019; Volume 1062, pp. 45–53. [Google Scholar] [CrossRef]
- Yousef, M.; Khalifa, W.; Abedallah, L. Ensemble Clustering Classification compete SVM and One-Class classifiers applied on plant microRNAs Data. J. Integr. Bioinform. 2016, 13, 304. [Google Scholar] [CrossRef] [PubMed]
- Kent, J.T. Information gain and a general measure of correlation. Biometrika 1983, 70, 163–173. [Google Scholar] [CrossRef]
- Brown, G.; Pocock, A.; Zhao, M.-J.; Lujan, M. Conditional Likelihood Maximisation: A Unifying Framework for Information Theoretic Feature Selection. J. Mach. Learn. Res. 2012, 13, 27–66. [Google Scholar]
- Fleuret, F. Fast Binary Feature Selection with Conditional Mutual Information. J. Mach. Learn. Res. 2004, 5, 1531–1555. [Google Scholar]
- Pedregosa, F. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Chen, T.; He, T. xgboost: eXtreme Gradient Boosting. Available online: https://cran.microsoft.com/snapshot/2017-12-11/web/packages/xgboost/vignettes/xgboost.pdf (accessed on 8 March 2022).
- Senliol, B.; Gulgezen, G.; Yu, L.; Cataltepe, Z. Fast Correlation Based Filter (FCBF) with a different search strategy. In Proceedings of the 2008 23rd International Symposium on Computer and Information Sciences, Istanbul, Turkey, 27–29 October 2008; pp. 1–4. [Google Scholar]
- Pirtskhalava, M.; Grigolava, M. Transmembrane and Antimicrobial Peptides. Hydrophobicity, Amphiphilicity and Propensity to Aggregation. arXiv 2013, arXiv:1307.6160. [Google Scholar]
- Kumar, P.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shai, Y. Mode of action of membrane active antimicrobial peptides. Biopolymers 2002, 66, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Osorio, D.; Rondón-Villarreal, P.; Torres, R.T.R. Peptides: A Package for Data Mining of Antimicrobial Peptides. R J. 2015, 7, 4–14. [Google Scholar] [CrossRef]
- Romestand, B.; Molina, F.; Richard, V.; Roch, P.; Granier, A.C. Key role of the loop connecting the two beta strands of mussel defensin in its antimicrobial activity. J. Biol. Inorg. Chem. 2003, 270, 2805–2813. [Google Scholar] [CrossRef]
- Bezerra, I.; Moreira, L.; Chiavone-Filho, O.; Mattedi, S. Effect of different variables in the solubility of ampicillin and corresponding solid phase. Fluid Phase Equilibria 2018, 459, 18–29. [Google Scholar] [CrossRef]
- Le, H.; Ting, L.; Jun, C.; Weng, W. Gelling properties of myofibrillar protein from abalone (Haliotis Discus Hannai Ino) muscle. Int. J. Food Prop. 2018, 21, 277–288. [Google Scholar] [CrossRef] [Green Version]
- Ni, N.; Wang, Z.; He, F.; Wang, L.; Pan, H.; Li, X.; Wang, Q.; Zhang, D. Gel properties and molecular forces of lamb myofibrillar protein during heat induction at different pH values. Process Biochem. 2014, 49, 631–636. [Google Scholar] [CrossRef]
- Ahn, H.-S.; Cho, W.; Kang, S.-H.; Ko, S.-S.; Park, M.-S.; Cho, H.; Lee, K.-H. Design and synthesis of novel antimicrobial peptides on the basis of α helical domain of Tenecin 1, an insect defensin protein, and structure-activity relationship study. Peptides 2006, 27, 640–648. [Google Scholar] [CrossRef]
- Pirtskhalava, M.; Vishnepolsky, B.; Grigolava, M. Physicochemical Features and Peculiarities of Interaction of Antimicrobial Peptides with the Membrane. Pharmaceuticals 2021, 14, 471. [Google Scholar] [CrossRef] [PubMed]
- Papo, N.; Shai, Y. Can we predict biological activity of antimicrobial peptides from their interactions with model phospholipid membranes? Peptides 2003, 24, 1693–1703. [Google Scholar] [CrossRef]
- Teixeira, V.; Feio, M.J.; Bastos, M. Role of lipids in the interaction of antimicrobial peptides with membranes. Prog. Lipid Res. 2012, 51, 149–177. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Guarnieri, M.T.; Vasil, A.I.; Vasil, M.L.; Mant, C.T.; Hodges, R.S. Role of Peptide Hydrophobicity in the Mechanism of Action of α-Helical Antimicrobial Peptides. Antimicrob. Agents Chemother. 2007, 51, 1398–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenberg, D.; Weiss, R.M.; Terwilliger, T. The helical hydrophobic moment: A measure of the amphiphilicity of a helix. Nature 1982, 299, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Yousef, M.; Levy, D.; Allmer, J. Species Categorization via MicroRNAs—Based on 3′UTR Target Sites using Sequence Features. In Proceedings of the 11th International Joint Conference on Biomedical Engineering Systems and Technologies, Funchal, Portugal, 19–21 January 2018; pp. 112–118. [Google Scholar]
- Yousef, M.; Khalifa, W.; Acar, I.E.; Allmer, J. Distinguishing between MicroRNA Targets from Diverse Species using Sequence Motifs and K-mers. In Proceedings of the 10th International Joint Conference on Biomedical Engineering Systems and Technologies, Porto, Portugal, 21–23 February 2017; pp. 133–139. [Google Scholar]
- Yousef, M.; Khalifa, W.; Acar, I.E.; Allmer, J. MicroRNA categorization using sequence motifs and k-mers. BMC Bioinform. 2017, 18, 170. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of Sequence | Sequence | Seq. Length | Norm. Hyd. Moment | Norm. Hyd. | Net Charge | Isoelectric Point | Penet. Depth | Tilt Angle | Disordered Conf. Propensity | Linear Moment | Propensity In Vitro Aggregation | Mean MIC | Class (AMP Category) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| XPF-B2 | GWASKIGTQLGKMAKVGLKEFVQS | 24 | 1.11 | −0.25 | 3 | 10.7 | 15 | 76 | 0.09 | 0.16 | 0 | 256.81 | 0 |
| Ovalbumin (271–290) | SNVMEERKIKVYLPRMKMEE | 20 | 0.13 | −0.28 | 1 | 9.38 | 30 | 67 | −0.11 | 0.29 | 0 | 800 | 0 |
| MBI 29 A1 | KWKSFIKKLTSVLKKVVTTALPALIS | 26 | 1.03 | −0.54 | 6 | 11.37 | 12 | 106 | 0.16 | 0.27 | 3.4 | 9.33 | 1 |
| Cyanophlyctin | FLNALKNFAKTAGKRLKSLLN | 21 | 1.69 | −0.24 | 5 | 11.74 | 15 | 88 | −0.03 | 0.25 | 0 | 12 | 1 |
| Model | Accuracy | Recall | Specificity | Precision | Area Under Curve | F1 | Balanced Acc. |
|---|---|---|---|---|---|---|---|
| AdaBoost | 0.85 ± 0.06 | 0.92 ± 0.06 | 0.72 ± 0.20 | 0.87 ± 0.07 | 0.88 ± 0.06 | 0.89 ± 0.04 | 0.82 ± 0.13 |
| Decision Tree | 0.79 ± 0.06 | 0.87 ± 0.07 | 0.66 ± 0.24 | 0.84 ± 0.07 | 0.78 ± 0.07 | 0.85 ± 0.04 | 0.76 ± 0.15 |
| LogitBoost | 0.86 ± 0.05 | 0.92 ± 0.06 | 0.74 ± 0.16 | 0.88 ± 0.06 | 0.89 ± 0.06 | 0.90 ± 0.03 | 0.83 ± 0.11 |
| RF | 0.89 ± 0.05 | 0.93 ± 0.04 | 0.79 ± 0.16 | 0.90 ± 0.06 | 0.92 ± 0.05 | 0.91 ± 0.03 | 0.86 ± 0.10 |
| SVM | 0.80 ± 0.05 | 0.93 ± 0.06 | 0.56 ± 0.21 | 0.81 ± 0.07 | 0.82 ± 0.06 | 0.86 ± 0.03 | 0.74 ± 0.13 |
| SVM + kNN | 0.80 ± 0.07 | 0.93 ± 0.05 | 0.56 ± 0.25 | 0.81 ± 0.08 | 0.82 ± 0.08 | 0.86 ± 0.04 | 0.74 ± 0.15 |
| LogitBoost + kNN | 0.80 ± 0.05 | 0.93 ± 0.06 | 0.56 ± 0.21 | 0.81 ± 0.07 | 0.82 ± 0.06 | 0.86 ± 0.03 | 0.74 ± 0.13 |
| Model | Accuracy | Recall | Specificity | Precision | Area Under Curve | F1 | Balanced Acc. |
|---|---|---|---|---|---|---|---|
| AdaBoost | 0.84 ± 0.06 | 0.85 ± 0.08 | 0.83 ± 0.14 | 0.83 ± 0.10 | 0.86 ± 0.06 | 0.83 ± 0.05 | 0.84 ± 0.11 |
| Decision Tree | 0.77 ± 0.07 | 0.77 ± 0.10 | 0.77 ± 0.16 | 0.769 ± 0.09 | 0.77 ± 0.06 | 0.76 ± 0.05 | 0.77 ± 0.13 |
| LogitBoost | 0.83 ± 0.06 | 0.84 ± 0.09 | 0.82 ± 0.15 | 0.83 ± 0.10 | 0.87 ± 0.05 | 0.83 ± 0.05 | 0.83 ± 0.12 |
| RF | 0.87 ± 0.04 | 0.87 ± 0.07 | 0.87 ± 0.08 | 0.87 ± 0.07 | 0.90 ± 0.04 | 0.87 ± 0.04 | 0.87 ± 0.07 |
| SVM | 0.77 ± 0.07 | 0.85 ± 0.11 | 0.71 ± 0.19 | 0.75 ± 0.12 | 0.81 ± 0.06 | 0.78 ± 0.05 | 0.78 ± 0.15 |
| SVM + kNN | 0.76 ± 0.08 | 0.81 ± 0.11 | 0.72 ± 0.21 | 0.76 ± 0.13 | 0.80 ± 0.07 | 0.77 ± 0.05 | 0.76 ± 0.16 |
| LogitBoost + kNN | 0.77 ± 0.07 | 0.85 ± 0.11 | 0.71 ± 0.19 | 0.75 ± 0.12 | 0.81 ± 0.06 | 0.78 ± 0.05 | 0.78 ± 0.15 |
| Features | Gram-Negative Dataset | Gram-Positive Dataset | ||
|---|---|---|---|---|
| Minimum Threshold | Maximum Threshold | Minimum Threshold | Maximum Threshold | |
| Hydrophobic Moment | 0.4 | 2 | 0.1 | 1.7 |
| Normalized Hydrophobicity | −0.9 | 0.55 | −0.8 | 1 |
| Net Charge | 5 | 13 | 4 | 13 |
| Isoelectric Point | 10.5 | 13 | 10 | 13 |
| Penetration Depth | 13 | 30 | 12 | 30 |
| Tilt Angle | 40 | 150 | 30 | 152 |
| Linear Moment | 0.1 | 0.4 | 0.15 | 0.32 |
| Propensity in vitro Aggregation | 0 | 250 | 0 | 87 |
| Disordered Conformation Propensity | −0.5 | 0.08 | −0.85 | 0.15 |
| Model | Accuracy | Recall | Specificity | Precision | Area Under Curve | F1 | Balanced Acc. |
|---|---|---|---|---|---|---|---|
| AdaBoost | 0.97 ± 0.03 | 0.99 ± 0.03 | 0.96 ± 0.04 | 0.95 ± 0.05 | 0.99 ± 0.01 | 0.97 ± 0.03 | 0.97 ± 0.04 |
| Decision Tree | 0.91 ± 0.06 | 0.92 ± 0.08 | 0.91 ± 0.08 | 0.89 ± 0.09 | 0.91 ± 0.06 | 0.90 ± 0.06 | 0.91 ± 0.08 |
| LogitBoost | 0.97 ± 0.03 | 0.99 ± 0.02 | 0.96 ± 0.05 | 0.95 ± 0.05 | 0.99 ± 0.01 | 0.97 ± 0.03 | 0.98 ± 0.03 |
| RF | 0.98 ± 0.02 | 0.99 ± 0.02 | 0.97 ± 0.04 | 0.97 ± 0.05 | 0.99 ± 0.01 | 0.98 ± 0.03 | 0.98 ± 0.03 |
| SVM | 0.98 ± 0.02 | 0.99 ± 0.03 | 0.97 ± 0.04 | 0.96 ± 0.04 | 0.98 ± 0.01 | 0.97 ± 0.03 | 0.98 ± 0.03 |
| SVM + kNN | 0.81 ± 0.11 | 0.82 ± 0.14 | 0.80 ± 0.24 | 0.81 ± 0.16 | 0.84 ± 0.10 | 0.80 ± 0.09 | 0.81 ± 0.19 |
| LogitBoost + kNN | 0.98 ± 0.02 | 0.99 ± 0.03 | 0.97 ± 0.04 | 0.96 ± 0.04 | 0.98 ± 0.01 | 0.97 ± 0.03 | 0.98 ± 0.03 |
| Model | Accuracy | Recall | Specificity | Precision | Area Under Curve | F1 | Balanced Acc. |
|---|---|---|---|---|---|---|---|
| AdaBoost | 0.93 ± 0.04 | 0.92 ± 0.08 | 0.94 ± 0.06 | 0.89 ± 0.09 | 0.96 ± 0.03 | 0.90 ± 0.05 | 0.93 ± 0.07 |
| Decision Tree | 0.88 ± 0.05 | 0.82 ± 0.12 | 0.91 ± 0.06 | 0.82 ± 0.11 | 0.86 ± 0.07 | 0.81 ± 0.09 | 0.86 ± 0.09 |
| LogitBoost | 0.93 ± 0.05 | 0.93 ± 0.09 | 0.93 ± 0.07 | 0.88 ± 0.11 | 0.96 ± 0.03 | 0.90 ± 0.07 | 0.93 ± 0.08 |
| RF | 0.95 ± 0.03 | 0.95 ± 0.07 | 0.95 ± 0.05 | 0.90 ± 0.09 | 0.97 ± 0.02 | 0.92 ± 0.05 | 0.95 ± 0.06 |
| SVM | 0.91 ± 0.04 | 0.90 ± 0.11 | 0.91 ± 0.06 | 0.85 ± 0.11 | 0.93 ± 0.04 | 0.86 ± 0.06 | 091 ± 0.09 |
| SVM + kNN | 0.77 ± 0.10 | 0.75 ± 0.16 | 0.78 ± 0.20 | 0.68 ± 0.17 | 0.81 ± 0.08 | 0.68 ± 0.08 | 0.76 ± 0.18 |
| LogitBoost + kNN | 0.91 ± 0.04 | 0.90 ± 0.11 | 0.91 ± 0.06 | 0.85 ± 0.11 | 0.93 ± 0.04 | 0.86 ± 0.04 | 0.91 ± 0.09 |
| Model | Accuracy | Recall | Specificity | Precision | Area Under Curve | F1 | Balanced Acc. |
|---|---|---|---|---|---|---|---|
| AdaBoost | 0.96 ± 0.03 | 0.98 ± 0.04 | 0.95 ± 0.05 | 0.94 ± 0.06 | 0.98 ± 0.02 | 0.96 ± 0.03 | 0.96 ± 0.05 |
| Decision Tree | 0.90 ± 0.06 | 0.90 ± 0.09 | 0.90 ± 0.08 | 0.88 ± 0.09 | 0.90 ± 0.06 | 0.88 ± 0.07 | 0.90 ± 0.08 |
| LogitBoost | 0.97 ± 0.03 | 0.98 ± 0.03 | 0.95 ± 0.06 | 0.95 ± 0.06 | 0.98 ± 0.01 | 0.96 ± 0.03 | 0.97 ± 0.04 |
| RF | 0.95 ± 0.04 | 0.98 ± 0.04 | 0.94 ± 0.06 | 0.93 ± 0.07 | 0.98 ± 0.02 | 0.95 ± 0.04 | 0.96 ± 0.05 |
| Model | Accuracy | Recall | Specificity | Precision | Area Under Curve | F1 | Balanced Acc. |
|---|---|---|---|---|---|---|---|
| AdaBoost | 0.89 ± 0.05 | 0.88 ± 0.10 | 0.90 ± 0.08 | 0.82 ± 0.11 | 0.93 ± 0.04 | 0.84 ± 0.07 | 0.89 ± 0.09 |
| Decision Tree | 0.82 ± 0.10 | 0.74 ± 0.14 | 0.86 ± 0.16 | 0.75 ± 0.13 | 0.80 ± 0.07 | 0.73 ± 0.10 | 0.80 ± 0.15 |
| LogitBoost | 0.90 ± 0.05 | 0.89 ± 0.09 | 0.91 ± 0.07 | 0.84 ± 0.11 | 0.94 ± 0.03 | 0.85 ± 0.06 | 0.90 ± 0.08 |
| RF | 0.92 ± 0.04 | 0.91 ± 0.09 | 0.92 ± 0.06 | 0.86 ± 0.10 | 0.95 ± 0.03 | 0.88 ± 0.06 | 0.92 ± 0.08 |
| Model | Accuracy | Recall | Specificity | Precision | Area Under Curve | F1 | Balanced Acc. |
|---|---|---|---|---|---|---|---|
| AdaBoost | 0.94 ± 0.05 | 0.97 ± 0.05 | 0.91 ± 0.09 | 0.91 ± 0.09 | 0.95 ± 0.06 | 0.93 ± 0.07 | 0.94 ± 0.07 |
| Decision Tree | 0.90 ± 0.07 | 0.90 ± 0.11 | 0.89 ± 0.09 | 0.87 ± 0.10 | 0.90 ± 0.08 | 0.88 ± 0.10 | 0.90 ± 0.10 |
| LogitBoost | 0.94 ± 0.05 | 0.98 ± 0.04 | 0.91 ± 0.09 | 0.90 ± 0.09 | 0.96 ± 0.06 | 0.94 ± 0.07 | 0.95 ± 0.06 |
| RF | 0.94 ± 0.05 | 0.97 ± 0.06 | 0.92 ± 0.08 | 0.91 ± 0.08 | 0.96 ± 0.05 | 0.93 ± 0.07 | 0.94 ± 0.07 |
| Model | Accuracy | Recall | Specificity | Precision | Area Under Curve | F1 | Balanced Acc. |
|---|---|---|---|---|---|---|---|
| AdaBoost | 0.86 ± 0.06 | 0.91 ± 0.10 | 0.83 ± 0.10 | 0.74 ± 0.12 | 0.90 ± 0.05 | 0.80 ± 0.07 | 0.87 ± 0.10 |
| Decision Tree | 0.83 ± 0.10 | 0.77 ± 0.12 | 0.86 ± 0.16 | 0.76 ± 0.14 | 0.82 ± 0.07 | 0.75 ± 0.10 | 0.82 ± 0.14 |
| LogitBoost | 0.87 ± 0.05 | 0.90 ± 0.10 | 0.86 ± 0.08 | 0.77 ± 0.11 | 0.91 ± 0.04 | 0.82 ± 0.06 | 0.88 ± 0.09 |
| RF | 0.90 ± 0.04 | 0.89 ± 0.10 | 0.91 ± 0.07 | 0.84 ± 0.11 | 0.94 ± 0.04 | 0.86 ± 0.06 | 0.90 ± 0.08 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Söylemez, Ü.G.; Yousef, M.; Kesmen, Z.; Büyükkiraz, M.E.; Bakir-Gungor, B. Prediction of Linear Cationic Antimicrobial Peptides Active against Gram-Negative and Gram-Positive Bacteria Based on Machine Learning Models. Appl. Sci. 2022, 12, 3631. https://doi.org/10.3390/app12073631
Söylemez ÜG, Yousef M, Kesmen Z, Büyükkiraz ME, Bakir-Gungor B. Prediction of Linear Cationic Antimicrobial Peptides Active against Gram-Negative and Gram-Positive Bacteria Based on Machine Learning Models. Applied Sciences. 2022; 12(7):3631. https://doi.org/10.3390/app12073631
Chicago/Turabian StyleSöylemez, Ümmü Gülsüm, Malik Yousef, Zülal Kesmen, Mine Erdem Büyükkiraz, and Burcu Bakir-Gungor. 2022. "Prediction of Linear Cationic Antimicrobial Peptides Active against Gram-Negative and Gram-Positive Bacteria Based on Machine Learning Models" Applied Sciences 12, no. 7: 3631. https://doi.org/10.3390/app12073631
APA StyleSöylemez, Ü. G., Yousef, M., Kesmen, Z., Büyükkiraz, M. E., & Bakir-Gungor, B. (2022). Prediction of Linear Cationic Antimicrobial Peptides Active against Gram-Negative and Gram-Positive Bacteria Based on Machine Learning Models. Applied Sciences, 12(7), 3631. https://doi.org/10.3390/app12073631