
Recognition of mRNA N4 Acetylcytidine (ac4C) by Using Non-Deep vs. Deep Learning

 ,
,  ,
,  ,
, 
 , , and
, , and
Abstract
:1. Introduction
2. Background
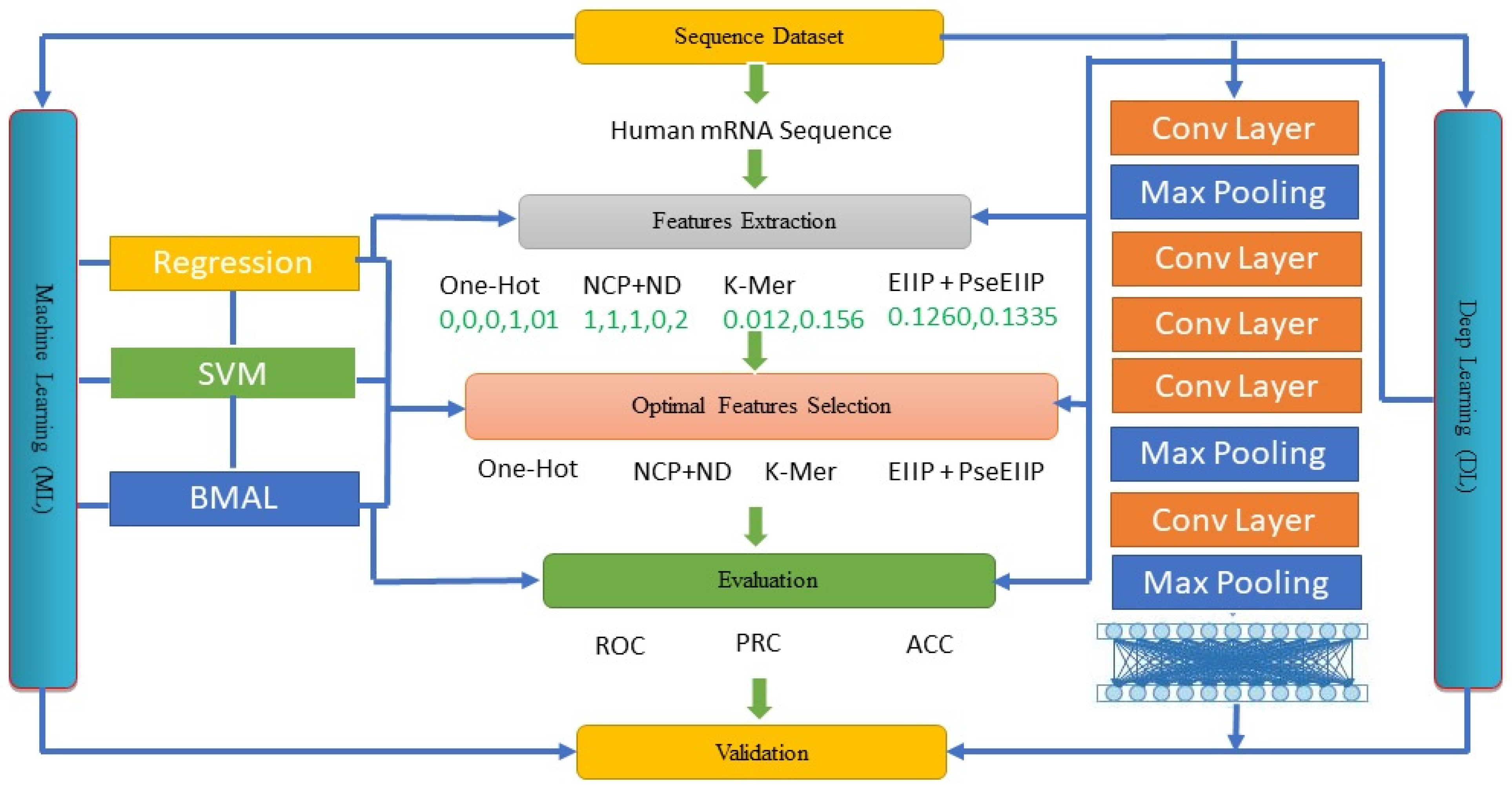
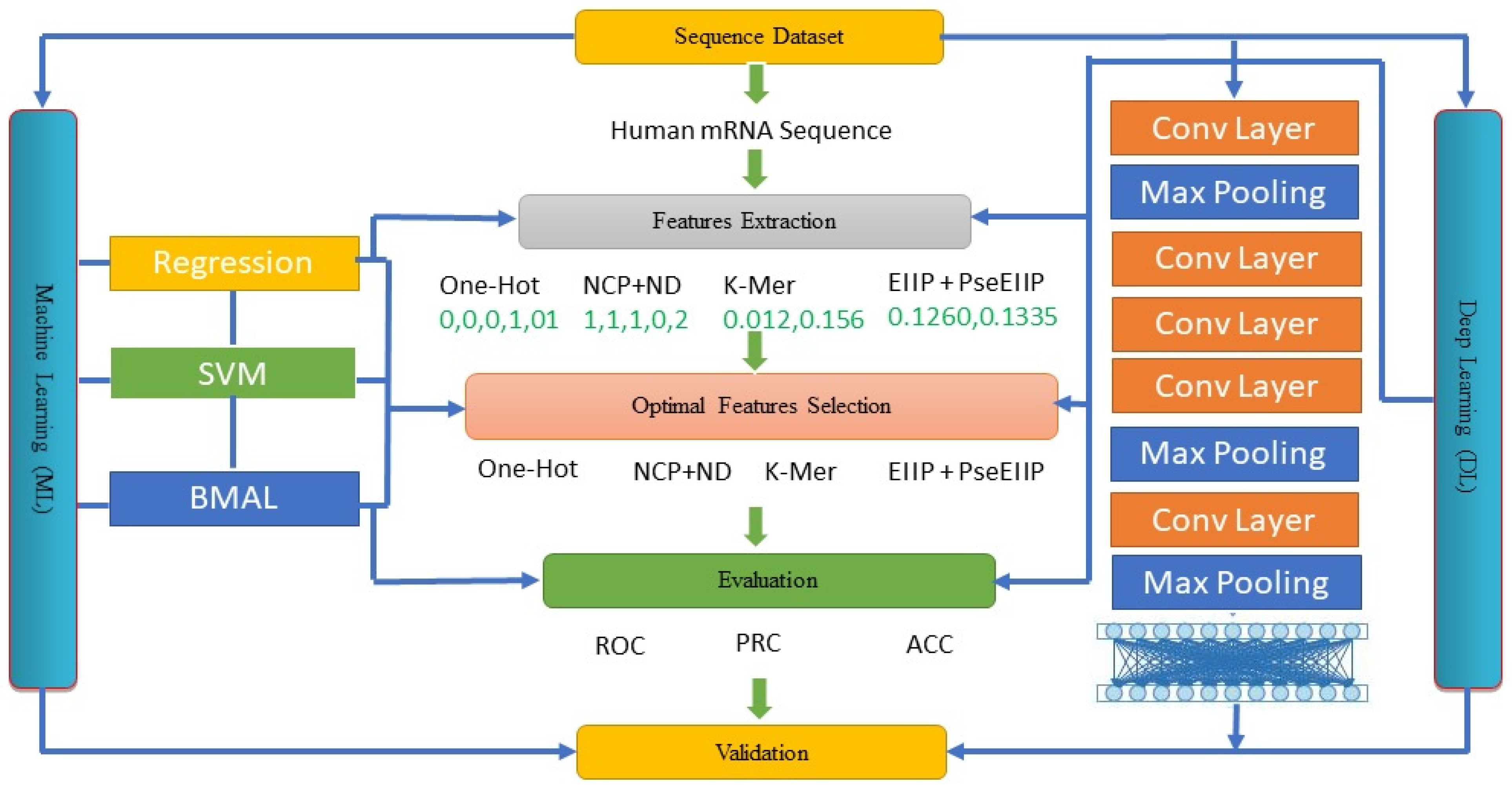
3. Materials and Methods
3.1. Regression
3.2. SVM
3.3. Bayesian MAML (BMAML)
3.4. DL-ac4C
3.5. The ac4C Deep Learning Model (ac4C-DL)
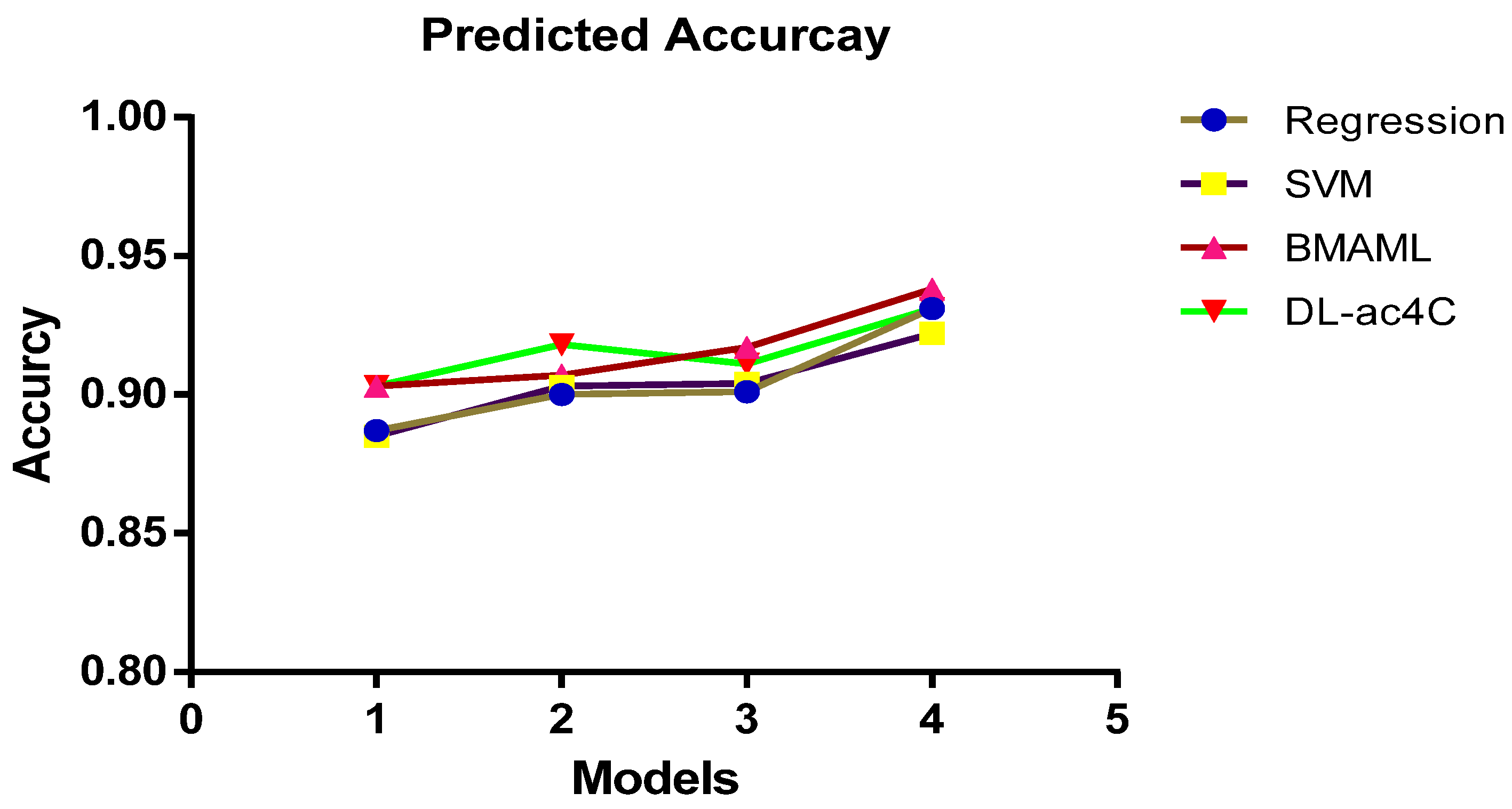
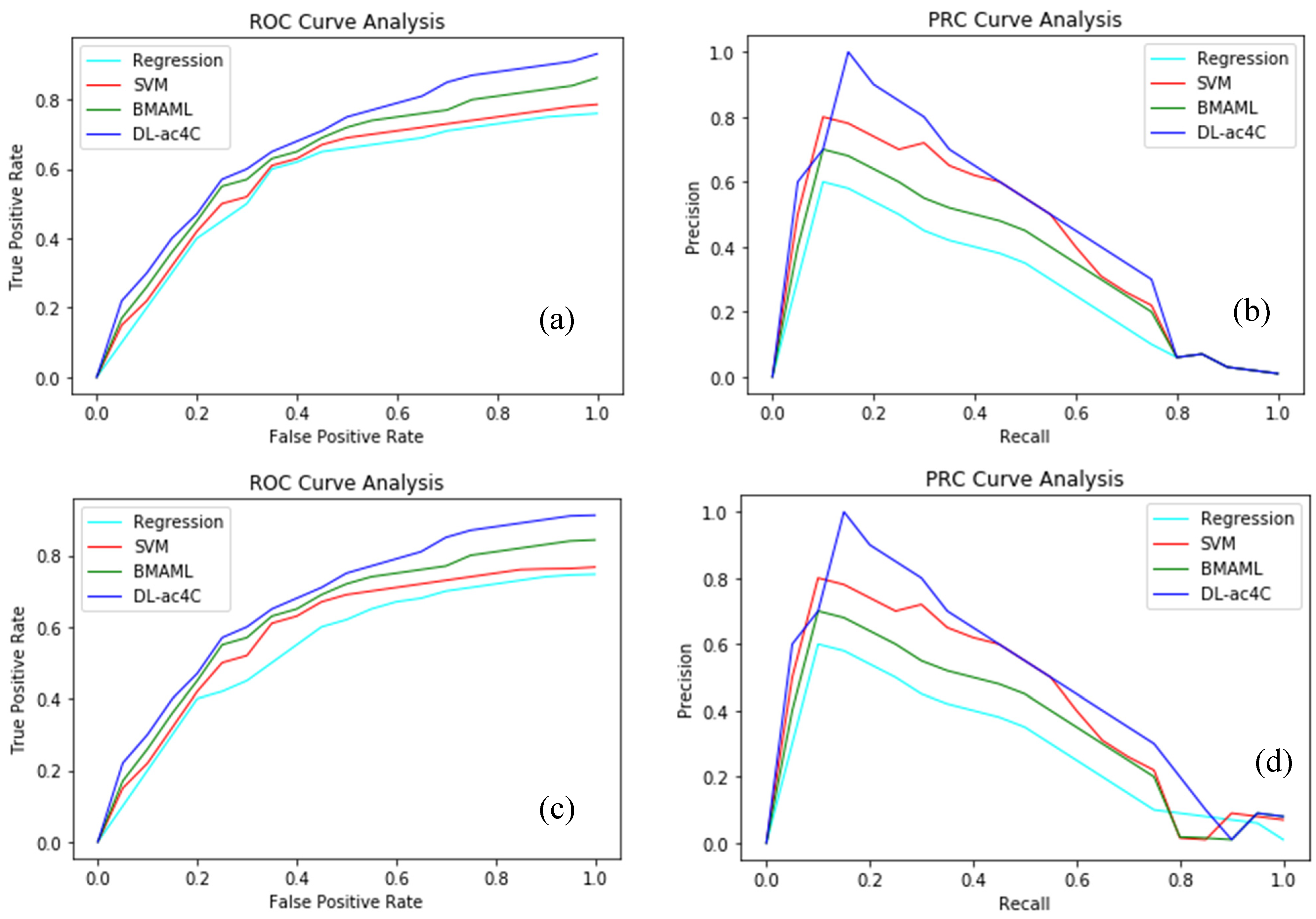
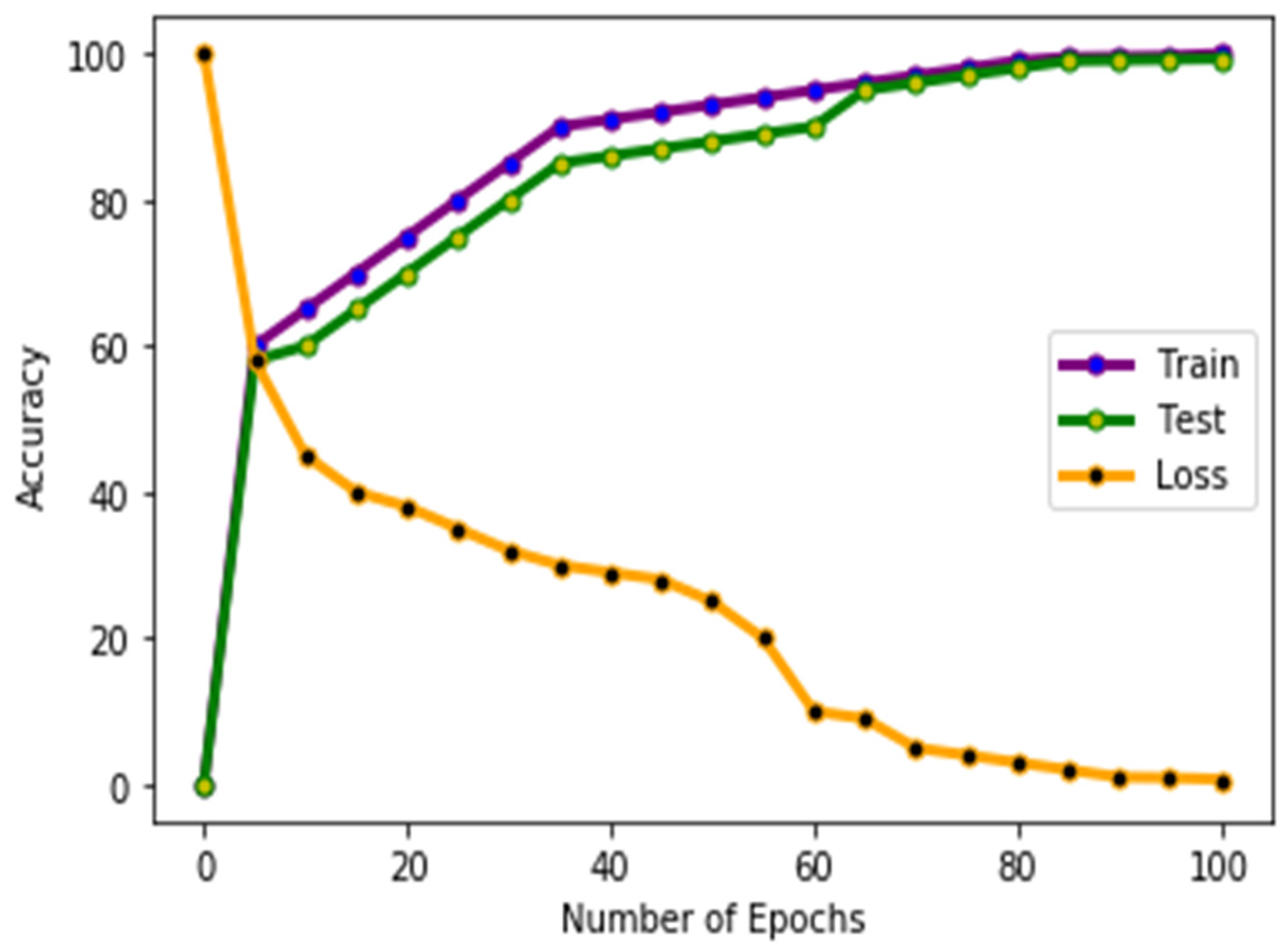
4. Results
4.1. Experiments and Results
4.2. Data Source and Data Preprocessing
4.3. Performance Metrics
5. Discussions
6. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yoon, J.; Kim, T.; Dia, O.; Kim, S.; Bengio, Y.; Ahn, S. Bayesian Model Agnostic Meta-Learning. In Proceedings of the 32nd International Conference on Neural Information Processing Systems, NIPS’18, Montreal, QC, Canada, 3–8 December 2018; pp. 7332–7342. [Google Scholar]
- Liu, Q.; Wang, D. Stein Variational Gradient Descent: A General-Purpose Bayesian Inference Algorithm. In Proceedings of the 30th International Conference on Neural Information Processing Systems, NIPS’16, Barcelona, Spain, 5–10 December 2016; pp. 2378–2386. [Google Scholar]
- Boccaletto, P.; Machnicka, M.A.; Purta, E.; Piątkowski, P.; Bagiński, B.; Wirecki, T.K.; Bujnicki, J.M. MODOMICS: A database of RNA modification pathways. Nucleic Acids Res. 2018, 46, D303–D307. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Langhendries, J.L.; Watzinger, P.; Kötter, P.; Entian, K.D.; Lafontaine, D.L. Yeast kre33 and human nat10 are conserved 18s rrna cytosine acetyltransferases that modify trnas assisted by the adaptor tan1/thumpd1. Nucleic Acids Res. 2015, 43, 2242–2258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arango, D.; Sturgill, D.; Alhusaini, N.; Dillman, A.A.; Sweet, T.J.; Hanson, G.; Hosogane, M.; Sinclair, W.R.; Nanan, K.K.; Mandler, M.D.; et al. Acetylation of cytidine in mrna promotes translation efciency. Cell 2018, 175, 1872–1886. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Zhou, Y.; Cui, Q.; Zhou, Y. PACES: Prediction of N4-acetylcytidine (ac4C) modification sites in mRNA. Sci. Rep. 2019, 9, 11112. [Google Scholar] [CrossRef] [Green Version]
- Tahir, M.; Hayat, M. iNuc-STNC: A sequence-based predictor for identification of nucleosome positioning in genomes by extending the concept of SAAC and Chou’s PseAAC. Mol. BioSyst. 2016, 12, 2587–2593. [Google Scholar] [CrossRef]
- Hayat, M.; Tahir, M. Psdentification: Identifcation of transmembrane helix segments using ensemble feature space by incorporated fuzzy support vector machine. Mol. BioSyst. 2015, 11, 2255–2262. [Google Scholar] [CrossRef] [PubMed]
- Tahir, M.; Hayat, M.; Chong, K.T. Prediction of n6-methyladenosine sites using convolution neural network model based on distributed feature representations. Neural Netw. 2020, 129, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Tayara, H.; Oubounyt, M.; Chong, K.T. Identifcation of promoters and their strength using deep learning. IBRO Rep. 2019, 6, S552–S553. [Google Scholar] [CrossRef]
- Tahir, M.; Hayat, M.; Ullah, I.; Chong, K.T. A deep learning-based computational approach for discrimination of dna n6-methyladenosine sites by fusing heterogeneous features. Chemomet. Intell. Lab. Syst. 2020, 206, 104151. [Google Scholar] [CrossRef]
- Chicco, D. Ten Quick tips for machine learning in computational biology. BioData Mining 2017, 10, 35. [Google Scholar] [CrossRef]
- Alam, W.; Tayara, H.; Chong, K.T. i4mC-Deep: An Intelligent Predictor of N4-Methylcytosine Sites Using a Deep Learning Approach with Chemical Properties. Genes 2021, 12, 1117. [Google Scholar] [CrossRef] [PubMed]
- Manyika, J.; Chui, M.; Brown, B.; Bughin, J.; Dobbs, R.; Roxburgh, C.; Hung Byers, A. Big Data: The Next Frontier for Innovation, Competition, and Productivity; McKinsey Global Institute: Washington, DC, USA, 2011. [Google Scholar]
- Ferrucci, D.; Brown, E.; Chu-Carroll, J.; Fan, J.; Gondek, D.; Kalyanpur, A.A.; Lally, A.; Murdock, J.W.; Nyberg, E.; Prager, J.; et al. Building Watson: An overview of the DeepQA project. AI Mag. 2010, 31, 59–79. [Google Scholar] [CrossRef] [Green Version]
- IBM and Oncology. Available online: https://www.ibm.com/watson-health/solutions/cancer-research-treatment (accessed on 3 January 2022).
- Silver, D.; Huang, A.; Maddison, C.J.; Guez, A.; Sifre, L.; Van Den Driessche, G.; Schrittwieser, J.; Antonoglou, I.; Panneershelvam, V.; Lanctot, M.; et al. Mastering the game of Go with deep neural networks and tree search. Nature 2016, 529, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Powles, J.; Hodson, H. Google DeepMind and healthcare in an age of algorithms. Health Technol. 2017, 7, 351–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, M.S.; Ahmad, I.; Bin, L.; Khan, S.; Rodrigues, J.J. Deep learning recognition of diseased and normal cell representation. Trans. Emerg. Telecommun. Technol. 2020, 32, e4017. [Google Scholar] [CrossRef]
- Iqbal, M.S.; Luo, B.; Mehmood, R.; Alrige, M.A.; Alharbey, R. Mitochondrial Organelle Movement Classification (Fission and Fusion) via Convolutional Neural Network Approach. IEEE Access 2019, 7, 86570–86577. [Google Scholar] [CrossRef]
- Iqbal, M.S.; Khan, T.; Hussain, S.; Mahmood, R.; El-Ashram, S.; Abbasi, R.; Luo, B. Cell Recognition of Microscopy Images of TPEF (Two Photon Excited Florescence) Probes. Procedia Comput. Sci. 2019, 147, 77–83. [Google Scholar] [CrossRef]
- Iqbal, M.S.; El-Ashram, S.; Hussain, S.; Khan, T.; Huang, S.; Mehmood, R.; Luo, B. Efficient cell classification of mitochondrial images by using deep learning. J. Opt. 2019, 48, 113–122. [Google Scholar] [CrossRef]
- Larrañaga, P.; Calvo, B.; Santana, R.; Bielza, C.; Galdiano, J.; Inza, I.; Lozano, J.A.; Armananzas, R.; Santafé, G.; Pérez, A.; et al. Machine learning in bioinformatics. Brief. Bioinform. 2006, 7, 86–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeCun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Jones, D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef] [Green Version]
- Ponomarenko, J.V.; Ponomarenko, M.P.; Frolov, A.S.; Vorobyev, D.G.; Overton, G.C.; Kolchanov, N.A. Conformational and physicochemical DNA features specific for transcription factor binding sites. Bioinformatics 1999, 15, 654–668. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.-D.; Lin, S.L. Support vector machines for predicting rRNA-, RNA-, and DNA-binding proteins from amino acid sequence. Biochim. Biophys. Acta BBA-Proteins Proteom. 2003, 1648, 127–133. [Google Scholar] [CrossRef]
- Atchley, W.R.; Zhao, J.; Fernandes, A.D.; Drüke, T. Solving the protein sequence metric problem. Proc. Natl. Acad. Sci. USA 2005, 102, 6395–6400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branden, C.I. Introduction to Protein Structure; Garland Science: New York, NY, USA, 1999. [Google Scholar]
- Richardson, J.S. The anatomy and taxonomy of protein structure. Adv. Protein Chem. 1981, 34, 167–339. [Google Scholar]
- Lyons, J.; Dehzangi, A.; Heffernan, R.; Sharma, A.; Paliwal, K.; Sattar, A.; Zhou, Y.; Yang, Y. Predicting backbone Cα angles and dihedrals from protein sequences by stacked sparse auto-encoder deep neural network. J. Comput. Chem. 2014, 35, 2040–2046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heffernan, R.; Paliwal, K.; Lyons, J.; Dehzangi, A.; Sharma, A.; Wang, J.; Sattar, A.; Yang, Y.; Zhou, Y. Improving prediction of secondary structure, local backbone angles, and solvent accessible surface area of proteins by iterative deep learning. Sci. Rep. 2015, 5, 11476. [Google Scholar] [CrossRef] [Green Version]
- Spencer, M.; Eickholt, J.; Cheng, J. A Deep Learning Network Approach to ab initio Protein Secondary Structure Prediction. Computational Biology and Bioinformatics. IEEE/ACM Trans. Comput. Biol. Bioinform. 2015, 12, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, S.P.; Shang, Y.; Xu, D. DL-PRO: A novel deep learning method for protein model quality assessment. In Proceedings of the 2014 International Joint Conference on Neural Networks (IJCNN), Beijing, China, 6–11 July 2014; pp. 2071–2078. [Google Scholar]
- Baldi, P.; Brunak, S.; Frasconi, P.; Soda, G.; Pollastri, G. Exploiting the past and the future in protein secondary structure prediction. Bioinformatics 1999, 15, 937–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldi, P.; Pollastri, G.; Andersen, C.A.; Brunak, S. Matching protein beta-sheet partners by feedforward and recurrent neural networks. In Proceedings of the 2000 Conference on Intelligent Systems for Molecular Biology (ISMB00), La Jolla, CA, USA, 19–23 August 2000; pp. 25–36. [Google Scholar]
- Sønderby, S.K.; Winther, O. Protein Secondary Structure Prediction with Long Short-Term Memory Networks. arXiv 2014, arXiv:1412.7828 2014. [Google Scholar]
- Lena, P.D.; Nagata, K.; Baldi, P.F. Deep spatio-temporal architectures and learning for protein structure prediction. In Advances in Neural Information Processing Systems; Massachusetts Institute of Technology Press: Cambridge, MA, USA, 2012; pp. 512–520. [Google Scholar]
- Lena, P.D.; Nagata, K.; Baldi, P. Deep architectures for protein contact map prediction. Bioinformatics 2012, 28, 2449–2457. [Google Scholar] [CrossRef] [Green Version]
- Baldi, P.; Pollastri, G. The principled design of large-scale recursive neural networ—Architectures—Dag-rnns and the protein structure prediction problem. J. Mach. Learn. Res. 2003, 4, 575–602. [Google Scholar]
- Leung, M.K.; Xiong, H.Y.; Lee, L.J.; Frey, B.J. Deep learning of the tissue-regulated splicing code. Bioinformatics 2014, 30, i121–i129. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.; Yoon, S. Boosted Categorical Restricted Boltzmann Machine for Computational Prediction of Splice Junctions. In Proceedings of the 32nd International Conference on Machine Learning, Lille, France, 7–9 July 2015; pp. 2483–2492. [Google Scholar]
- Zhang, S.; Zhou, J.; Hu, H.; Gong, H.; Chen, L.; Cheng, C.; Zeng, J. A deep learning framework for modeling structural features of RNA-binding protein targets. Nucleic Acids Res. 2015, 44, e32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Li, Y.; Narayan, R.; Subramanian, A.; Xie, X. Gene expression inference with deep learning. Bioinformatics 2016, 32, 1832–1839. [Google Scholar] [CrossRef] [Green Version]
- Denas, O.; Taylor, J. Deep modeling of gene expression regulation in an Erythropoiesis model. In Proceedings of the International Conference on Machine Learning workshop on Representation Learning, Atlanta, GA, USA, 2–4 May 2013. [Google Scholar]
- Alipanahi, B.; Delong, A.; Weirauch, M.T.; Frey, B.J. Predicting the sequence specificities of DNAand RNA-binding proteins by deep learning. Nat. Biotechnol. 2015, 33, 831–838. [Google Scholar] [CrossRef]
- Zhou, J.; Troyanskaya, O.G. Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods 2015, 12, 931–934. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.; Lee, T.; Na, B.; Yoon, S. DNA-Level Splice Junction Prediction using Deep Recurrent Neural Networks. arXiv 2015, arXiv:1512.05135. [Google Scholar]
- Hochreiter, S.; Heusel, M.; Obermayer, K. Fast model-based protein homology detection without alignment. Bioinformatics 2007, 23, 1728–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sønderby, S.K.; Sønderby, C.K.; Nielsen, H.; Winther, O. Convolutional LSTM Networks for Subcellular Localization of Proteins. arXiv 2015, arXiv:1503.01919. [Google Scholar]
- Fakoor, R.; Ladhak, F.; Nazi, A.; Huber, M. Using deep learning to enhance cancer diagnosis and classification. In Proceedings of the International Conference on Machine Learning, Washington, DC, USA, 4–7 December 2013. [Google Scholar]
- Do, D.T.; Le, T.Q.T.; Le, N.Q.K. Using deep neural networks and biological subwords to detect protein S-sulfenylation sites. Brief. Bioinform. 2021, 22, bbaa128. [Google Scholar] [CrossRef]
- Tng, S.S.; Le, N.Q.K.; Yeh, H.Y.; Chua, M.C.H. Improved Prediction Model of Protein Lysine Crotonylation Sites Using Bidirectional Recurrent Neural Networks. J. Proteome Res. 2021, 21, 265–273. [Google Scholar] [CrossRef]
- Bin Heyat, M.B.; Akhtar, F.; Khan, M.H.; Ullah, N.; Gul, I.; Khan, H.; Lai, D. Detection, Treatment Planning, and Genetic Predisposition of Bruxism: A Systematic Mapping Process and Network Visualization Technique. CNS Neurol. Disord.-Drug Targets 2020, 20, 755–775. [Google Scholar] [CrossRef] [PubMed]
- Bin Heyat, M.B.; Lai, D.; Khan, F.I.; Zhang, Y. Sleep Bruxism Detection Using Decision Tree Method by the Combination of C4-P4 and C4-A1 Channels of Scalp EEG. IEEE Access 2019, 7, 102542–102553. [Google Scholar] [CrossRef]
- Bin Heyat, M.B.; Akhtar, F.; Khan, A.; Noor, A.; Benjdira, B.; Qamar, Y.; Abbas, S.J.; Lai, D. A Novel Hybrid Machine Learning Classification for the Detection of Bruxism Patients Using Physiological Signals. Appl. Sci. 2020, 10, 7410. [Google Scholar] [CrossRef]
- Khan, H.; Bin Heyat, M.B.; Lai, D.; Akhtar, F.; Ansari, M.A.; Khan, A.; Alkahtani, F. Progress in Detection of Insomnia Sleep Disorder: A Comprehensive Review. Current Drug Targets 2021, 22, 672–684. [Google Scholar] [CrossRef]
- Abbasi, R.; Xu, L.; Wang, Z.; Chughtai, G.R.; Amin, F.; Luo, B. Dynamic weighted histogram equalization for contrast enhancement using for Cancer Progression Detection in medical imaging. In Proceedings of the 2018 International Conference on Signal Processing and Machine Learning, Shanghai, China, 28–30 November 2018; pp. 93–98. [Google Scholar]
- Abbasi, R.; Chen, J.; Al-Otaibi, Y.; Rehman, A.; Abbas, A.; Cui, W. RDH-based dynamic weighted histogram equalization using for secure transmission and cancer prediction. Multimed. Syst. 2021, 27, 177–189. [Google Scholar] [CrossRef]
- Khan, A.R.; Khan, S.; Harouni, M.; Abbasi, R.; Iqbal, S.; Mehmood, Z. Brain tumor segmentation using K-means clustering and deep learning with synthetic data augmentation for classification. Microsc. Res. Technol. 2021, 84, 1389–1399. [Google Scholar] [CrossRef]
- Alam, W.; Tayara, H.; Chong, K.T. XG-ac4C: Identification of N4-acetylcytidine (ac4C) in mRNA using eXtreme gradient boosting with electron-ion interaction pseudopotentials. Sci. Rep. 2020, 10, 20942. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Numbers | Types | In_Put Size | Out_Put Size |
|---|---|---|---|
| 1 | INPUT LAYER | … | 134 × 4 |
| 2 | CONV LAYER | 16 × 4 × 4 | 131 × 16 |
| 3 | RELU LAYER | .. | 131 × 16 |
| 4 | DROPOUT LAYER | .. | 131 × 16 |
| 5 | CONV LAYER | 16 × 4 × 4 | 128 × 64 |
| 6 | RELU LAYER | .. | 128 × 64 |
| 7 | POOL LAYER | 4 × 2 | 64 × 64 |
| 8 | BDLSTM LAYER | 64 × 4 × 6 | 128 × 64 |
| 9 | RELU LAYER | .. | 128 × 64 |
| 10 | FULLY CONNECTED LAYER | 128 | 128 |
| 11 | DROPOUT LAYER | .. | 128 × 64 |
| 12 | FULLY CONNECTED LAYER | One | One |
| 13 | SIGMOID LAYER | One | One |
| Classifier | Features | ROC | PRC | ACC |
|---|---|---|---|---|
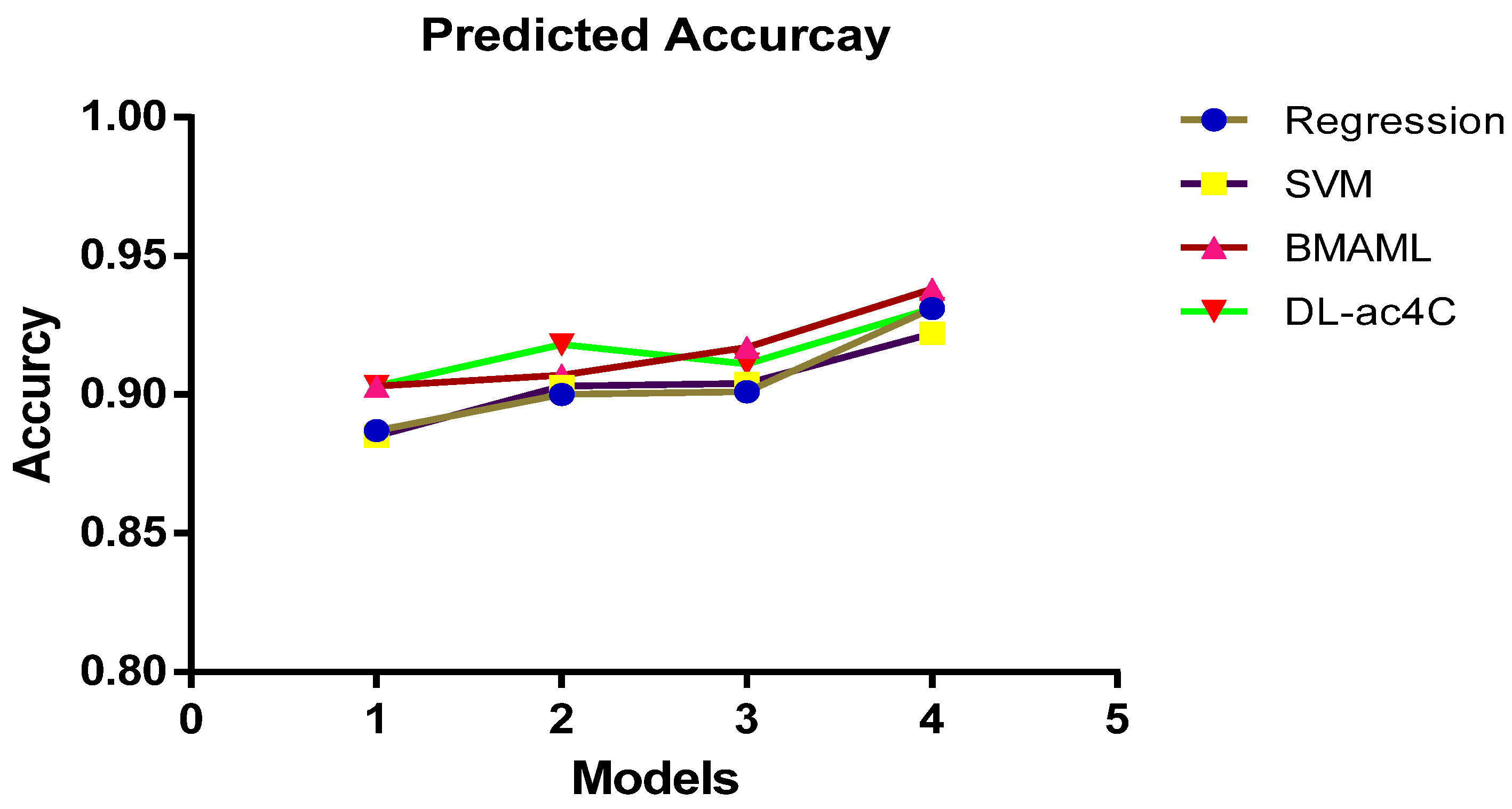
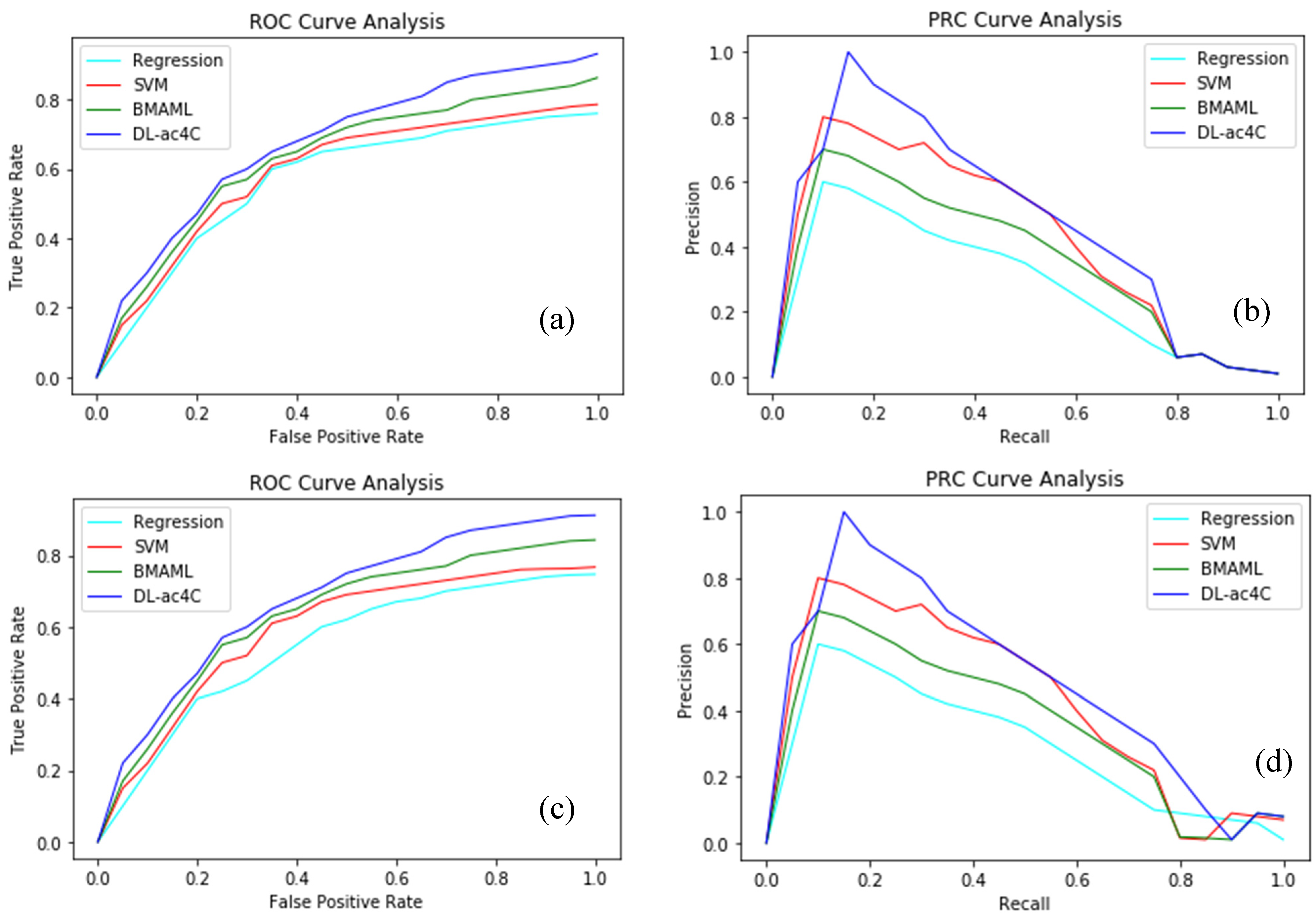
| Regression | One-Hot | 0.812 | 0.381 | 0.887 |
| NCP + ND | 0.976 | 0.392 | 0.885 | |
| K-mer | 0.842 | 0.274 | 0.903 | |
| EIIP + PseEIIP | 0.780 | 0.354 | 0.903 | |
| SVM | One-Hot | 0.784 | 0.361 | 0.900 |
| NCP+ND | 0.821 | 0.384 | 0.903 | |
| K-mer | 0.847 | 0.429 | 0.907 | |
| EIIP + PseEIIP | 0.849 | 0.527 | 0.918 | |
| BMAML | One-Hot | 0.787 | 0.364 | 0.901 |
| NCP+ND | 0.799 | 0.348 | 0.904 | |
| K-mer | 0.847 | 0.502 | 0.917 | |
| EIIP + PseEIIP | 0.863 | 0.514 | 0.911 | |
| DL-ac4C | One-Hot | 0.881 | 0.569 | 0.931 |
| NCP+ND | 0.914 | 0.606 | 0.922 | |
| K-mer | 0.901 | 0.559 | 0.938 | |
| EIIP + PseEIIP | 0.932 | 0.663 | 0.931 |
| Dataset | Methods | ROC | PRC |
|---|---|---|---|
| Cross Validation | PACES | 0.885 | 0.559 |
| XG-acc4C | 0.91 | 0.653 | |
| DL-acc4C | 0.93 | 0.673 | |
| Independent | PACES | 0.874 | 0.485 |
| XG-acc4C | 0.889 | 0.581 | |
| DL-acc4C | 0.912 | 0.621 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iqbal, M.S.; Abbasi, R.; Bin Heyat, M.B.; Akhtar, F.; Abdelgeliel, A.S.; Albogami, S.; Fayad, E.; Iqbal, M.A. Recognition of mRNA N4 Acetylcytidine (ac4C) by Using Non-Deep vs. Deep Learning. Appl. Sci. 2022, 12, 1344. https://doi.org/10.3390/app12031344
Iqbal MS, Abbasi R, Bin Heyat MB, Akhtar F, Abdelgeliel AS, Albogami S, Fayad E, Iqbal MA. Recognition of mRNA N4 Acetylcytidine (ac4C) by Using Non-Deep vs. Deep Learning. Applied Sciences. 2022; 12(3):1344. https://doi.org/10.3390/app12031344
Chicago/Turabian StyleIqbal, Muhammad Shahid, Rashid Abbasi, Md Belal Bin Heyat, Faijan Akhtar, Asmaa Sayed Abdelgeliel, Sarah Albogami, Eman Fayad, and Muhammad Atif Iqbal. 2022. "Recognition of mRNA N4 Acetylcytidine (ac4C) by Using Non-Deep vs. Deep Learning" Applied Sciences 12, no. 3: 1344. https://doi.org/10.3390/app12031344
APA StyleIqbal, M. S., Abbasi, R., Bin Heyat, M. B., Akhtar, F., Abdelgeliel, A. S., Albogami, S., Fayad, E., & Iqbal, M. A. (2022). Recognition of mRNA N4 Acetylcytidine (ac4C) by Using Non-Deep vs. Deep Learning. Applied Sciences, 12(3), 1344. https://doi.org/10.3390/app12031344