
Automation System for the Flexible Sample Preparation for Quantification of Δ9-THC-D3, THC-OH and THC-COOH from Serum, Saliva and Urine



Abstract
1. Introduction
1.1. Serum, Plasma, Whole Blood
1.2. Urine
1.3. Saliva
2. Materials and Methods
2.1. Chemicals and Reagents
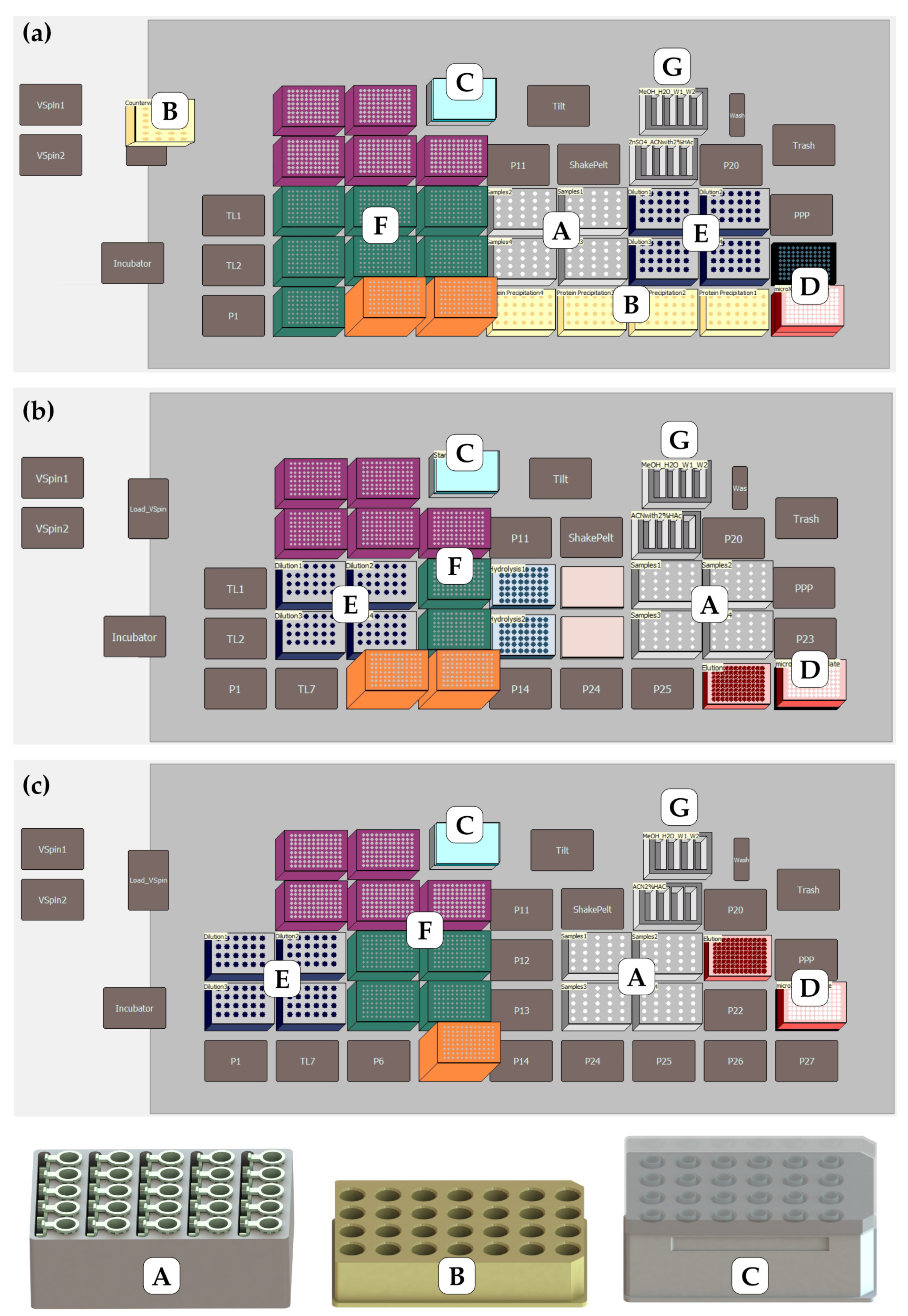
2.2. Instrumentation for Automatic Sample Preparation
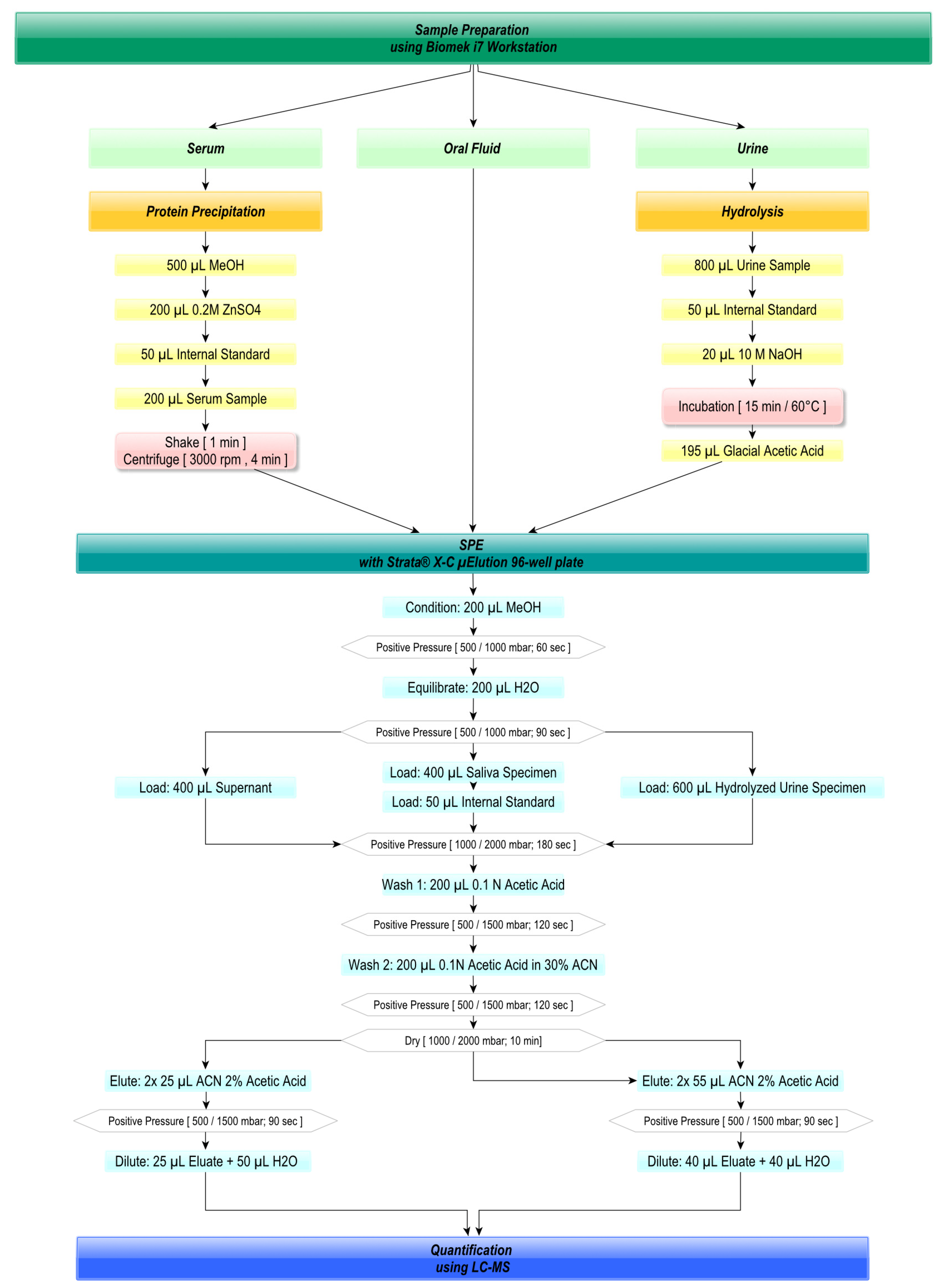
2.3. Automated Sample Preparation
2.4. Liquid Chromatography Conditions
2.5. Mass Spectrometry
2.6. Method Validation
2.7. Evaluation under Economic Criteria
3. Results
3.1. Validation Results
3.2. Results of Economic Evaluation
4. Discussions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Europäischer Drogenbericht 2021: Trends und Entwicklungen; Europäische Beobachtungsstelle für Drogen und Drogensucht, Amt für Veröffentlichungen der Europäischen Union: Luxembourg, 2021.
- Pellati, F.; Borgonetti, V.; Brighenti, V.; Biagi, M.; Benvenuti, S.; Corsi, L. Cannabis sativa L. and Nonpsychoactive Cannabinoids: Their Chemistry and Role against Oxidative Stress, Inflammation, and Cancer. BioMed Res. Int. 2018, 2018, 1691428. [Google Scholar] [CrossRef] [PubMed]
- Abraham, T.T.; Lowe, R.H.; Pirnay, S.O.; Darwin, W.D.; Huestis, M.A. Simultaneous GC-EI-MS determination of Delta9-tetrahydrocannabinol, 11-hydroxy-Delta9-tetrahydrocannabinol, and 11-nor-9-carboxy-Delta9-tetrahydrocannabinol in human urine following tandem enzyme-alkaline hydrolysis. J. Anal. Toxicol. 2007, 31, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Buchanan, B.; Zuccolo, J.; Poulin, M.M.; Gabriele, J.; Baranowski, D.C. A reliable and validated LC-MS/MS method for the simultaneous quantification of 4 cannabinoids in 40 consumer products. PLoS ONE 2018, 13, e0196396. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, R.A.; Moolchan, E.T.; Barnes, A.; Levine, B.; Huestis, M.A. Validated method for the simultaneous determination of Delta 9-tetrahydrocannabinol (THC), 11-hydroxy-THC and 11-nor-9-carboxy-THC in human plasma using solid phase extraction and gas chromatography-mass spectrometry with positive chemical ionization. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2003, 798, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Yamaori, S.; Funahashi, T.; Kimura, T.; Yamamoto, I. Cytochrome P450 enzymes involved in the metabolism of tetrahydrocannabinols and cannabinol by human hepatic microsomes. Life Sci. 2007, 80, 1415–1419. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, L.K.; Hasselstrøm, J.B. Sensitive Determination of Cannabinoids in Whole Blood by LC-MS-MS After Rapid Removal of Phospholipids by Filtration. J. Anal. Toxicol. 2017, 41, 382–391. [Google Scholar] [CrossRef]
- Ferreirós, N.; Labocha, S.; Walter, C.; Lötsch, J.; Geisslinger, G. Simultaneous and sensitive LC-MS/MS determination of tetrahydrocannabinol and metabolites in human plasma. Anal. Bioanal. Chem. 2013, 405, 1399–1406. [Google Scholar] [CrossRef]
- Huestis, M.A.; Cone, E.J. Differentiating new marijuana use from residual drug excretion in occasional marijuana users. J. Anal. Toxicol. 1998, 22, 445–454. [Google Scholar] [CrossRef]
- Teixeira, H.; Verstraete, A.; Proença, P.; Corte-Real, F.; Monsanto, P.; Vieira, D.N. Validated method for the simultaneous determination of Delta9-THC and Delta9-THC-COOH in oral fluid, urine and whole blood using solid-phase extraction and liquid chromatography-mass spectrometry with electrospray ionization. Forensic Sci. Int. 2007, 170, 148–155. [Google Scholar] [CrossRef]
- Lee, D.; Huestis, M.A. Current knowledge on cannabinoids in oral fluid. Drug Test. Anal. 2014, 6, 88–111. [Google Scholar] [CrossRef]
- Soares Nunes, L.A.; Mussavira, S.; Sukumaran Bindhu, O. Clinical and diagnostic utility of saliva as a non-invasive diagnostic fluid: A systematic review. Biochem. Med. 2015, 25, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Aps, J.K.; Martens, L.C. Review: The physiology of saliva and transfer of drugs into saliva. Forensic Sci. Int. 2005, 150, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Lemberger, L.; Silberstein, S.D.; Axelrod, J.; Kopin, I.J. Marihuana: Studies on the disposition and metabolism of delta-9-tetrahydrocannabinol in man. Science 1970, 170, 1320–1322. [Google Scholar] [CrossRef] [PubMed]
- Wall, M.E.; Sadler, B.M.; Brine, D.; Taylor, H.; Perez-Reyes, M. Metabolism, disposition, and kinetics of delta-9-tetrahydrocannabinol in men and women. Clin. Pharmacol. Ther. 1983, 34, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Kemp, P.M.; Abukhalaf, I.K.; Manno, J.E.; Manno, B.R.; Alford, D.D.; Mcwilliams, M.E.; Nixon, F.E.; Fitzgerald, M.J.; Reeves, R.R.; Wood, M.J. Cannabinoids in humans. II. The influence of three methods of hydrolysis on the concentration of THC and two metabolites in urine. J. Anal. Toxicol. 1995, 19, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Milman, G.; Barnes, A.J.; Schwope, D.M.; Schwilke, E.W.; Darwin, W.D.; Goodwin, R.S.; Kelly, D.L.; Gorelick, D.A.; Huestis, M.A. Disposition of Cannabinoids in Oral Fluid after Controlled Around-the-Clock Oral THC Administration. Clin. Chem. 2010, 56, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.L.; Moffat, A.C. Identification in human urine of Δ9-tetrahydrocannabinol-11-oic acid glucuronide: A tetrahydrocannabinol metabolite. J. Pharm. Pharmacol. 2011, 32, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Valen, A.; Leere Øiestad, Å.M.; Strand, D.H.; Skari, R.; Berg, T. Determination of 21 drugs in oral fluid using fully automated supported liquid extraction and UHPLC-MS/MS. Drug Test. Anal. 2017, 9, 808–823. [Google Scholar] [CrossRef] [PubMed]
- Caplan, Y.H.; Goldberger, B.A. Alternative specimens for workplace drug testing. J. Anal. Toxicol. 2001, 25, 396–399. [Google Scholar] [CrossRef]
- Andersen, D.; Rasmussen, B.; Linnet, K. Validation of a fully automated robotic setup for preparation of whole blood samples for LC-MS toxicology analysis. J. Anal. Toxicol. 2012, 36, 280–287. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dziadosz, M.; Klintschar, M.; Teske, J. Simple protein precipitation-based analysis of Δ9-tetrahydrocannabinol and its metabolites in human serum by liquid chromatography–tandem mass spectrometry. Forensic Toxicol. 2017, 35, 190–194. [Google Scholar] [CrossRef]
- Hädener, M.; König, S.; Fabritius, M.M.; Weinmann, W. Using THC-COOH cut-off concentrations for assessing cannabis consumption frequency: A recently detected THC-COOH isomer poses an important analytical problem. Toxichem. Krimtech. 2017, 84, 168. [Google Scholar]
- Toennes, S.W.; Hanisch, S.; Pogoda, W.; Wunder, C.; Paulke, A. Pitfall in cannabinoid analysis—Detection of a previously unrecognized interfering compound in human serum. Anal. Bioanal. Chem. 2015, 407, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Kneisel, S.; Auwärter, V. Analysis of 30 synthetic cannabinoids in serum by liquid chromatography-electrospray ionization tandem mass spectrometry after liquid-liquid extraction. J. Mass Spectrom. 2012, 47, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Jagerdeo, E.; Schaff, J.E.; Montgomery, M.A.; Lebeau, M.A. A semi-automated solid-phase extraction liquid chromatography/tandem mass spectrometry method for the analysis of tetrahydrocannabinol and metabolites in whole blood. Rapid Commun. Mass Spectrom. 2009, 23, 2697–2705. [Google Scholar] [CrossRef] [PubMed]
- Schwope, D.M.; Scheidweiler, K.B.; Huestis, M.A. Direct quantification of cannabinoids and cannabinoid glucuronides in whole blood by liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2011, 401, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- Kristoffersen, L.; Langødegård, M.; Gaare, K.I.; Amundsen, I.; Terland, M.N.; Strand, D.H. Determination of 12 commonly found compounds in DUID cases in whole blood using fully automated supported liquid extraction and UHPLC-MS/MS. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2018, 1093, 8–23. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, J.A.; Smith, B.E.; Sobolesky, P.M.; Kim, S.; Hoffman, M.A.; Stone, J.; Huestis, M.A.; Grelotti, D.J.; Grant, I.; Marcotte, T.D.; et al. Validation of a liquid chromatography tandem mass spectrometry (LC-MS/MS) method to detect cannabinoids in whole blood and breath. Clin. Chem. Lab. Med. 2020, 58, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Tiscione, N.B.; Miller, R.; Shan, X.; Sprague, J.; Yeatman, D.T. An Efficient, Robust Method for the Determination of Cannabinoids in Whole Blood by LC-MS-MS. J. Anal. Toxicol. 2016, 40, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Purschke, K.; Heinl, S.; Lerch, O.; Erdmann, F.; Veit, F. Development and validation of an automated liquid-liquid extraction GC/MS method for the determination of THC, 11-OH-THC, and free THC-carboxylic acid (THC-COOH) from blood serum. Anal. Bioanal. Chem. 2016, 408, 4379–4388. [Google Scholar] [CrossRef]
- Giroud, C.; Ménétrey, A.; Augsburger, M.; Buclin, T.; Sanchez-Mazas, P.; Mangin, P. Delta(9)-THC, 11-OH-Delta(9)-THC and Delta(9)-THCCOOH plasma or serum to whole blood concentrations distribution ratios in blood samples taken from living and dead people. Forensic Sci. Int. 2001, 123, 159–164. [Google Scholar] [CrossRef]
- Weller, J.P.; Wolf, M.; Szidat, S. Enhanced selectivity in the determination of delta9-tetrahydrocannabinol and two major metabolites in serum using ion-trap GC-MS-MS. J. Anal. Toxicol. 2000, 24, 359–364. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Aizpurua-Olaizola, O.; Zarandona, I.; Ortiz, L.; Navarro, P.; Etxebarria, N.; Usobiaga, A. Simultaneous quantification of major cannabinoids and metabolites in human urine and plasma by HPLC-MS/MS and enzyme-alkaline hydrolysis. Drug Test. Anal. 2017, 9, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Gundersen, P.O.M.; Spigset, O.; Josefsson, M. Screening, quantification, and confirmation of synthetic cannabinoid metabolites in urine by UHPLC-QTOF-MS. Drug Test. Anal. 2019, 11, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.; Shin, I.; Kim, J.; Yang, W. Simultaneous quantification of 37 synthetic cannabinoid metabolites in human urine by liquid chromatography-tandem mass spectrometry. Forensic Toxicol. 2015, 33, 221–234. [Google Scholar] [CrossRef]
- Kleis, J.N.; Hess, C.; Germerott, T.; Roehrich, J. Sensitive screening of synthetic cannabinoids using liquid chromatography quadrupole time-of-flight mass spectrometry after solid phase extraction. Drug Test. Anal. 2021, 13, 1535–1551. [Google Scholar] [CrossRef] [PubMed]
- Breindahl, T.; Andreasen, K. Determination of 11-nor-delta9-tetrahydrocannabinol-9-carboxylic acid in urine using high-performance liquid chromatography and electrospray ionization mass spectrometry. J. Chromatogr. B Biomed. Sci. Appl. 1999, 732, 155–164. [Google Scholar] [CrossRef]
- Freijo, T.D.; Harris, S.E.; Kala, S.V. A rapid quantitative method for the analysis of synthetic cannabinoids by liquid chromatography-tandem mass spectrometry. J. Anal. Toxicol. 2014, 38, 466–478. [Google Scholar] [CrossRef]
- Sempio, C.; Scheidweiler, K.B.; Barnes, A.J.; Huestis, M.A. Optimization of recombinant β-glucuronidase hydrolysis and quantification of eight urinary cannabinoids and metabolites by liquid chromatography tandem mass spectrometry. Drug Test. Anal. 2018, 10, 518–529. [Google Scholar] [CrossRef]
- Automated Extraction of Synthetic Cannabinoids (SPICE) from Urine Using ISOLUTE® SLE + Prior to LC-MS/MS; AN793; Biotage: Uppsala, Sweden, 2013.
- Scheidweiler, K.B.; Desrosiers, N.A.; Huestis, M.A. Simultaneous quantification of free and glucuronidated cannabinoids in human urine by liquid chromatography tandem mass spectrometry. Clin. Chim. Acta 2012, 413, 1839–1847. [Google Scholar] [CrossRef]
- Sample Preparation with the AC Extraction Plate: Extraction of Urinary THC Metabolites for LC-MSMS Quantification; Tecan Trading AG: Männedorf, Switzerland, 2014.
- Weinmann, W.; Goerner, M.; Vogt, S.; Goerke, R.; Pollak, S. Fast confirmation of 11-nor-9-carboxy-Delta(9)-tetrahydrocannabinol (THC-COOH) in urine by LC/MS/MS using negative atmospheric-pressure chemical ionisation (APCI). Forensic Sci. Int. 2001, 121, 103–107. [Google Scholar] [CrossRef]
- Montesano, C.; Sergi, M.; Odoardi, S.; Simeoni, M.C.; Compagnone, D.; Curini, R. A μ-SPE procedure for the determination of cannabinoids and their metabolites in urine by LC-MS/MS. J. Pharm. Biomed. Anal. 2014, 91, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Conti, M.; Tazzari, V.; Bertona, M.; Brambilla, M.; Brambilla, P. Surface-activated chemical ionization combined with electrospray ionization and mass spectrometry for the analysis of cannabinoids in biological samples. Part I: Analysis of 11-nor-9-carboxytetrahydro-cannabinol. Rapid Commun. Mass Spectrom. 2011, 25, 1552–1558. [Google Scholar] [CrossRef] [PubMed]
- Rumpler, M.J. Quantitative analysis of 11-nor-9-carboxy-tetrahydrocannbinol (THC–COOH) in urine by LC–MS/MS following a simple filtration. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 957, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Robandt, P.V.; Klette, K.L.; Sibum, M. Automated solid-phase extraction-liquid chromatography-tandem mass spectrometry analysis of 11-nor-Delta9-tetrahydrocannabinol-9-carboxylic acid in human urine specimens: Application to a high-throughput urine analysis laboratory. J. Anal. Toxicol. 2009, 33, 456–460. [Google Scholar] [CrossRef][Green Version]
- Jagerdeo, E.; Montgomery, M.A.; Karas, R.P.; Sibum, M. A fast method for screening and/or quantitation of tetrahydrocannabinol and metabolites in urine by automated SPE/LC/MS/MS. Anal. Bioanal. Chem. 2010, 398, 329–338. [Google Scholar] [CrossRef]
- Huq, S.; Dixon, A.; Kelly, K.; Kallury, K.M. Novel solid-phase extraction protocol for 11-nor-9-carboxy-delta9-tetrahydrocannabinol from urine samples employing a polymeric mixed-mode cation-exchange resin, Strata-X-C, suitable for gas chromatography-mass spectrometry or liquid chromatography-mass spectrometry analysis. J. Chromatogr. A 2005, 1073, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Pope, J.D.; Black, M.J.; Drummer, O.H.; Schneider, H.G. Urine toxicology screening by liquid chromatography time-of-flight mass spectrometry in a quaternary hospital setting. Clin. Biochem. 2021, 95, 66–72. [Google Scholar] [CrossRef]
- Kronstrand, R.; Brinkhagen, L.; Birath-Karlsson, C.; Roman, M.; Josefsson, M. LC-QTOF-MS as a superior strategy to immunoassay for the comprehensive analysis of synthetic cannabinoids in urine. Anal. Bioanal. Chem. 2014, 406, 3599–3609. [Google Scholar] [CrossRef] [PubMed]
- Cabrices, O.G.; Foster, F.D.; Taylor, A.M.; Lee, L.A. “Prep-and-Shoot”: The Completely Automated Workflow for the Hydrolysis and Analysis of Urine Samples by LC/MS/MS; Gerstel: Linthicum, MD, USA, 2014. [Google Scholar]
- Badawi, N.; Simonsen, K.W.; Steentoft, A.; Bernhoft, I.M.; Linnet, K. Simultaneous screening and quantification of 29 drugs of abuse in oral fluid by solid-phase extraction and ultraperformance LC-MS/MS. Clin. Chem. 2009, 55, 2004–2018. [Google Scholar] [CrossRef]
- Concheiro, M.; De Castro, A.; Quintela, Ó.; Cruz, A.; López-Rivadulla, M. Determination of illicit and medicinal drugs and their metabolites in oral fluid and preserved oral fluid by liquid chromatography–tandem mass spectrometry. Anal. Bioanal. Chem. 2008, 391, 2329–2338. [Google Scholar] [CrossRef] [PubMed]
- Coulter, C.; Garnier, M.; Moore, C. Analysis of Tetrahydrocannabinol and its Metabolite, 11-Nor-Δ9-Tetrahydrocannabinol-9-Carboxylic Acid, in Oral Fluid using Liquid Chromatography with Tandem Mass Spectrometry. J. Anal. Toxicol. 2012, 36, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Quintela, O.; Andrenyak, D.M.; Hoggan, A.M.; Crouch, D.J. A validated method for the detection of Delta 9-tetrahydrocannabinol and 11-nor-9-carboxy-Delta 9-tetrahydrocannabinol in oral fluid samples by liquid chromatography coupled with quadrupole-time-of-flight mass spectrometry. J. Anal. Toxicol. 2007, 31, 157–164. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lund, H.M.E.; Øiestad, E.L.; Gjerde, H.; Christophersen, A.S. Drugs of abuse in oral fluid collected by two different sample kits—Stability testing and validation using ultra performance tandem mass spectrometry analysis. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 3367–3377. [Google Scholar] [CrossRef] [PubMed]
- Bakke, E.; Høiseth, G.; Furuhaugen, H.; Berg, T.; Arnestad, M.; Gjerde, H. Oral Fluid to Blood Concentration Ratios of Different Psychoactive Drugs in Samples from Suspected Drugged Drivers. Ther. Drug Monit. 2020, 42, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Sergi, M.; Montesano, C.; Odoardi, S.; Mainero Rocca, L.; Fabrizi, G.; Compagnone, D.; Curini, R. Micro extraction by packed sorbent coupled to liquid chromatography tandem mass spectrometry for the rapid and sensitive determination of cannabinoids in oral fluids. J. Chromatogr. A 2013, 1301, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Milman, G.; Barnes, A.J.; Schwope, D.M.; Schwilke, E.W.; Goodwin, R.S.; Kelly, D.L.; Gorelick, D.A.; Huestis, M.A. Cannabinoids and metabolites in expectorated oral fluid after 8 days of controlled around-the-clock oral THC administration. Anal. Bioanal. Chem. 2011, 401, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.D.; Chang, Y.J.; Lin, K.L.; Chang, Y.Z. Simultaneous determination of Δ9-tetrahydrocannabinol and 11-nor-9-carboxy-Δ9-tetrahydrocannabinol in oral fluid using isotope dilution liquid chromatography tandem mass spectrometry. Anal. Bioanal. Chem. 2012, 402, 851–859. [Google Scholar] [CrossRef]
- Sergi, M.; Bafile, E.; Compagnone, D.; Curini, R.; D’ascenzo, G.; Romolo, F.S. Multiclass analysis of illicit drugs in plasma and oral fluids by LC-MS/MS. Anal. Bioanal. Chem. 2009, 393, 709–718. [Google Scholar] [CrossRef]
- Choi, H.; Baeck, S.; Kim, E.; Lee, S.; Jang, M.; Lee, J.; Choi, H.; Chung, H. Analysis of cannabis in oral fluid specimens by GC-MS with automatic SPE. Sci. Justice 2009, 49, 242–246. [Google Scholar] [CrossRef]
- Cerillant Corporation Certificate of Analysis (COA). Available online: https://www.cerilliant.com/products/searchCOA.aspx (accessed on 8 March 2022).
- Bach, A.; Fleischer, H.; Wijayawardena, B.; Thurow, K. Optimization of Automated Sample Preparation for Vitamin D Determination on a Biomek i7 Workstation. SLAS Technol. 2021, 26, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Fleischer, H.; Thurow, K. Automation Solutions for Analytical Measurements; Wiley-VCH: Weinheim, Germany, 2017; p. 272. [Google Scholar]
- Otto, M. Analytische Chemie; Wiley-VCH: Weinheim, Germany, 2019; p. 552. [Google Scholar]
- Christophersen, A.S. Tetrahydrocannabinol stability in whole blood: Plastic versus glass containers. J. Anal. Toxicol. 1986, 10, 129–131. [Google Scholar] [CrossRef] [PubMed]
- Kneisel, S.; Speck, M.; Moosmann, B.; Auwärter, V. Stability of 11 prevalent synthetic cannabinoids in authentic neat oral fluid samples: Glass versus polypropylene containers at different temperatures. Drug Test. Anal. 2013, 5, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Sanli, S.; Ghorbani-Zamani, F.; Moulahoum, H.; Gumus, Z.P.; Coskunol, H.; Odaci Demirkol, D.; Timur, S. Application of Biofunctionalized Magnetic Nanoparticles Based-Sensing in Abused Drugs Diagnostics. Anal. Chem. 2020, 92, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Scurati, S.; Brambilla, M.; D’Amato, L.; Brambilla, P. THC-COOH Quantification in Urine Using Dilute and Shoot LC-MS/MS Method for Forensic Toxicology; Thermo Fisher Scientific: Rodano, Italy, 2011; p. 548. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Serum | Δ9-THC-D3 | THC-OH | THC-COOH | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Range | Range | Range | ||||||||||||||
| No. of Samples | Mean Value [%] | CV [%] | Min [%] | - | Max [%] | Mean Value [%] | CV [%] | Min [%] | - | Max [%] | Mean Value [%] | CV [%] | Min [%] | - | Max [%] | |
| Recovery | 25 | 101.21 | 0.86 | 100.12 | - | 104.15 | 99.54 | 1.51 | 96.41 | - | 101.45 | 100.65 | 0.52 | 99.69 | - | 101.75 |
| Within-laboratory Precision 1 | 10 | 94.28 | 2.73 | 98.38 | 2.99 | 101.28 | 1.61 | |||||||||
| Within-laboratory Precision 2 | 10 | 101.01 | 0.51 | 100.42 | 0.44 | 100.79 | 0.62 | |||||||||
| Within-laboratory Precision 3 | 10 | 100.66 | 0.58 | 99.93 | 0.96 | 100.62 | 1.52 | |||||||||
| Within-laboratory Precision 4 | 10 | 99.11 | 1.24 | 103.11 | 0.80 | 102.25 | 3.11 | |||||||||
| Within-laboratory Precision 5 | 10 | 102.74 | 2.00 | 101.29 | 2.05 | 100.72 | 1.84 | |||||||||
| Measurement Precision | 25 | 101.21 | 0.30 | 99.08 | 0.53 | 101.25 | 0.43 | |||||||||
| LOD | 10 | 0.087 | 0.196 | 0.503 | ||||||||||||
| LOQ | 10 | 0.099 | 0.215 | 0.554 | ||||||||||||
| LOD (method) | 10 | 0.156 | 0.349 | 0.896 | ||||||||||||
| LOQ (method) | 10 | 0.176 | 0.384 | 0.987 | ||||||||||||
| Urine | THC-D3 | THC-OH | THC-COOH | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Range | Range | Range | ||||||||||||||
| No. of Samples | Mean Value [%] | CV [%] | Min [%] | - | Max [%] | Mean Value [%] | CV [%] | Min [%] | - | Max [%] | Mean Value [%] | CV [%] | Min [%] | - | Max [%] | |
| Recovery | 25 | 83.86 | 3.64 | 77.54 | - | 87.53 | 91.09 | 1.85 | 88.91 | - | 94.53 | 83.54 | 35.7 | 79.04 | - | 92.66 |
| Within-laboratory Precision 1 | 10 | 96.02 | 2.97 | 88.46 | 1.03 | 77.85 | 1.66 | |||||||||
| Within-laboratory Precision 2 | 10 | 90.09 | 2.22 | 86.43 | 2.46 | 80.53 | 2.78 | |||||||||
| Within-laboratory Precision 3 | 10 | 85.34 | 2.82 | 90.97 | 1.74 | 84.09 | 3.89 | |||||||||
| Within-laboratory Precision 4 | 10 | 98.24 | 3.42 | 88.91 | 1.60 | 83.84 | 2.51 | |||||||||
| Within-laboratory Precision 5 | 10 | 99.64 | 2.92 | 85.95 | 2.03 | 84.96 | 2.76 | |||||||||
| Measurement Precision | 25 | 99.95 | 1.22 | 87.52 | 0.83 | 87.01 | 1.59 | |||||||||
| LOD | 10 | 0.081 | 0.232 | 0.482 | ||||||||||||
| LOQ | 10 | 0.228 | 0.571 | 1.089 | ||||||||||||
| LOD (method) | 10 | 0.040 | 0.113 | 0.235 | ||||||||||||
| LOQ (method) | 10 | 0.111 | 0.279 | 0.531 | ||||||||||||
| Saliva | THC-D3 | THC-OH | THC-COOH | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Range | Range | Range | ||||||||||||||
| No. of Samples | Mean Value [%] | CV [%] | Min [%] | - | Max [%] | Mean Value [%] | CV [%] | Min [%] | - | Max [%] | Mean Value [%] | CV [%] | Min [%] | - | Max [%] | |
| Recovery | 25 | 98.15 | 3.85 | 89.62 | - | 104.85 | 103.22 | 1.36 | 100.68 | - | 105.47 | 104.08 | 2.48 | 98.22 | - | 108.34 |
| Within-laboratory Precision 1 | 10 | 106.48 | 3.02 | 99.67 | 1.45 | 104.33 | 1.94 | |||||||||
| Within-laboratory Precision 2 | 10 | 99.27 | 2.90 | 103.31 | 1.32 | 103.38 | 2.76 | |||||||||
| Within-laboratory Precision 3 | 10 | 99.36 | 3.74 | 107.73 | 1.79 | 108.29 | 2.66 | |||||||||
| Within-laboratory Precision 4 | 10 | 103.02 | 3.97 | 106.68 | 2.22 | 104.53 | 3.12 | |||||||||
| Within-laboratory Precision 5 | 10 | 107.08 | 2.69 | 106.63 | 2.69 | 108.62 | 2.70 | |||||||||
| Measurement Precision | 25 | 100.45 | 0.50 | 101.62 | 0.41 | 100.59 | 0.78 | |||||||||
| LOD | 10 | 0.068 | 0.596 | 1.712 | ||||||||||||
| LOQ | 10 | 0.149 | 1.405 | 4.086 | ||||||||||||
| LOD (method) | 10 | 0.037 | 0.328 | 0.942 | ||||||||||||
| LOQ (method) | 10 | 0.082 | 0.773 | 2.247 | ||||||||||||
| Serum | Urine | Oral Fluid | |||||
|---|---|---|---|---|---|---|---|
| Lab Technician | Biomek | Lab Technician | Biomek | Lab Technician | Biomek | Unit | |
| No. of samples per run | 96 | 96 | 96 | 96 | 96 | 96 | [unit] |
| Operating costs | 1,095,758.29 € | 546,602.36 € | 1,058,167.50 € | 478,749.00 € | 1,104,136.61 € | 1,107,571.18 € | [€] |
| Annual depreciation (10 years) | 0.00 € | 39,637.46 € | 0.00 € | 40,079.86 € | 0.00 € | 29,727.16 € | [€] |
| Annual imputed interest | 0.00 € | 1981.87 € | 0.00 € | 2003.99 € | 0.00 € | 1486.36 € | [€] |
| Total costs per year | 1,095,758.29 € | 588,221.69 € | 1,058,167.50 € | 520,832.85 € | 1,104,136.61 € | 1,138,784.70 € | [€] |
| Turnaround time | 5.63 | 7.20 | 5.95 | 7.09 | 4.88 | 2.43 | [h] |
| No. of samples per year | 100,800 | 72,000 | 96,000 | 72,000 | 115,200 | 192,000 | [unit] |
| Cost per sample | 10.87 € | 7.59 € | 11.02 € | 6.65 € | 9.58 € | 5.93 € | [€] |
| Net present value (NPV) | 166,056.24 € | 1,533,671.83 € | −16,585.04 € | 2,167,508.14 € | 1,247,705.42 € | 8,920,346.90 € | [€] |
| Payback period | 0 | 1.95 | never | 1.48 | 0 | 0.31 | [years] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bach, A.; Fleischer, H.; Wijayawardena, B.; Thurow, K. Automation System for the Flexible Sample Preparation for Quantification of Δ9-THC-D3, THC-OH and THC-COOH from Serum, Saliva and Urine. Appl. Sci. 2022, 12, 2838. https://doi.org/10.3390/app12062838
Bach A, Fleischer H, Wijayawardena B, Thurow K. Automation System for the Flexible Sample Preparation for Quantification of Δ9-THC-D3, THC-OH and THC-COOH from Serum, Saliva and Urine. Applied Sciences. 2022; 12(6):2838. https://doi.org/10.3390/app12062838
Chicago/Turabian StyleBach, Anna, Heidi Fleischer, Bhagya Wijayawardena, and Kerstin Thurow. 2022. "Automation System for the Flexible Sample Preparation for Quantification of Δ9-THC-D3, THC-OH and THC-COOH from Serum, Saliva and Urine" Applied Sciences 12, no. 6: 2838. https://doi.org/10.3390/app12062838
APA StyleBach, A., Fleischer, H., Wijayawardena, B., & Thurow, K. (2022). Automation System for the Flexible Sample Preparation for Quantification of Δ9-THC-D3, THC-OH and THC-COOH from Serum, Saliva and Urine. Applied Sciences, 12(6), 2838. https://doi.org/10.3390/app12062838