
A Brief Review of Machine Learning-Based Bioactive Compound Research


{kind=link}
{kind=link}
Abstract
1. Introduction
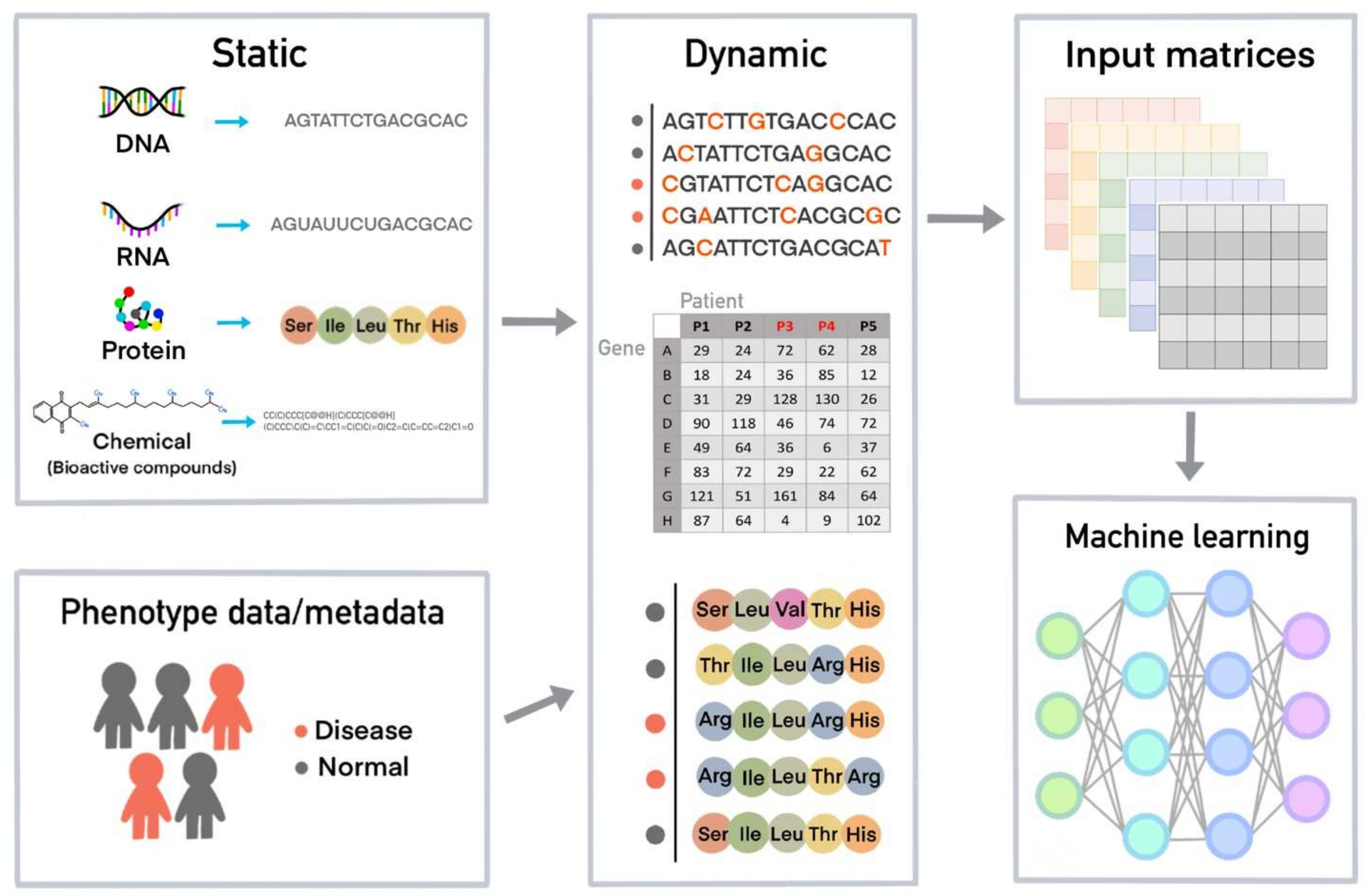
2. Cheminformatics, Bioinformatics, and Databases for Machine Learning
3. Chemical Space Where Unidentified Bioactive Compounds Exist
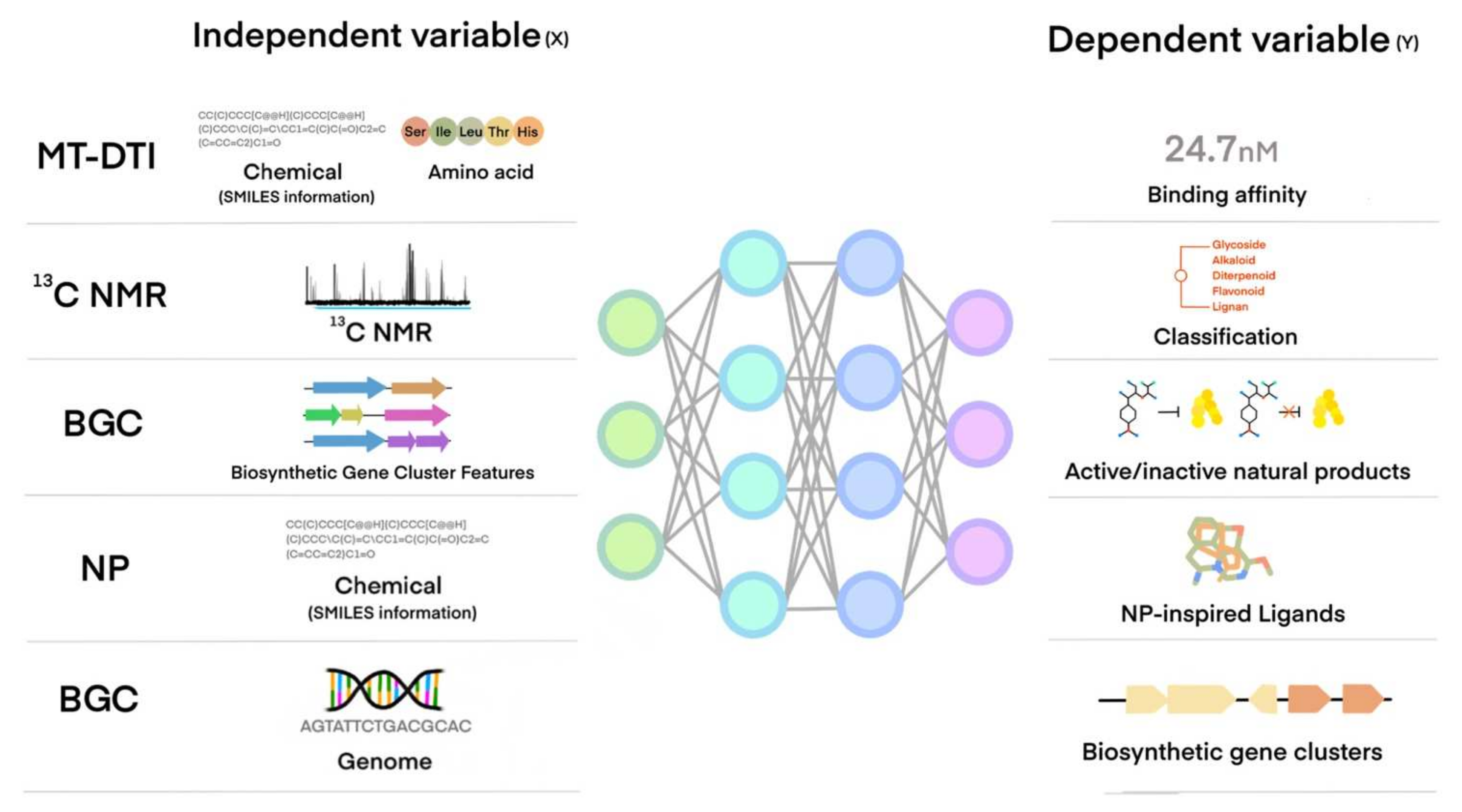
4. How a Machine Learns from Data and Creates a Model for a Task Using Machine Learning Algorithms
5. Machine Learning Application of NP or NP-Like Chemical Compounds Discovery for Cardiovascular and Metabolic Diseases
6. Conclusions
7. Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef]
- Thomford, N.E.; Senthebane, D.A.; Rowe, A.; Munro, D.; Seele, P.; Maroyi, A.; Dzobo, K. Natural products for drug discovery in the 21st century: Innovations for novel drug discovery. Int. J. Mol. Sci. 2018, 19, 1578. [Google Scholar] [CrossRef]
- Atanasov, A.G.; Waltenberger, B.; Pferschy-Wenzig, E.-M.; Linder, T.; Wawrosch, C.; Uhrin, P.; Temml, V.; Wang, L.; Schwaiger, S.; Heiss, E.H.; et al. Discovery and resupply of pharmacologically active plant-derived natural products: A review. Biotechnol. Adv. 2015, 33, 1582–1614. [Google Scholar] [CrossRef]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef] [PubMed]
- Waltenberger, B.; Mocan, A.; Šmejkal, K.; Heiss, E.H.; Atanasov, A.G. Natural products to counteract the epidemic of cardiovascular and metabolic disorders. Molecules 2016, 21, 807. [Google Scholar] [CrossRef] [PubMed]
- Fraenkel, G.S. The raison d’ĕtre of secondary plant substances; These odd chemicals arose as a means of protecting plants from insects and now guide insects to food. Science 1959, 129, 1466–1470. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Lounkine, E.; Bajorath, J. Many approved drugs have bioactive analogs with different target annotations. AAPS J. 2014, 16, 847–859. [Google Scholar] [CrossRef][Green Version]
- Yu, M.J.; Zheng, W.; Seletsky, B.M. From micrograms to grams: Scale-up synthesis of eribulin mesylate. Nat. Prod. Rep. 2013, 30, 1158–1164. [Google Scholar] [CrossRef]
- Eder, J.; Sedrani, R.; Wiesmann, C. The discovery of first-in-class drugs: Origins and evolution. Nat. Rev. Drug Discov. 2014, 13, 577–587. [Google Scholar] [CrossRef]
- Doak, B.C.; Over, B.; Giordanetto, F.; Kihlberg, J. Oral druggable space beyond the rule of 5: Insights from drugs and clinical candidates. Chem. Biol. 2014, 21, 1115–1142. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.; Hopkins, A. Navigating chemical space for biology and medicine. Nature 2004, 432, 855–861. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Bohacek, R.S.; McMartin, C.; Guida, W.C. The art and practice of structure-based drug design: A molecular modeling perspective. Med. Res. Rev. 1996, 16, 3–50. [Google Scholar] [CrossRef]
- Zabolotna, Y.; Ertl, P.; Horvath, D.; Bonachera, F.; Marcou, G.; Varnek, A. NP navigator: A new look at the natural product chemical space. Mol. Inform. 2021, 40, e2100068. [Google Scholar] [CrossRef]
- Nelson, A.; Karageorgis, G. Natural product-informed exploration of chemical space to enable bioactive molecular discovery. RSC Med. Chem. 2021, 12, 353–362. [Google Scholar] [CrossRef]
- Larsson, J.; Gottfries, J.; Muresan, S.; Backlund, A. ChemGPS-NP: Tuned for navigation in biologically relevant chemical space. J. Nat. Prod. 2007, 70, 789–794. [Google Scholar] [CrossRef]
- Grazina, L.; Rodrigues, P.J.; Igrejas, G.; Nunes, M.A.; Mafra, I.; Arlorio, M.; Oliveira, M.B.P.P.; Amaral, J.S. Machine learning approaches applied to GC-FID fatty acid profiles to discriminate wild from farmed salmon. Foods 2020, 9, 1622. [Google Scholar] [CrossRef]
- Paul, D.; Sanap, G.; Shenoy, S.; Kalyane, D.; Kalia, K.; Tekade, R.K. Artificial intelligence in drug discovery and development. Drug Discov. Today 2021, 26, 80–93. [Google Scholar] [CrossRef]
- Mak, K.-K.; Balijepalli, M.K.; Pichika, M.R. Success stories of AI in drug discovery—Where do things stand? Expert Opin. Drug Discov. 2022, 17, 79–92. [Google Scholar] [CrossRef]
- Jiménez-Luna, J.; Grisoni, F.; Weskamp, N.; Schneider, G. Artificial intelligence in drug discovery: Recent advances and future perspectives. Expert Opin. Drug Discov. 2021, 16, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Öztürk, H.; Özgür, A.; Ozkirimli, E. DeepDTA: Deep drug–target binding affinity prediction. Bioinformatics 2018, 34, i821–i829. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.; Zhung, W.; Yang, S.; Lim, J.; Kim, W.Y. PIGNet: A physics-informed deep learning model toward generalized drug-target interaction predictions. Chem. Sci. 2022, in press. [Google Scholar] [CrossRef]
- Gentile, F.; Agrawal, V.; Hsing, M.; Ton, A.-T.; Ban, F.; Norinder, U.; Gleave, M.E.; Cherkasov, A. Deep docking: A deep learning platform for augmentation of structure based drug discovery. ACS Cent. Sci. 2020, 6, 939–949. [Google Scholar] [CrossRef]
- Francoeur, P.G.; Masuda, T.; Sunseri, J.; Jia, A.; Iovanisci, R.B.; Snyder, I.; Koes, D.R. Three-dimensional convolutional neural networks and a cross-docked data set for structure-based drug design. J. Chem. Inf. Model. 2020, 60, 4200–4215. [Google Scholar] [CrossRef]
- Venkatraman, V. FP-ADMET: A compendium of fingerprint-based ADMET prediction models. J. Cheminform. 2021, 13, 75. [Google Scholar] [CrossRef]
- Ferreira, L.L.G.; Andricopulo, A.D. ADMET modeling approaches in drug discovery. Drug Discov. Today 2019, 24, 1157–1165. [Google Scholar] [CrossRef]
- Göller, A.H.; Kuhnke, L.; Montanari, F.; Bonin, A.; Schneckener, S.; ter Laak, A.; Wichard, J.; Lobell, M.; Hillisch, A. Bayer’s in silico ADMET platform: A journey of machine learning over the past two decades. Drug Discov. Today 2020, 25, 1702–1709. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Model. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL Database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15-ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Woods, J.M.; Bethell, R.C.; Coates, J.A.; Healy, N.; Hiscox, S.A.; Pearson, B.A.; Ryan, D.M.; Ticehurst, J.; Tilling, J.; Walcott, S.M. 4-guanidino-2,4-dideoxy-2,3-dehydro-N-acetylneuraminic acid is a highly effective inhibitor both of the sialidase (neuraminidase) and of growth of a wide range of influenza A and B viruses in vitro. Antimicrob. Agents Chemother. 1993, 37, 1473–1479. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide protein data bank. Nat. Struct. Biol. 2003, 10, 980. [Google Scholar] [CrossRef]
- Sheynkman, G.M.; Shortreed, M.R.; Cesnik, A.J.; Smith, L.M. Proteogenomics: Integrating next-generation sequencing and mass spectrometry to characterize human proteomic variation. Annu. Rev. Anal. Chem. 2016, 9, 521–545. [Google Scholar] [CrossRef]
- NCBI. Resource coordinators database resources of the national center for biotechnology information. Nucleic Acids Res. 2018, 46, D8–D13. [Google Scholar] [CrossRef]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. UniProt: The Universal Protein knowledgebase. Nucleic Acids Res. 2018, 46, 2699. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Sorokina, M.; Steinbeck, C. Review on natural products databases: Where to find data in 2020. J. Cheminform. 2020, 12, 20. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Lachmann, A.; Torre, D.; Keenan, A.B.; Jagodnik, K.M.; Lee, H.J.; Wang, L.; Silverstein, M.C.; Ma’ayan, A. Massive mining of publicly available RNA-seq data from human and mouse. Nat. Commun. 2018, 9, 1366. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M. Chemical space and biology. Nature 2004, 432, 824–828. [Google Scholar] [CrossRef]
- Chen, Y.; Garcia de Lomana, M.; Friedrich, N.-O.; Kirchmair, J. Characterization of the chemical space of known and readily obtainable natural products. J. Chem. Inf. Model. 2018, 58, 1518–1532. [Google Scholar] [CrossRef] [PubMed]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef]
- Durán-Iturbide, N.A.; Díaz-Eufracio, B.I.; Medina-Franco, J.L. In silico ADME/tox profiling of natural products: A focus on BIOFACQUIM. ACS Omega 2020, 5, 16076–16084. [Google Scholar] [CrossRef]
- Dong, J.; Wang, N.-N.; Yao, Z.-J.; Zhang, L.; Cheng, Y.; Ouyang, D.; Lu, A.-P.; Cao, D.-S. ADMETlab: A platform for systematic ADMET evaluation based on a comprehensively collected ADMET database. J. Cheminform. 2018, 10, 29. [Google Scholar] [CrossRef]
- Bocci, G.; Carosati, E.; Vayer, P.; Arrault, A.; Lozano, S.; Cruciani, G. ADME-space: A new tool for medicinal chemists to explore ADME properties. Sci. Rep. 2017, 7, 6359. [Google Scholar] [CrossRef]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef]
- Van de Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef]
- Feinberg, E.N.; Joshi, E.; Pande, V.S.; Cheng, A.C. Improvement in ADMET prediction with multitask deep featurization. J. Med. Chem. 2020, 63, 8835–8848. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Cáceres, E.L.; Tudor, M.; Cheng, A.C. Deep learning approaches in predicting ADMET properties. Future Med. Chem. 2020, 12, 1995–1999. [Google Scholar] [CrossRef] [PubMed]
- Greener, J.G.; Kandathil, S.M.; Moffat, L.; Jones, D.T. A guide to machine learning for biologists. Nat. Rev. Mol. Cell Biol. 2021, 23, 40–55. [Google Scholar] [CrossRef]
- Artrith, N.; Butler, K.T.; Coudert, F.-X.; Han, S.; Isayev, O.; Jain, A.; Walsh, A. Best practices in machine learning for chemistry. Nat. Chem. 2021, 13, 505–508. [Google Scholar] [CrossRef]
- Moving towards reproducible machine learning. Nat. Comput. Sci. 2021, 1, 629–630. [CrossRef]
- Patel, L.; Shukla, T.; Huang, X.; Ussery, D.W.; Wang, S. Machine learning methods in drug discovery. Molecules 2020, 25, 5277. [Google Scholar] [CrossRef]
- Mirza, B.; Wang, W.; Wang, J.; Choi, H.; Chung, N.C.; Ping, P. Machine learning and integrative analysis of biomedical big data. Genes 2019, 10, 87. [Google Scholar] [CrossRef]
- Eraslan, G.; Avsec, Ž.; Gagneur, J.; Theis, F.J. Deep learning: New computational modelling techniques for genomics. Nat. Rev. Genet. 2019, 20, 389–403. [Google Scholar] [CrossRef]
- Shin, B.; Park, S.; Kang, K.; Ho, J.C. Self-attention based molecule representation for predicting drug-target interaction. arXiv 2019, arXiv:1908.06760. [Google Scholar]
- Tang, J.; Szwajda, A.; Shakyawar, S.; Xu, T.; Hintsanen, P.; Wennerberg, K.; Aittokallio, T. Making sense of large-scale kinase inhibitor bioactivity data sets: A comparative and integrative analysis. J. Chem. Inf. Model. 2014, 54, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Treviño, S.H.; Uc-Cetina, V.; Fernández-Herrera, M.A.; Merino, G. Prediction of natural product classes using machine learning and 13C NMR spectroscopic data. J. Chem. Inf. Model. 2020, 60, 3376–3386. [Google Scholar] [CrossRef] [PubMed]
- López-Pérez, J.L.; Therón, R.; del Olmo, E.; Díaz, D. NAPROC-13: A database for the dereplication of natural product mixtures in bioassay-guided protocols. Bioinformatics 2007, 23, 3256–3257. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.S.; Clardy, J. A machine learning bioinformatics method to predict biological activity from biosynthetic gene clusters. J. Chem. Inf. Model. 2021, 61, 2560–2571. [Google Scholar] [CrossRef]
- Hannigan, G.D.; Prihoda, D.; Palicka, A.; Soukup, J.; Klempir, O.; Rampula, L.; Durcak, J.; Wurst, M.; Kotowski, J.; Chang, D.; et al. A deep learning genome-mining strategy for biosynthetic gene cluster prediction. Nucleic Acids Res. 2019, 47, e110. [Google Scholar] [CrossRef]
- Hochreiter, S.; Schmidhuber, J. Long short-term memory. Neural Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef]
- Mikolov, T.; Sutskever, I.; Chen, K.; Corrado, G.S.; Dean, J. Distributed representations of words and phrases and their compositionality. Adv. Neural Inf. Process. Syst. 2013, 26, 9. [Google Scholar]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Grisoni, F.; Merk, D.; Friedrich, L.; Schneider, G. Design of natural-product-inspired multitarget ligands by machine learning. ChemMedChem 2019, 14, 1129–1134. [Google Scholar] [CrossRef]
- Sharma, K. Cholinesterase inhibitors as Alzheimer’s therapeutics (review). Mol. Med. Rep. 2019, 20, 1479–1487. [Google Scholar] [CrossRef] [PubMed]
- Grisoni, F.; Merk, D.; Consonni, V.; Hiss, J.A.; Tagliabue, S.G.; Todeschini, R.; Schneider, G. Scaffold hopping from natural products to synthetic mimetics by holistic molecular similarity. Commun. Chem. 2018, 1, 44. [Google Scholar] [CrossRef]
- Reker, D.; Rodrigues, T.; Schneider, P.; Schneider, G. Identifying the macromolecular targets of de novo-designed chemical entities through self-organizing map consensus. Proc. Natl. Acad. Sci. USA 2014, 111, 4067–4072. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Schneider, G. A computational method for unveiling the target promiscuity of pharmacologically active compounds. Angew. Chem. Int. Ed. 2017, 56, 11520–11524. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F. Machine learning methods to predict the terrestrial and marine origin of natural products. Mol. Inform. 2021, 40, e2060034. [Google Scholar] [CrossRef]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef]
- Lehrke, M.; Lazar, M.A. The many faces of PPARγ. Cell 2005, 123, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Rupp, M.; Schroeter, T.; Steri, R.; Zettl, H.; Proschak, E.; Hansen, K.; Rau, O.; Schwarz, O.; Müller-Kuhrt, L.; Schubert-Zsilavecz, M.; et al. From machine learning to natural product derivatives that selectively activate transcription factor PPARγ. ChemMedChem 2010, 5, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, M.; Rayan, M.; Zeidan, N.; Falah, M.; Rayan, A. Indexing natural products for their potential anti-diabetic activity: Filtering and mapping discriminative physicochemical properties. Molecules 2017, 22, 1563. [Google Scholar] [CrossRef]
- Yoo, S.; Yang, H.C.; Lee, S.; Shin, J.; Min, S.; Lee, E.; Song, M.; Lee, D. A deep learning-based approach for identifying the medicinal uses of plant-derived natural compounds. Front. Pharmacol. 2020, 11, 584875. [Google Scholar] [CrossRef]
- Lolmède, K.; Duffaut, C.; Zakaroff-Girard, A.; Bouloumié, A. Immune cells in adipose tissue: Key players in metabolic disorders. Diabetes Metab. 2011, 37, 283–290. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Aswad, M.; Rayan, M.; Abu-Lafi, S.; Falah, M.; Raiyn, J.; Abdallah, Z.; Rayan, A. Nature is the best source of anti-inflammatory drugs: Indexing natural products for their anti-inflammatory bioactivity. Inflamm. Res. 2018, 67, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Galvez-Llompart, M.; del Carmen Recio Iglesias, M.; Gálvez, J.; García-Domenech, R. Novel potential agents for ulcerative colitis by molecular topology: Suppression of IL-6 production in Caco-2 and RAW 264.7 cell lines. Mol. Divers. 2013, 17, 573–593. [Google Scholar] [CrossRef]
- Chagas-Paula, D.; Oliveira, T.; Zhang, T.; Edrada-Ebel, R.; Da Costa, F. Prediction of anti-inflammatory plants and discovery of their biomarkers by machine learning algorithms and metabolomic studies. Planta Med. 2015, 81, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Linardatos, P.; Papastefanopoulos, V.; Kotsiantis, S. Explainable AI: A review of machine learning interpretability methods. Entropy 2020, 23, 18. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zhao, K.; Chu, X. AutoML: A survey of the state-of-the-art. Knowl.-Based Syst. 2021, 212, 106622. [Google Scholar] [CrossRef]
- Detsi, A.; Kontogiorgis, C.; Hadjipavlou-Litina, D. Coumarin derivatives: An updated patent review (2015–2016). Expert Opin. Ther. Pat. 2017, 27, 1201–1226. [Google Scholar] [CrossRef]
- Hu, Y.; Shen, Y.; Wu, X.; Tu, X.; Wang, G.-X. Synthesis and biological evaluation of coumarin derivatives containing imidazole skeleton as potential antibacterial agents. Eur. J. Med. Chem. 2018, 143, 958–969. [Google Scholar] [CrossRef]
- Park, S.; Ko, Y.H.; Lee, B.; Shin, B.; Beck, B.R. Abstract 35: Molecular optimization of phase III trial failed anticancer drugs using target affinity and toxicity-centered multiple properties reinforcement learning. In Proceedings of the Poster Presentations—Proffered Abstracts, Ljubljana, Slovenia, 15 June 2020; p. 35. [Google Scholar]
- Zhou, Z.; Kearnes, S.; Li, L.; Zare, R.N.; Riley, P. Optimization of molecules via deep reinforcement learning. Sci. Rep. 2019, 9, 10752. [Google Scholar] [CrossRef]
- Zhavoronkov, A.; Ivanenkov, Y.A.; Aliper, A.; Veselov, M.S.; Aladinskiy, V.A.; Aladinskaya, A.V.; Terentiev, V.A.; Polykovskiy, D.A.; Kuznetsov, M.D.; Asadulaev, A.; et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 2019, 37, 1038–1040. [Google Scholar] [CrossRef] [PubMed]
- Shin, B.; Park, S.; Bak, J.; Ho, J.C. Controlled molecule generator for optimizing multiple chemical properties. In Proceedings of the Conference on Health, Inference, and Learning, Virtual Event, 8 April 2021; pp. 146–153. [Google Scholar]
- Li, Y.; Pei, J.; Lai, L. Structure-based de novo drug design using 3D deep generative models. Chem. Sci. 2021, 12, 13664–13675. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.; Beck, B.R.; Kim, H.H.; Lee, S.; Kang, K. A Brief Review of Machine Learning-Based Bioactive Compound Research. Appl. Sci. 2022, 12, 2906. https://doi.org/10.3390/app12062906
Park J, Beck BR, Kim HH, Lee S, Kang K. A Brief Review of Machine Learning-Based Bioactive Compound Research. Applied Sciences. 2022; 12(6):2906. https://doi.org/10.3390/app12062906
Chicago/Turabian StylePark, Jihye, Bo Ram Beck, Hoo Hyun Kim, Sangbum Lee, and Keunsoo Kang. 2022. "A Brief Review of Machine Learning-Based Bioactive Compound Research" Applied Sciences 12, no. 6: 2906. https://doi.org/10.3390/app12062906
APA StylePark, J., Beck, B. R., Kim, H. H., Lee, S., & Kang, K. (2022). A Brief Review of Machine Learning-Based Bioactive Compound Research. Applied Sciences, 12(6), 2906. https://doi.org/10.3390/app12062906